

Abstract
Among the μ-conotoxins that block vertebrate voltage-gated sodium channels (VGSCs), some have been shown to be potent analgesics following systemic administration in mice. We have determined the solution structure of a new representative of this family, μ-BuIIIB, and established its disulfide connectivities by direct mass spectrometric collision induced dissociation fragmentation of the peptide with disulfides intact. The major oxidative folding product adopts a 1-4/2-5/3-6 pattern with the following disulfide bridges: Cys5-Cys17, Cys6-Cys23 and Cys13-Cys24. The solution structure reveals that the unique N-terminal extension in μ-BuIIIB, which is also present in μ-BuIIIA and μ-BuIIIC but absent in other μ-conotoxins, forms part of a short α-helix encompassing Glu3 to Asn8. This helix is packed against the rest of the toxin and stabilized by the Cys5-Cys17 and Cys6-Cys23 disulfide bonds. As such, the side chain of Val1 is located close to the aromatic rings of Trp16 and His20, which are located on the canonical helix that displays several residues found to be essential for VGSC blockade in related μ-conotoxins. Mutations of residues 2 and 3 in the N-terminal extension enhanced the potency of μ-BuIIIB for NaV1.3. One analog, [d-Ala2]BuIIIB, showed a 40-fold increase, making it the most potent peptide blocker of this channel characterized to date and thus a useful new tool with which to characterize this channel. Based on previous results for related μ-conotoxins, the dramatic effects of mutations at the N-terminus were unanticipated, and suggest that further gains in potency might be achieved by additional modifications of this region.
Cone snails elaborate a variety of peptide toxins that have proven to be valuable probes of ion channel function.(1-3) In particular, the μ-conotoxin family targets voltage-gated sodium channels (VGSCs), binding to the extracellular side of the pore (site 1) and preventing the passage of sodium ions.(4, 5) In mammals, nine different VGSC subtypes of the α-subunit (NaV1.1-1.9) have been identified, each with different distributions in the body.(4) Of particular interest are the neuronal subtypes implicated in the perception of pain (6, 7) as modulators of these subtypes may have potential therapeutic use as analgesics.(7, 8)
Of the nine α-subunits found in mammals, six bind tetrodotoxin (TTX) with high affinity (IC50 in the nM range), and three, NaV1.5, NaV1.8, and NaV1.9, are classified as TTX-resistant (IC50 > 1 μM). The first μ-conotoxin characterized, μ-GIIIA from Conus geographus, targets mainly the TTX-sensitive skeletal muscle subtype NaV1.4.(9, 10) μ-PIIIA also has a strong preference for NaV1.4, but can block other TTX-sensitive subtypes as well, albeit with lower affinities.(11-13) More recently, a group of μ-conotoxins was discovered that targets neuronal sodium channels with greater selectivity and potency than μ-GIIIA or μ-PIIIA.(14-18) This group includes μ-SmIIIA, which irreversibly inhibited TTX-resistant sodium currents in amphibian sympathetic and dorsal root ganglion (DRG) neurons,(14) as well as μ-KIIIA and μ-SIIIA.(16, 19) The selectivity of these and several other μ-conotoxins for NaV1.1 through NaV1.8 has recently been described in detail, and combinations of these peptides could be used to define NaV1 contributions to action potentials in sciatic nerve.(20)
The μ-conotoxins also have potential in the treatment of chronic pain. Micromolar concentrations of μ-SIIIA slowly blocked TTX-sensitive currents in mouse DRG neurons, and 1 μM of the peptide almost completely blocked A-compound action potentials in mouse sciatic nerve preparations after exposure for an hour.(21) Moreover, μ-SIIIA significantly reduced the acute and the inflammatory pain responses in the formalin assay in mice at doses < 1 mg/kg following intraperitoneal administration.(21) μ-KIIIA blocked several subtypes of mammalian neuronal VGSCs and displayed potent analgesic activity following its systemic administration in mice.(17) Studies of structure-activity relationships in μ-KIIIA (17, 22) identified key residues important for its activity on the mammalian neuronal NaV1.2 and skeletal muscle NaV1.4 subtypes, and demonstrated that the engineering of μ-KIIIA could provide subtype-selective therapeutics against mammalian VGSCs for the potential treatment of pain.
Much of the focus on VGSCs as analgesic targets has been on NaV1.7, although other subtypes such as NaV1.3 and NaV1.8 are also potentially important targets.(7, 8) In this context, the recently isolated μ-conotoxin, μ-BuIIIB,(23) is of interest as it is one of the more potent blockers of NaV1.3 among the 11 tested by Wilson et al.(20) As a basis for understanding this potency, we have determined the solution structure of μ-BuIIIB and compared it with those of other μ-conotoxins. We also mapped the disulfide connectivities by direct mass spectrometric collision induced dissociation fragmentation. Unexpectedly, the N-terminal region of μ-BuIIIB (Figure 1) adopts an ordered structure that, moreover, interacts with functionally important residues on the helical C-terminal region. To probe the importance of the N-terminal region we have constructed a number of synthetic analogs, one of which [d-Ala2]BuIIIB shows dramatically enhanced NaV1.3 blocking activity and, aside from the guanidinium alkaloids tetrodotoxin and saxitoxin,(24) represents the most potent blocker of this channel described to date.
Figure 1.

(A) Amino acid sequences of μ-conotoxins. Disulfide connectivities are indicated. Z: pyroglutamate; O: 4-Hydroxyproline; #: C-terminal amidation. * for μ-KIIIA indicates that the disulfide pattern differs from that determined here for μ-BuIIIB, with the major product from oxidative refolding in vitro adopting a 1-5/2-4/3-6 pattern rather than the 1-4/2-5/3-6 pattern for μ-BuIIIB.(38) ** for μ-SIIIA and μ-SmIIIA indicates that the disulfide patterns for the major products from oxidative refolding in vitro have not been determined experimentally as yet. (B) Amino acid sequences of μ-BuIIIB and [d-Ala2]BuIIIB for this study. The d-Ala substitution is in bold and underlined.
Results and Discussion
Determination of Disulfide Connectivity in μ-BuIIIB
The cysteine pairing of μ-BuIIIB was determined through direct mass spectrometric collision induced dissociation (CID) fragmentation of the intact peptide. This method, described elsewhere,(25) relies on the modes of backbone and disulfide fragmentation under positive ion CID conditions, a procedure that does not require any prior chemical steps of partial reductions of disulfide bonds. The fragmentation data generated under these conditions are analyzed through a custom built program DisConnect.(26) In μ-BuIIIB, presence of a large number of tryptic sites encourages a proteolysis step to cleave some of the peptide bonds prior to mass spectrometric fragmentation. Upon trypsin digestion a peptide fragment with mass of 1423.6Da was obtained (Inset to Figure 2A), corresponding to a peptide in which three tetra-peptide segments (VGER, NGKR and DHSR) have been excised by trypsin cleavage at the R-C5, K-N, R-G, R-D and R-C23 peptide bonds. In addition, a proteolytic cleavage at the R-W bond was also observed. This meant that the tryptic peptide consisted of four individual peptide chains held together by three disulfide bonds, as shown in Figure 2D. Figure 2A shows the MS2 CID fragmentation spectrum of the doubly-charged species of this tryptic peptide. All the structures were obtained by directly querying the m/z and assigned charge state values against the native sequence of μ-BuIIIB, through DisConnect. The structures of the key ions, as obtained by DisConnect, are shown in Figure 2D. All the experimental masses, rounded off to first decimal place, are within 0.1 Da of the theoretical values of the obtained structures; indeed, in determining the structures of the experimentally obtained ions through DisConnect, even with a window range as high as 0.5 Da, the assigned structures were the unique hits for each of the experimental values. Direct determination of the connectivity pattern was achieved by the MS3 fragmentation of two MS2 ions. Figure 2B shows the CID MS3 fragmentation spectrum of 645.3. The structures of the obtained product ions are shown in Figure 2D. Among these product ions, the structure of 510.2 directly establishes the C13-C24 connectivity. To resolve the remaining connectivity patterns we turned to the CID MS3 spectrum of 995.3, as shown in Figure 2C. The structures of the obtained MS3 product ions, as derived through DisConnect, are shown in Figure 2D. The structure of 860.3 contains three peptides chains held by two disulfide bonds between four cysteine residues (C6, C13, C23 and C24). Since a connectivity between C13-C24 is already established, the other disulfide bond in 860.3 must be between C6-C23. Thus the overall connectivity of μ-BuIIIB is C5-C17/C6-C23/C13-C24.
Figure 2.
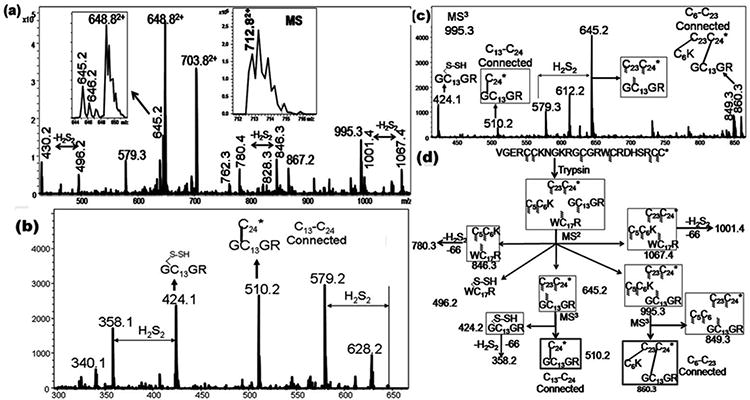
(A) CID MS2 spectra of the doubly charged species (712.8, (M+2H)2+) of the tryptic peptide of μ‐BuIIIB. Inset shows the MS of the precursor ion. (B) CID MS3 spectra of 645.5. (C) CID MS3 spectra of 995.5. (D) Assignments of the key MSn fragment ions of tryptic Bu-IIIB.The m/z values of each of the ions are indicated against the respective structures and they correspond to the singly charged values, unless otherwise specified. For every structure, Cys residues with indeterminate connectivity are indicated with the wavy lines. Subsequently, the structures from which a particular Cys connectivity is evident, the connected Cys residues are joined through a line. In this case, while the C13‐C24 connectivity is established by the MS3 ion 510.2, the ion 860.3 confirms the C6‐C23 connectivity.
NMR Spectroscopy
Good quality spectra were obtained for both μ-BuIIIB and [d-Ala2]BuIIIB. Broad NH chemical shift dispersion for both peptides indicated that they were well structured and adopted a single major conformation in solution. The amide and aromatic regions of 1D spectra of μ-BuIIIB at different temperatures are shown in Supplementary Figure S2A and complete 1D spectra of [Ala2]BuIIIB and [d-Ala2]BuIIIB are shown in Supplementary Figure S2B-D. Chemical shift assignments and structural constraints have been deposited in the BioMagResBank (27) with accession numbers 18203 and 18206 for μ-BuIIIB and [d-Ala2]BuIIIB, respectively. Distance constraints were obtained from the intensities of the NOE cross-peaks at 5 °C and pH 5.5 for both μ-BuIIIB and [d-Ala2]BuIIIB, with a NOESY spectrum of μ-BuIIIB at 15 °C being used to resolve peak overlap. NOE statistics and connectivities plotted as a function of residue number are shown in Supplementary Figure S3.
Solution Structures
Table 1 summarizes the experimental constraints and structural statistics for μ-BuIIIB and [d-Ala2]BuIIIB. The structures fit well with experimentally derived distance and angle constraints, and are well-defined over the entire length of the peptide. The angular order parameters for φ and ψ angles in the final ensemble of 20 structures for μ-BuIIIB were >0.8 except for the φ angle of Gly2 (Supplementary Figure S4), indicating that backbone dihedral angles are well-defined across the family of structures. The mean pairwise RMSD of backbone heavy atoms over all residues for the families of μ-BuIIIB and [d-Ala2]BuIIIB were 0.54 and 0.59 Å, respectively. The ensemble of the μ-BuIIIB structures is shown in Figure 3A, and the closest-to-average structure is shown in Figure 3B. The final structures have been deposited in the Protein Data Bank (PDB)(28) under ID codes 2LO9 and 2LOC for μ-BuIIIB and [d-Ala2]BuIIIB, respectively.
Table 1. Structural statistics for μ-BuIIIB and [d-Ala2]BuIIIB.
| μ-BuIIIB | [d-Ala2]BuIIIB | |
|---|---|---|
| Number of distance constraints | 446 | 422 |
| Intra-residue (i = j) | 210 | 203 |
| Sequential (|i – j| = 1) | 115 | 108 |
| Short (1< |i – j| < 6) | 77 | 70 |
| Long | 44 | 41 |
| Number of dihedral constraints | 34 | 30 |
|
| ||
| Energy (kcal/mol) a | ||
| ENOE | 13.5 ± 1.1 | 12.4 ± 0.7 |
|
| ||
| Deviations from ideal geometry b | ||
| Bonds (Å) | 0.0048 ± 0.0002 | 0.0049 ± 0.0002 |
| Angles (deg) | 0.6955 ± 0.0205 | 0.7201 ± 0.0372 |
| Impropers (deg) | 0.5855 ± 0.0410 | 0.6005 ± 0.0549 |
|
| ||
| Mean global rmsd (Å) c | ||
| Backbone heavy atoms (N, Cα, C′) | 0.54 ± 0.10 | 0.59 ± 0.13 |
| All heavy atoms | 1.62 ± 0.24 | 1.48 ± 0.22 |
|
| ||
| Ramachandran plot d | ||
| Most favored (%) | 85.6 | 88.6 |
| Allowed (%) | 14.4 | 11.4 |
| Additionally allowed (%) | 0 | 0 |
| Disallowed (%) | 0 | 0 |
The values for ENOE are calculated from a square well potential with force constants of 50 kcal mol-1 Å2.
The values for the bonds, angles, and impropers show the deviations from ideal values based on perfect stereochemistry.
Mean pairwise RMSD over all residues calculated in MOLMOL.
As determined by the program PROCHECK-NMR for all residues except Gly in BuIIIB, and except D-Ala2 and Gly in [d-Ala2]BuIIIB.
Figure 3.
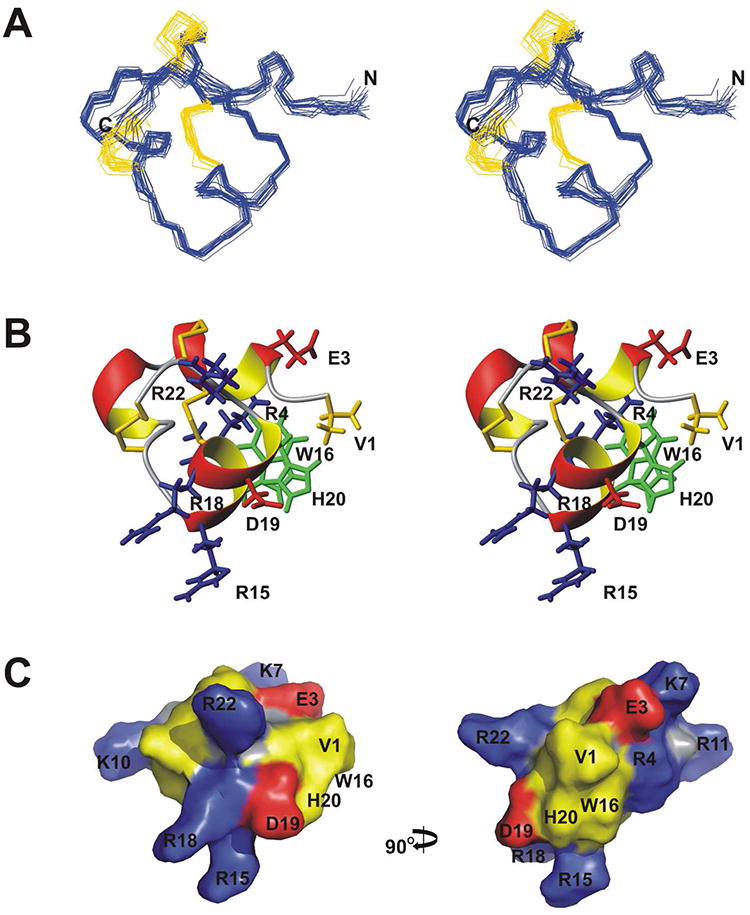
μ-BuIIIB Structure. (A) Stereo views of the family of 20 final structures, superimposed over backbone heavy atoms. Disulfide bonds are colored gold. (B) Stereo ribbon views of the closest-to-average structure. Disulfide bonds are colored gold. Side chains of selective residues are shown and labeled. (C) Surface representation of μ-BuIIIB, with Arg and Lys residues colored blue, Asp and Glu residues colored red, Cys, His, Trp and Val residues colored yellow, and all other residues colored grey. Two views rotated by 90° about the vertical axis are shown.
The unique N-terminal extension in μ-BuIIIB, which is also present in μ-BuIIIA and μ-BuIIIC but absent in other μ-conotoxins (Figure 1), forms part of a short α-helix in μ-BuIIIB (residues Glu3 to Asn8). The presence of medium-range NOEs [dαN(i, i+3), dαβ(i, i+3) and dαN(i, i+4)] in this region supports the helix observed (Supplementary Figure S3). This helix is packed against the remaining part of the toxin, and is stabilized by the Cys5-Cys17 and Cys6-Cys23 disulfide bonds. As such, the side chain of Val1 is located at the outer edge of a π-π stacking formed by the aromatic rings of Trp16 and His20 (Figure 3B). Methyl NOEs of Val1 and Cα proton NOEs of Gly2 to the Trp16 ring protons were observed. In the final 20 structures, the φ angle of Gly2 has a broad range distribution (140 ± 42°) and is predominantly (16 structures) positive (Supplementary Figure S5). This indicates that the N-terminal residues Val1 and Gly2 have a preferred conformation but still retain a degree of flexibility. In the surface model of μ-BuIIIB (Figure 3C), Val1 and the aromatic rings of Trp16 and His20 form a prominent solvent-exposed hydrophobic patch that is adjacent to charged residues Arg15, Arg18, Asp19 and Arg22, equivalent residues of which were found to be critical in VGSC blocking of μ-KIIIA.(17) Superimposition of the μ-BuIIIB structure with structures determined previously for μ-KIIIA, μ-SIIIA, μ-SmIIIA and μ-TIIIA reveals that none of these toxins has structural equivalents of Val1 and Gly2 in μ-BuIIIB (Figure 4). Both μ-SIIIA and μ-SmIIIA have two residues N-terminal to the first Cys, and Arg2 of μ-SmIIIA corresponds to Arg4 of μ-BuIIIB (Figure 1); the structure of the N-terminal residues in μ-SIIIA shows no similarity to that of μ-BuIIIB (Figure 4B) although the structure of μ-SmIIIA is more similar (Figure 4C). While μ-TIIIA has three residues in its N-terminus before the first cysteine, only one residue less than μ-BuIIIB (Figure 1), it has large structural differences in the N-terminal region and the C-terminal α-helix region compared to μ-BuIIIB (Figure 4D).
Figure 4.
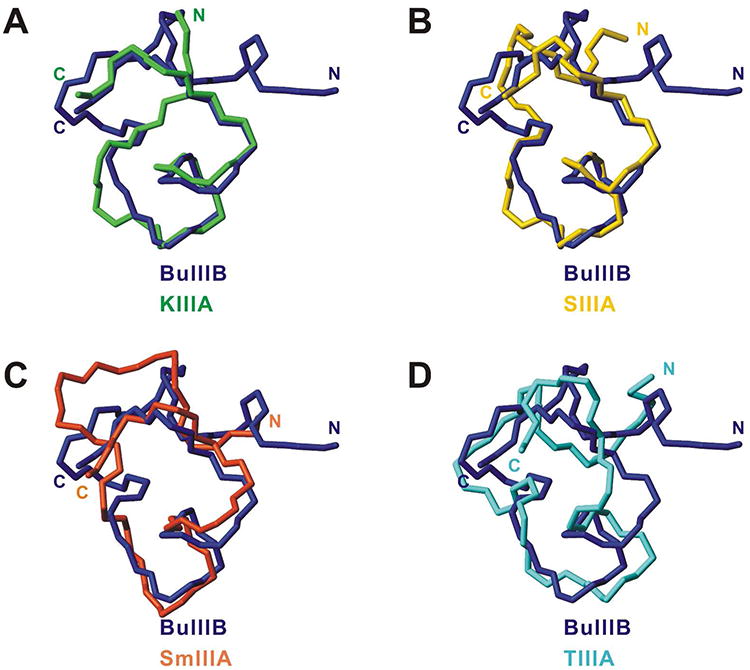
Comparison of μ-BuIIIB and other μ-conotoxin structures. The closest-to-average structures of μ-BuIIIB (blue) and (A) μ-KIIIA (green,(38) PDB 2LXG), (B) μ-SIIIA (yellow,(19) BMRB 20023), (C) μ-SmIIIA (orange,(15) PDB 1Q2J), (D) μ-TIIIA (cyan,(18) BMRB 20024) are superimposed over backbone heavy atoms (N, Cα, and C′) of residues 5,6,13,14-24 for μ-BuIIIB (residues 1,2,4,6-16 for μ-KIIIA, residues 3,4,8,10-20 for μ-SIIIA, residues 3,4,10,12-22 for μ-SmIIIA, and residues 4,5,11,12-22 for μ-TIIIA). N- and C-termini of the molecules are labeled.
μ-BuIIIB residues Gly9 to Gly12 form the second helical (310-like) region, containing the side chains of two basic residues (Lys10 and Arg11). Gly9 has a positive φ value whereas that of Gly12 is negative. Interestingly, while the residue numbers between the second and third cysteines vary from five to eight among μ-BuIIIA, μ-BuIIIB and μ-BuIIIC, Lys10 and Arg11 are conserved (Figure 1). These two residues do not have basic residue counterparts in μ-KIIIA or μ-SIIIA, which only have one and three residues, respectively, between the second and third cysteines (Figures 1 and 4). μ-conotoxins SmIIIA, GIIIA, PIIIA, and TIIIIA have five residues in this loop and have one to two basic residues (Figure 1).
From residues Cys13 to Cys24, μ-BuIIIB shares high sequence conservation with other μ-conotoxins, including μ-KIIIA, μ-SIIIA and μ-SmIIIA, but less conservation with μ-GIIIA, μ-PIIIA and μ-TIIIA (Figure 1). Structurally this part of the toxin forms a C-terminal α-helix (residues 15-20). μ-BuIIIB, as well as μ-BuIIIA and μ-BuIIIC, has three residues but other μ-conotoxins have four residues between the third and the fourth cysteine residues (Cys13 and Cys17 in μ-BuIIIB, Figure 1), resulting in a shorter loop in μ-BuIIIB structure preceding the C-terminal α-helix (Figure 4). The large structural differences in the C-terminal α-helix between μ-BuIIIB and μ-TIIIA (Figure 4D) are probably due to the fact that μ-TIIIA lacks the Trp and His residues conserved in μ-BuIIIB (Trp16 and His20), μ-KIIIA, μ-SIIIA and μ-SmIIIA, and that the loop between the fourth and fifth cysteines is one residue shorter (Figure 1). Basic residues Arg15 (the homologous residue in μ-KIIIA, μ-SIIIA, and μ-SmIIIA is Lys) and Arg18, as well as the acidic residue Asp19, reside on the C-terminal α-helix. Another basic residue, Arg22, is nearby. These charged residues are conserved in μ-conotoxins and corresponding residues have been found to be important for activities against VGSCs.(2, 4, 5, 8) μ-BuIIIB is a highly basic polypeptide as a result of its five Arg and two Lys residues. Consequently, the surface of μ-BuIIIB is highly positively charged (Figure 3C).
Comparison of μ-BuIIIB and [D-Ala2]BuIIIB
Backbone HN and Hα proton chemical shift differences between μ-BuIIIB and [d-Ala2]BuIIIB (Figure 5A) indicated that the d-Ala substitution of Gly2 affected predominantly the chemical shifts of residues 1-6, as well as the HN chemical shift of Ser21, which is distant from Gly2 in the amino acid sequence but is in very close proximity in the three-dimensional structure. Interestingly, the backbone amide proton chemical shifts of Gly9, Gly12, and Gly14 were all affected, even though these residues are structurally distant from the substitution site; these perturbations further emphasize that the N-terminal region is coupled structurally to the rest of the molecule and that changes there are sensed throughout the molecule. An overlay of the fingerprint regions of 2D NOESY spectra of μ-BuIIIB and [d-Ala2]BuIIIB showed that NOE connections in μ-BuIIIB are maintained in [d-Ala2]BuIIIB (Supplementary Figure S6). For example, methyl NOEs of Val1 to the aromatic ring protons of Trp16 were found in both μ-BuIIIB and [d-Ala2]BuIIIB, whereas Hα NOEs of Gly2 to Trp16 in μ-BuIIIB are replaced by the Hα and methyl NOEs of d-Ala2 to Trp16 in [d-Ala2]BuIIIB. As suggested by the NMR data, superimposition of the final families (Figure 5B) or closest-to-average structures (Figure 5C) of μ-BuIIIB and [d-Ala2]BuIIIB over backbone heavy atoms showed that the backbones align very well, with RMSD values of 0.79 Å over all residues and 0.66 Å over residues 3-24 (for the closest-to-average structures). The two structures have only minor conformational differences in the region of residues 1-6 and the aromatic side chains of Trp16 and His20, while the rest of the structures are essentially identical.
Figure 5.
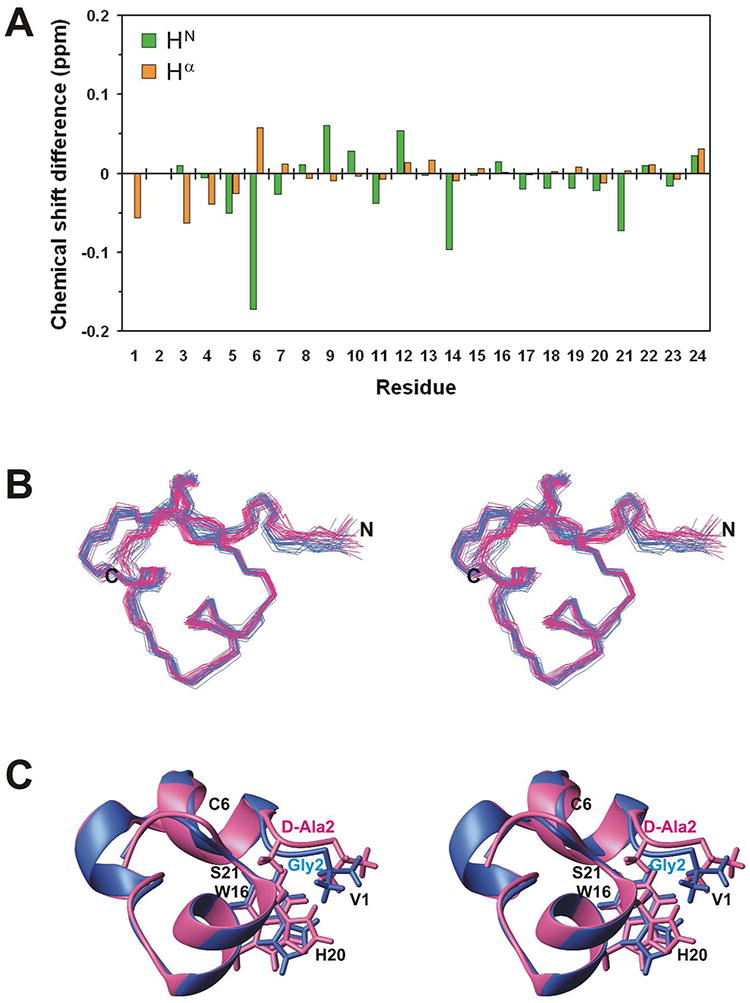
Comparison of μ-BuIIIB and [d-Ala2]BuIIIB structures. (A) Backbone HN and Hα chemical shift differences between μ-BuIIIB and [d-Ala2]BuIIIB. (B) Stereo views of 20 final structures of μ-BuIIIB (blue) and [d-Ala2]BuIIIB (pink) superimposed over backbone heavy atoms. (C) Stereo views of the closest-to-average structures of μ-BuIIIB (blue) and [d-Ala2]BuIIIB (pink) superimposed over backbone heavy atoms. Side chains of Val1, Trp16, His20, and the d-Ala2 substitution are shown and labeled.
Functional Activities of [D-Ala2]BuIIIB and Other N-terminal Analogs in Blocking NaV1.3
Synthetic μ-BuIIIB and N-terminal analogs thereof were tested for their ability to inhibit voltage-gated sodium currents of voltage-clamped Xenopus oocytes expressing NaV1.3, as described in Methods. Representative control and peptide-blocked sodium current traces for μ-BuIIIB and [D-Ala2]BuIIIB are illustrated in Figure 6A & C, and corresponding plots of the time course of inhibition are shown in Figure 6B & D. For each peptide, the onset of block was fit to a single exponential to obtain the observed rate constant, kobs, and kobs values were determined at different peptide concentrations. The plots of kobs versus [peptide] are shown in Supplementary Figure S7, from which the value of kon for each peptide was obtained from the slope of the line (see Methods), and these are given in Table 2. The koff values obtained by direct measurements of the rates of washout of the toxins (see Methods) are listed in Table 2 along with the corresponding Kd values. In principle, the Y-intercepts in Supplementary Figure S7 can provide koff values; however, those obtained were (mean ± SD): -0.084 ± 0.832 and 0.021 ± 0.085 min-1 for [d-Ala2]BuIIIB and μ-BuIIIB, respectively, and are not statistically meaningful.
Figure 6.
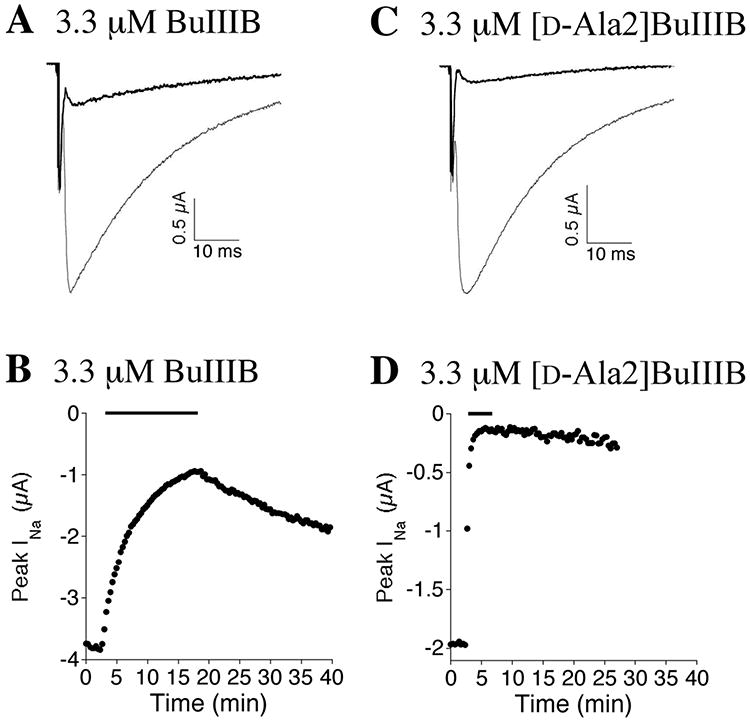
Block of NaV1.3 by μ-BuIIIB and [d-Ala2]BuIIIB. Oocytes expressing NaV1.3 were voltage clamped at -80 mV and sodium channels were activated with a voltage step to -10 mV as described in Methods. Representative sodium currents before (gray trace) and during (black trace) exposure to 3.3 μM of μ-BuIIIB (A) or [d-Ala2]BuIIIB (C). Representative time course of block and recovery, where the bar above each plot indicates when 3.3 μM of μ-BuIIIB (B) or [d-Ala2]BuIIIB (C) was present. The traces in the presence of toxin in panels A and C were obtained at the peak of block in panels B and D, respectively.
Table 2. Comparison of the inhibition of NaV1.3 by μ-BuIIIB and N-terminal analogsa.
| Peptide |
kon (μM•min)-1 |
koff (min1) |
Kd (μM) |
|---|---|---|---|
| μ-BuIIIB | 0.085 ± 0.01 | 0.017 ± 0.007 | 0.2 ± 0.086 |
| [Ala1]BuIIIB | 0.014 ± 0.004 | 0.01 ± 0.003 | 0.71 ± 0.3 |
| [d-Ala2]BuIIIB | 0.83 ± 0.05 | 0.004 ± 0.0017 | 0.0048 ± 0.002 |
| [Ala2]BuIIIB | 0.25 ± 0.03 | 0.009 ± 0.002 | 0.036 ± 0.009 |
| [Ala3]BuIIIB | 0.46 ± 0.03 | 0.007 ± 0.0013 | 0.015 ± 0.003 |
| [Ala4]BuIIIB | 0.056 ± 0.008 | 0.01 ± 0.002 | 0.18 ± 0.044 |
| [des1-4]BuIIIB | 0.47 ± 0.06 | 0.025 ± 0.005 | 0.053 ± 0.013 |
Rate constants were determined as described in Methods. Kd was determined from koff/kon. Data for μ-BuIIIB are from Wilson et al.(20)
Block by 3.3 μM [d-Ala2]BuIIIB is incomplete (e.g., Fig. 6D), with a block of 94 ± 2% (mean ± SD, N = 6 oocytes). However, with a Kd of 4.8 nM (Table 2), >99% block is expected, assuming a dose-response relationship described by the Langmuir adsorption isotherm: fraction blocked = 1/(1 + (Kd/[toxin])). This suggests that a “residual current” remains at saturating peptide concentrations; indeed, when tests were conducted at a ten-fold higher concentration of 33 μM, the residual current persisted (not shown). At least two possible explanations may account for such a residual current: A) there are two populations of channels, with the majority being sensitive to the peptide and a small minority being insensitive; B) the blocking efficacy of the peptide is less than 100%, such as that observed when μ-KIIIA and its derivatives were tested on NaV1.2 and NaV1.4,(29-31) and when μ-CnIIIA was tested on NaV1.4.(32) Further tests, which are beyond the scope of the present study, are necessary to pinpoint the mechanism responsible for the small (∼5%) residual current observed with [d-Ala2]BuIIIB and NaV1.3.
Five additional N-terminal analogs of μ-BuIIIB were synthesized and tested on NaV1.3. Four of these represent an alanine scan of the four N-terminal residues of μ-BuIIIB, and the fifth, [des1-4]BuIIIB, was μ-BuIIIB bereft of its N-terminal four residues. The activities of these analogs, summarized in Table 2, show that the rank order of their Kd values in blocking NaV1.3 was [d-Ala2]BuIIIB < [Ala3]BuIIIB < [Ala2]BuIIIB < [des1-4]BuIIIB < [Ala4]BuIIIB < BuIIIB < [Ala1]BuIIIB. [d-Ala2]BuIIIB was the most potent in blocking NaV1.3 and had the fastest on-rate and slowest off-rate.
Conclusions
The solution structures determined in this study for μ-BuIIIB and [d-Ala2]BuIIIB show, rather unexpectedly, that the N-terminal residues in μ-BuIIIB are in close proximity to a region of these peptides that has been shown previously to be important for VGSC-blocking activity. The functional significance of this close proximity is underscored by the dramatic effects of modifications of the N-terminal extension on activity. Mutations in Gly2 or Glu3 significantly enhance the potency of μ-BuIIIB for NaV1.3, with one analog, [d-Ala2]BuIIIB, showing a 40-fold increase. Indeed, [d-Ala2]BuIIIB appears to be the most potent peptide blocker of the NaV1.3 channel characterized to date.
The eight-fold difference in Kd between the d-Ala2 and l-Ala2 analogs also highlights the importance of the local conformation in this region of the molecule in generating high potency against NaV1.3. The d-Ala2 substitution is more likely to stabilize a type II β-turn conformation, with D-Ala mimicking the positive φ-angle of Gly2 in μ-BuIIIB (Supplementary Figure S5) in a way that l-Ala cannot.(33, 34) Moreover, this reinforces the view that the N-terminus of μ-BuIIIB adopts a well-defined structure, as indicated by our NMR data. The 13-fold enhancement in Kd for NaV1.3 generated by the substitution of Glu3 by Ala is less likely to have a structural basis and may indicate that Glu3 engages in unfavorable interactions with NaV1.3, implying that further manipulation of this position in μ-BuIIIB may have even more beneficial effects on the potency and perhaps also the selectivity of this toxin.
Intriguingly, deletion of all four N-terminal residues in μ-BuIIIB causes a four-fold improvement in the Kd for NaV1.3. This implies that some of the other substitutions in μ-BuIIIB relative to μ-KIIIA, in particular those in the first inter-cysteine loop (Figure 1), are favorable for NaV1.3 blockade, and suggests that a combination of those substitutions in μ-BuIIIB with further manipulation of the N-terminal four residues would be beneficial in generating an even more potent and selective NaV1.3 blocker from this peptide.
μ-BuIIIA, μ-BuIIIB, and vBuIIIC were shown recently to block NaV1.4 exogenously expressed in oocytes.(23) In more recent experiments, μ-BuIIIA and μ-BuIIIB were tested on NaV1.1 through 1.8 expressed in oocytes, where it was observed that μ-BuIIIA was by and large less potent than μ-BuIIIB, and the latter's rank order of affinity (Kd or IC50, prefix “r” and “m” represent rat and mouse) was rNaV1.4 ≈ rNaV1.2 < rNaV1.3 ≈ rNaV1.1 < mNaV1. 6 < rNaV1.5, with no block of rNaV1.7 or rNaV1.8 observed when tested at a concentration of 100 μM.(20) The experiments described here focused on NaV1.3, where the [Gly2d-Ala] mutation was observed to increase μ-BuIIIB's kon by ten-fold and decrease its koff by four fold, such that the Kd was decreased 40-fold by the substitution of d-Ala for Gly at position 2 of μ-BuIIIB (Table 2). Experiments are underway to examine how the mutation affects μ-BuIIIB's affinity for the other NaV1 isoforms.
Specific disulfide mapping for μ-BuIIIB, which belongs to the M5 branch of the μ-conotoxin family (so called because of the presence of five residues in the third and final inter-cysteine loop, as shown in Figure 1), had not been conducted until now. A 1-4/2-5/3-6 pattern of disulfide connectivities was assumed based on sequence alignment with the closely related μ-conotoxins in the M4 branch, and solution structures of μ-SmIIIA,(15) μ-SIIIA,(19) μ-KIIIA(35) and most recently μ-CnIIIC(36) were solved assuming the canonical disulfide connectivity pattern. In this study we have verified that the 1-4/2-5/3-6 pattern is correct for the major product of in vitro oxidative refolding. In contrast, recent studies on μ-KIIIA (37, 38) showed that the thermodynamically favored product of oxidative folding in vitro has 1-5/2-4/3-6 disulfide connectivity. We note that μ-KIIIA has a shorter first loop compared with other M5 μ-conotoxins, with only one amino acid residue between the second and third cysteine residues (Figure 1), and this is likely to influence folding and thus disulfide bridge formation.
Our observations for μ-BuIIIB also have important consequences for attempts to recapitulate the activity of the μ-conotoxins in truncated and stabilized peptide analogs. Recent attempts along these lines (39, 40) have focused on incorporating the residues identified as important for μ-KIIIA blockade of VGSC (17, 22) on a helical scaffold as found in the full-length toxin.(35) The significant effects of modifications at the N-terminus of μ-BuIIIB suggest that elements of this region of the μ-conotoxins might be incorporated to optimize the blockade of certain VGSC subtypes such as NaV1.3.
Methods
All materials and methods are described in the Supporting Information.
Supplementary Material
Acknowledgments
We thank A. Goldin (University of California, Irvine) for the rat NaV1.3, rat NaV1.4, and mouse NaV1.6 clones, G. Mandel (Howard Hughes Medical Institute, Portland, OR) for the rat NaV1.7 clone, and L. Azam (University of Utah) for producing cRNA from these clones. We also thank B. Green and K. Gowd for helpful discussions, and A. Redding, J. Erchegyi, C. Miller and W. Low for technical assistance. This work was supported in part by grants from the Australian Research Council (DP1094212 to R.S.N, B.M.O and G.B) and the National Institute of General Medical Sciences grants GM 48677 (to G.B., B.M.O. and D.Y.). K.G. acknowledges CSIR, Government of India, for a senior research fellowship. The work at Bangalore (IISc, India) is supported by grants from the Department of Biotechnology, Government of India. R.S.N. acknowledges fellowship support from the Australian National Health and Medical Research Council.
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. Full details of Methods, as well as Supplementary Figures S1-Supplementary S6 and Supplementary Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes. Chemical shift assignments for μ-BuIIIB and [d-Ala2]BuIIIB have been deposited in the BioMagResBank with accession numbers 18203 and 18206, respectively. NMR solution structures of μ-BuIIIB and [d-Ala2]BuIIIB have been deposited in the Protein Data Bank (PDB) under ID codes 2LO9 and 2LOC, respectively.
References
- 1.Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- 2.Norton RS, Olivera BM. Conotoxins down under. Toxicon. 2006;48:780–798. doi: 10.1016/j.toxicon.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 3.Han TS, Teichert RW, Olivera BM, Bulaj G. Conus venoms - a rich source of peptide-based therapeutics. Curr Pharm Des. 2008;14:2462–2479. doi: 10.2174/138161208785777469. [DOI] [PubMed] [Google Scholar]
- 4.French RJ, Terlau H. Sodium channel toxins--receptor targeting and therapeutic potential. Curr Med Chem. 2004;11:3053–3064. doi: 10.2174/0929867043363866. [DOI] [PubMed] [Google Scholar]
- 5.French RJ, Yoshikami D, Sheets MF, Olivera BM. The tetrodotoxin receptor of voltage-gated sodium channels--perspectives from interactions with μ-conotoxins. Mar Drugs. 2010;8:2153–2161. doi: 10.3390/md8072153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131:243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 8.Norton RS. μ-conotoxins as leads in the development of new analgesics. Molecules. 2010;15:2825–2844. doi: 10.3390/molecules15042825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem. 1985;260:9280–9288. [PubMed] [Google Scholar]
- 10.Li RA, Ennis IL, Xue T, Nguyen HM, Tomaselli GF, Goldin AL, Marban E. Molecular basis of isoform-specific μ-conotoxin block of cardiac, skeletal muscle, and brain Na+ channels. J Biol Chem. 2003;278:8717–8724. doi: 10.1074/jbc.M210882200. [DOI] [PubMed] [Google Scholar]
- 11.Shon KJ, Olivera BM, Watkins M, Jacobsen RB, Gray WR, Floresca CZ, Cruz LJ, Hillyard, Brink A, Terlau H, Yoshikami D. μ-Conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J Neurosci. 1998;18:4473–4481. doi: 10.1523/JNEUROSCI.18-12-04473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen KJ, Watson M, Adams DJ, Hammarstrom AK, Gage PW, Hill JM, Craik DJ, Thomas L, Adams D, Alewood PF, Lewis RJ. Solution structure of μ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J Biol Chem. 2002;277:27247–27255. doi: 10.1074/jbc.M201611200. [DOI] [PubMed] [Google Scholar]
- 13.Safo P, Rosenbaum T, Shcherbatko A, Choi DY, Han E, Toledo-Aral JJ, Olivera BM, Brehm P, Mandel G. Distinction among neuronal subtypes of voltage-activated sodium channels by μ-conotoxin PIIIA. J Neurosci. 2000;20:76–80. doi: 10.1523/JNEUROSCI.20-01-00076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. μ-conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry. 2002;41:15388–15393. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]
- 15.Keizer DW, West PJ, Lee EF, Yoshikami D, Olivera BM, Bulaj G, Norton RS. Structural basis for tetrodotoxin-resistant sodium channel binding by μ-conotoxin SmIIIA. J Biol Chem. 2003;278:46805–46813. doi: 10.1074/jbc.M309222200. [DOI] [PubMed] [Google Scholar]
- 16.Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, Smith BJ, Yoshikami D, Olivera BM. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry. 2005;44:7259–7265. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- 17.Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur, French RJ, Gulyas J, Rivier JE, Smith BJ, Norton RS, Olivera BM, Yoshikami D, Bulaj G. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J Biol Chem. 2007;282:30699–30706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 18.Lewis RJ, Schroeder CI, Ekberg J, Nielsen KJ, Loughnan M, Thomas L, Adams DA, Drinkwater R, Adams DJ, Alewood PF. Isolation and structure-activity of μ-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol Pharmacol. 2007;71:676–685. doi: 10.1124/mol.106.028225. [DOI] [PubMed] [Google Scholar]
- 19.Yao S, Zhang MM, Yoshikami D, Azam L, Olivera BM, Bulaj G, Norton RS. Structure, dynamics, and selectivity of the sodium channel blocker μ-conotoxin SIIIA. Biochemistry. 2008;47:10940–10949. doi: 10.1021/bi801010u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson MJ, Yoshikami D, Azam L, Gajewiak J, Olivera BM, Bulaj G, Zhang MM. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc Natl Acad Sci U S A. 2011;108:10302–10307. doi: 10.1073/pnas.1107027108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green BR, Catlin P, Zhang MM, Fiedler B, Bayudan W, Morrison A, Norton RS, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Conotoxins containing nonnatural backbone spacers: cladistic-based design, chemical synthesis, and improved analgesic activity. Chemistry and Biology. 2007;14:399–407. doi: 10.1016/j.chembiol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 22.McArthur, Singh G, McMaster D, Winkfein R, Tieleman DP, French RJ. Interactions of key charged residues contributing to selective block of neuronal sodium channels by μ-conotoxin KIIIA. Mol Pharmacol. 2011;80:573–584. doi: 10.1124/mol.111.073460. [DOI] [PubMed] [Google Scholar]
- 23.Holford M, Zhang MM, Gowd KH, Azam L, Green BR, Watkins M, Ownby JP, Yoshikami D, Bulaj G, Olivera BM. Pruning nature: Biodiversity-derived discovery of novel sodium channel blocking conotoxins from Conus bullatus. Toxicon. 2009;53:90–98. doi: 10.1016/j.toxicon.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacological reviews. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 25.Gupta K, Kumar M, Balaram P. Disulfide bond assignments by mass spectrometry of native natural peptides: cysteine pairing in disulfide bonded conotoxins. Anal Chem. 2010;82:8313–8319. doi: 10.1021/ac101867e. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharyya M, Gupta K, Gowd KH, Balaram P. Rapid mass spectrometric determination disulfide connectivity in peptides and proteins. Molecular Biosystems. 2013 doi: 10.1039/C1033MB25534D. [DOI] [PubMed] [Google Scholar]
- 27.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang MM, McArthur, Azam L, Bulaj G, Olivera BM, French RJ, Yoshikami D. Synergistic and antagonistic interactions between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 2009;3:32–38. doi: 10.4161/chan.3.1.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang MM, Han TS, Olivera BM, Bulaj G, Yoshikami D. μ-conotoxin KIIIA derivatives with divergent affinities versus efficacies in blocking voltage-gated sodium channels. Biochemistry. 2010;49:4804–4812. doi: 10.1021/bi100207k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang MM, Gruszczynski P, Walewska A, Bulaj G, Olivera BM, Yoshikami D. Cooccupancy of the outer vestibule of voltage-gated sodium channels by μ-conotoxin KIIIA and saxitoxin or tetrodotoxin. J Neurophysiol. 2010;104:88–97. doi: 10.1152/jn.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markgraf R, Leipold E, Schirmeyer J, Paolini-Bertrand M, Hartley O, Heinemann SH. Mechanism and molecular basis for the sodium channel subtype specificity of μ-conopeptide CnIIIC. Br J Pharmacol. 2012;167:576–586. doi: 10.1111/j.1476-5381.2012.02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pallaghy PK, Duggan BM, Pennington MW, Norton RS. Three-dimensional structure in solution of the calcium channel blocker ω-conotoxin. J Mol Biol. 1993;234:405–420. doi: 10.1006/jmbi.1993.1595. [DOI] [PubMed] [Google Scholar]
- 34.Venkatachalam CM. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers. 1968;6:1425–1436. doi: 10.1002/bip.1968.360061006. [DOI] [PubMed] [Google Scholar]
- 35.Khoo KK, Feng ZP, Smith BJ, Zhang MM, Yoshikami D, Olivera BM, Bulaj G, Norton RS. Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry. 2009;48:1210–1219. doi: 10.1021/bi801998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Favreau P, Benoit E, Hocking HG, Carlier L, D'hoedt D, Leipold E, Markgraf R, Schlumberger S, Cordova MA, Gaertner H, Paolini-Bertrand M, Hartley O, Tytgat J, Heinemann SH, Bertrand D, Boelens R, Stocklin R, Molgo J. A novel μ-conopeptide, CnIIIC, exerts potent and preferential inhibition of NaV1.2/1.4 channels and blocks neuronal nicotinic acetylcholine receptors. Br J Pharmacol. 2012;166:1654–1668. doi: 10.1111/j.1476-5381.2012.01837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poppe L, Hui JO, Ligutti J, Murray JK, Schnier PD. PADLOC: a powerful tool to assign disulfide bond connectivities in peptides and proteins by NMR spectroscopy. Anal Chem. 2012;84:262–266. doi: 10.1021/ac203078x. [DOI] [PubMed] [Google Scholar]
- 38.Khoo KK, Gupta K, Green BR, Zhang MM, Watkins M, Olivera BM, Balaram P, Yoshikami D, Bulaj G, Norton RS. Distinct disulfide isomers of μ-conotoxins KIIIA and KIIIB block voltage-gated sodium channels. Biochemistry accepted for publication. 2012 doi: 10.1021/bi301256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khoo KK, Wilson MJ, Smith BJ, Zhang MM, Gulyas J, Yoshikami D, Rivier JE, Bulaj G, Norton RS. Lactam-stabilized helical analogues of the analgesic μ-conotoxin KIIIA. J Med Chem. 2011;54:7558–7566. doi: 10.1021/jm200839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens M, Peigneur S, Dyubankova N, Lescrinier E, Herdewijn P, Tytgat J. Design of bioactive peptides from naturally occurring μ-conotoxin structures. J Biol Chem. 2012;287:31382–31392. doi: 10.1074/jbc.M112.375733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.