

Abstract
In Pavlovian fear conditioning, the lateral amygdala (LA) has been highlighted as a key brain site for association between sensory cues and aversive stimuli. However, learning-related changes are also found in upstream sensory regions such as thalamus and cortex. To isolate the essential neural circuit components for fear memory association, we tested whether direct activation of presynaptic sensory inputs in LA, without the participation of upstream activity, is sufficient to form fear memory in mice. Photostimulation of axonal projections from the two main auditory brain regions, the medial geniculate nucleus of the thalamus and the secondary auditory cortex, was paired with aversive footshock. Twenty-four hours later the same photostimulation induced robust conditioned freezing and this fear memory formation was disrupted when glutamatergic synaptic transmission was locally blocked in the LA. Therefore, our results prove for the first time that synapses between sensory input areas and the LA, previously implicated as a crucial brain site for fear memory formation, actually are sufficient to serve as a conditioned stimulus. Our results strongly support the idea that the LA may be sufficient to encode and store associations between neutral cue and aversive stimuli during natural fear conditioning as a critical part of a broad fear memory engram.
Classical fear conditioning has provided a useful model to study associative learning and memory (Maren 2001; Kim and Jung 2006; Pape and Pare 2010; Johansen et al. 2011). During fear conditioning, a subject learns to associate a neutral sensory stimulus (the conditioned stimulus, CS), such as a tone, with an aversive stimulus, such as a footshock (the unconditioned stimulus, US). After learning, the previously neutral CS alone is sufficient to elicit a conditioned fear response. It has been suggested that the memory for such conditioning is represented by learning-related changes within the neuronal pathways that transmit the CS.
In the case of auditory fear conditioning, sound is delivered to the lateral amygdala (LA) through two parallel pathways: the medial geniculate nucleus of the thalamus and the auditory cortex (LeDoux et al. 1990; Romanski and LeDoux 1992, 1993; McDonald 1998). The LA is known to receive neuronal inputs for both the CS and the US (Romanski et al. 1993). Based on this anatomy, a large number of studies have indicated that the LA is the site where these CS and US inputs are integrated to form associative fear memory (McKernan and Shinnick-Gallagher 1997; Rogan et al. 1997; Blair et al. 2001; Johansen et al. 2010). However, changes in response to fear learning have also been found in other subcortical and cortical regions (Fanselow and Poulos 2005; Letzkus et al. 2011; Gdalyahu et al. 2012). In particular, the thalamic and cortical auditory regions upstream of the LA show increased neuronal activity and synaptic changes in response to the CS (Gerren and Weinberger 1983; Weinberger 1998; Cahill et al. 1999; Kwon et al. 2012), suggesting that plasticity at these auditory afferents might be necessary to form associative fear memory, rather than to simply deliver sensory information (Parsons et al. 2006).
Concurrent learning-related changes at multiple sites within the broad circuitry related to fear make it difficult to define precisely which circuit elements are sufficient to form an associative memory between the CS and the US. Using electrical stimulation, a previous study searched for circuit components sufficient to form fear memory and reported that electrical stimulation of the medial part of the medial geniculate nucleus (MGm) can serve as a CS to form fear memory (Kwon and Choi 2009). However, due to the possible nonspecific effects of electrical stimulation, it is unclear precisely which circuit elements were manipulated and thus were sufficient for fear memory formation. For example, electrical stimulation of MGm could yield mixed activation of input synapses and somata within MGm, as well as neurons in the auditory cortex receiving inputs from MGm, all output pathways from auditory brain regions, and axonal projections from regions unrelated to the auditory CS information that pass through MGm (Lanuza et al. 2004). Therefore, despite the clear importance of LA, it remains unknown whether the LA is a sufficient neural substrate to represent the association between the CS and the US. If this is the case, then it is predicted that activating the sensory inputs to the LA should be sufficient to serve as a CS in forming an associative memory. However, until now, this idea has never been directly tested.
We have performed such a test by using optogenetic photostimulation to specifically activate auditory axons and their presynaptic terminals in the mouse LA in a temporally and spatially restricted manner. We found that such selective activation of auditory synaptic inputs to the LA could induce robust freezing after pairing with an aversive stimulus. Therefore, our results directly demonstrate that the activity of auditory synaptic inputs to the LA is sufficient to serve as a CS in induction of learning-related changes and to support associative memory. Our findings indicate that associative learning occurs, and the memory for association between the CS and the US is formed, at this specific brain site during natural fear conditioning, thereby identifying the circuit components for a specific memory trace.
Results
AAV-mediated ChR2-Venus expression in auditory projections in the LA
To selectively evoke synaptic transmission between presynaptic auditory inputs and postsynaptic target neurons in the LA, we used ChR2-based photostimulation (Cardin et al. 2010; Johansen et al. 2012). ChR2 fused to the Venus fluorescent protein was expressed by an AAV2/1-hSyn-ChR2-Venus viral construct (AAV-ChR2). An AAV-EGFP construct was used to control for possible nonspecific effects related to viral infection or light exposure (Fig. 1A). These viruses were injected into the two major auditory brain regions that send direct projections to the LA: the MGm and the ventral part of the secondary auditory cortex (AuV) (Fig. 1B). Four weeks after virus injection, brains were sectioned and ChR2-Venus expression was observed under a fluorescence microscope. We detected ChR2 fluorescence on the membranes of neurons only in the targeted brain regions, MGm and the adjacent posterior intralaminar nucleus (PIN), as well as the AuV (Fig. 1C). Importantly, strong ChR2-Venus expression was also detected in the amygdala, indicating that ChR2 was efficiently expressed in the auditory projections and presynaptic terminals innervating the LA through the external and internal capsule (Fig. 1D).
Figure 1.
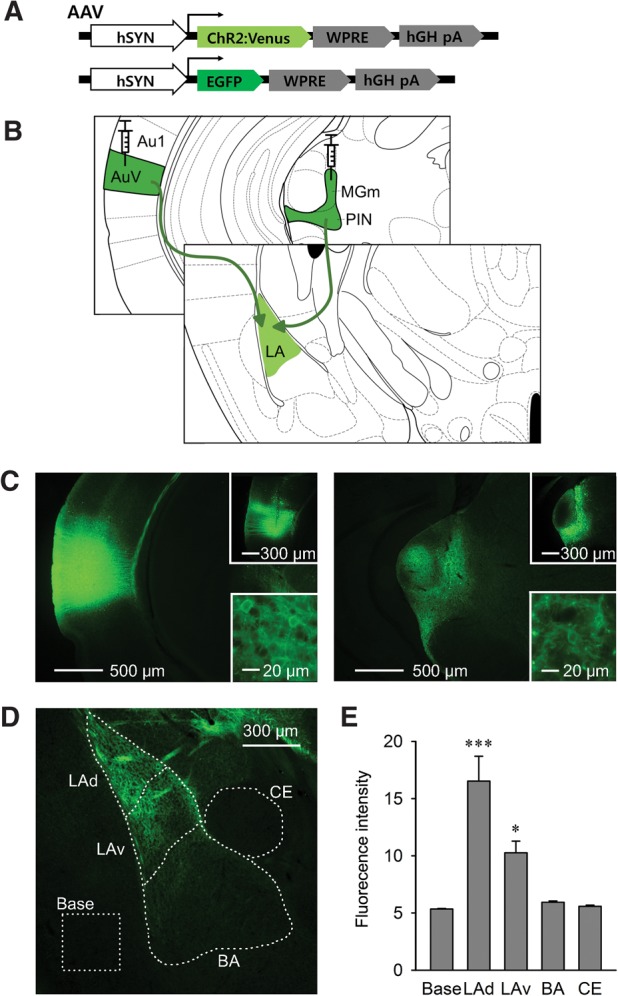
ChR2-expression in the auditory pathways projecting from auditory thalamus and cortex to the LA. (A) AAV constructs expressing ChR2-Venus or control EGFP. (B) AAV viruses were injected into ipsilateral MGm and AuV, which directly project to the LA. (C) ChR2-Venus expression was observed in the auditory thalamus (right) and cortex (left) with reciprocal projections from each other. Single injections of AAV-EGFP into either AuV or MGm specifically yielded infected neurons without reciprocal axonal projection (top insets). ChR2-Venus expression was detected on the membranes of infected neurons (bottom insets). (D) Amygdala showed ChR2-Venus expression representing axonal projections and terminals from auditory afferents. (E) Fluorescence in amygdala subdivisions was analyzed in the dorsal and ventral LA, BA, and CE (n = 10). Data are expressed as mean ± SEM. (***) P < 0.001, (*) P < 0.05. (Au1) Primary auditory cortex, (AuV) ventral part of secondary auditory cortex, (MGm/PIN) medial division of medial geniculate/posterior intralaminar nucleus, (LAd) dorsal part of lateral amygdala, (LAv) ventral part of lateral amygdala, (BA) basal amygdala, (CE) central amygdala, and Base is background fluorescence measured in piriform cortex.
To analyze the distribution of auditory projections within the amygdala, we quantified ChR2-Venus fluorescence intensity in various subdivisions of the amygdala: dorsal LA (LAd) and ventral LA (LAv), basal amygdala (BA) and the central nucleus of the amygdala (CE). The LA displayed substantial fluorescence, particularly in its dorsal division. A few axonal projections were also observed in the BA, while we barely detected fluorescence in the CE. One-way ANOVA revealed significant effect among subdivisions (F(4,45) = 20.2, P < 0.0001), and Bonferroni post hoc test confirmed statistical significance in LAd and LAv (P < 0.0001 and P = 0.02132, respectively), but not in BA and CE, in comparison with the adjacent piriform cortex that showed no ChR2-expression and thus was used to define the background fluorescence level (Fig. 1E).
Photostimulation of presynaptic auditory inputs in the LA
To characterize the functionality of ChR2 expressed in AuV and MGm neurons, we performed whole-cell patch-clamp recordings in brain slices prepared from mice in which AAV-ChR2 had been injected into the AuV and MGm. Brief photostimuli could reliably induce action potentials in both AuV and MGm neurons (Fig. 2A, top and middle). These responses were not blocked by a broad-spectrum glutamate receptor antagonist, kynurenic acid (2 mM), indicating that they were a direct result of photostimulation of these neurons rather than being an indirect consequence of activating excitatory synaptic input. Thus, both AuV and MGm neurons expressed ChR2 and could be photostimulated.
Figure 2.

ChR2-mediated photostimulation in vitro and in vivo. (A) Whole-cell patch-clamp recordings during photostimulation of AuV, MGm, and LA neurons. (Top) In AuV and MGm, light pulses induced action potentials. These action potentials were not blocked by a glutamate receptor blocker, kynurenic acid (2 mM). (Bottom) Light pulses induced responses in the LA that were blocked by kynurenic acid, indicating that they are excitatory postsynaptic currents resulting from photostimulation of presynaptic inputs from AuV and MGm. (B) In vivo field recording in ChR2-expressing mice. Evoked field potentials (EFPs) were recorded with optrodes inserted into AuV, MGm, and LA. EFPs induced by photostimulation of each region were recorded from the three areas. Gray trace shows the absence of responses in control mice injected with AAV-EGFP. Blue bars indicate 10-msec duration light pulses.
We next asked whether photostimulation of ChR2-expressing auditory afferent axons and terminals within the LA could evoke synaptic transmission. For this purpose, we measured excitatory postsynaptic currents (EPSCs) in LA neurons. Local conjoint photostimulation of AuV and MGm fibers within the LA induced robust EPSCs in 84% of our recordings from LA pyramidal neurons (N = 50). In control experiments, where only AuV or MGm were infected with AAV-ChR2, we found that a similar percentage of LA pyramidal neurons responded with EPSCs following photostimulation of either AuV (79%) or MGm (89%) inputs. Thus, we conclude that LA pyramidal neurons receive inputs from both AuV and MGm and that both these inputs were likely photostimulated in our experiments. These optically evoked EPSCs were blocked by kynurenic acid (Fig. 2A, bottom), indicating that photostimulation of ChR2-expressing auditory projections could evoke glutamatergic excitatory synaptic transmission in the LA.
To determine whether ChR2 enabled photostimulation of these inputs in the intact brain, we conducted in vivo field recordings in anesthetized mice. Optrodes, consisting of an optical fiber attached to a tungsten electrode, were placed into the LA and into one of the auditory brain regions, either MGm or AuV. Consistent with our results from brain slices, photostimulation of MGm or AuV elicited robust-evoked field potentials (EFPs) in each of these regions. These direct responses exhibited a very brief latency (negative peak within 2–3 msec) and had mean amplitudes (baseline-to-negative peak) of 1.38 ± 0.38 mV for MGm recordings and 2.01 ± 0.71 mV for AuV. We also measured EFPs in the LA in response to photostimulation of MGm or AuV. These synaptic responses had a latency of 12 msec and were ∼60% smaller compared with responses evoked by photostimulation within the LA (Fig. 2B): Photostimulation of ChR2-expressing auditory axons in the LA evoked robust and reliable EFPs with a mean amplitude of 1.19 ± 0.33 mV and a latency to peak of ∼6–7 msec. This response depended upon ChR2-Venus expression because mice injected with the control EGFP virus showed no EFPs in response to photostimulation of LA.
It is possible that photostimulation of axons in the LA could generate back-propagating action potentials that spread to upstream auditory brain regions. However, we observed virtually no EFP responses in either MGm or AuV in response to LA photostimulation (Fig. 2B; mean EFP amplitude: 0.05 ± 0.03 mV for MGm, 0.19 ± 0.03 mV for AuV), indicating that photostimulation of auditory axons in the LA did not induce antidromic firing of MGm or AuV. This is consistent with a previous optogenetic study of the amygdala microcircuit, which reported that photostimulation of BLA axon terminals projecting to CE produces no reliable antidromic firing in the BLA nucleus (Tye et al. 2011). Taken together, these data reveal that photostimulation of auditory axons and terminals in the LA rapidly and strongly activated presynaptic auditory inputs to the LA with no relevant activation of upstream MGm or AuV. Such photostimuli were therefore used as a CS in the behavioral experiments described in the following section.
Fear conditioning with the optogenetic CS
We next asked whether selective photostimulation of auditory presynaptic inputs in the LA can serve as a CS to form fear memories. For this purpose, virus-injected mice with an implanted guide cannula were subjected to fear conditioning. These mice received six pairings of photostimuli applied to the LA (Fig. 3A; also see Materials and Methods for details) along with a footshock that served as the US. To determine whether this optical conditioning could form long-term fear memory, we examined retention of responses to the same photostimuli by putting the mice in a novel chamber 24 h after fear conditioning (Fig. 3A). Freezing behavior elicited by the photostimuli was then used as an index of fear memory. Mice injected with AAV-ChR2 exhibited robust freezing in response to photostimulation during such testing, indicating that activation of auditory presynaptic input in the LA could act as a CS for fear conditioning and could form long-term fear memories (Fig. 3B; see also Supplemental movie). There was significant interaction between the group and the CS presentation (F(3,29) = 6.51, P = 0.00167, two-way repeated-measures ANOVA); only the group expressing ChR2 showed significant induction of freezing compared with pre-CS levels (P < 0.0001, Bonferroni post hoc test), whereas control mice injected with AAV-EGFP did not.
Figure 3.

Fear conditioning with photostimulation of auditory synaptic input in LA. (A) Diagram of experimental strategy. During training, photostimulation of ChR2-expressing axons and terminals in the LA was presented as a CS and was followed by a footshock that served as a US. After 24 h, retention was tested by giving mice photostimulation alone in a context-shifted chamber. Stimulus parameters during training and retention are shown (inset; see Materials and Methods for details). (B) ChR2-paired group (n = 14) showed robust freezing in response to presentation of the optogenetic CS compared with pre-CS, while the control GFP-paired group (n = 8) did not. The ChR2-CS only group (n = 4), in which the US was not presented during training, did not show any freezing during either training or retention testing. ChR2-expressing mice trained with an unpaired protocol (n = 6) failed to evince significant freezing in response to presentation of the optogenetic CS compared with the pre-CS. (C) Mice in the ChR2-paired group were reclassified into two groups, Success (n = 10) and fail (n = 4), based on the criterion of 30% CS-induced freezing. (D) The correlation between fluorescent intensity in LA and freezing responses was analyzed in the Success (red symbols) and Fail (black symbols) groups. Data are expressed as mean ± SEM. (***) P < 0.001. (ACp) auditory cortical projection, (MGp) medial geniculate projection, (LAn) LA neuron.
As another control, ChR2-expressing mice were given the CS alone during training, without the US. These mice did not show any noticeable behavioral responses to the light during either training or retention periods (Fig. 3B). This result is important because it demonstrates that simply activating auditory inputs to the LA is insufficient to elicit unconditioned freezing behavior. Moreover, ChR2-expressing mice trained with an unpaired protocol, in which photostimuli and electrical footshocks were explicitly unpaired, failed to show significant freezing in response to subsequent presentation of photostimuli (Fig. 3B). This suggests that information about the contingency of the CS and the US can be represented at LA synapses without requiring processes in any other brain areas.
Because pairing of the CS and the US did not show significant freezing compared with the baseline in a few ChR2-expressing mice, we further analyzed ChR2-Venus expression in these mice. For this analysis, mice were reclassified into two groups: those exhibiting >30% freezing (Success) and those with <30% freezing (Fail) (Fig. 3C). We found that in the Fail group all mice showed low ChR2-Venus fluorescence (<7 arbitrary units, a.u.), whereas in the Success group all showed higher fluorescence (10 a.u. or greater). There was no significant correlation between freezing percentage and fluorescence intensity within either the Success or Fail groups (Fig. 3D), suggesting that the amount of auditory inputs activated affected the threshold level for a CS to form fear memory rather than the strength of the memory.
Optogenetic fear memory is mediated by glutamatergic synaptic transmission in LA
To establish that fear conditioning was exclusively mediated by synaptic transmission between the presynaptic auditory inputs and postsynaptic LA neurons, we locally blocked glutamatergic synaptic transmission in the LA in anesthetized mice by in vivo microinfusion of kynurenic acid (2 mM). Ten minutes after administration of kynurenic acid, the peak amplitude of EFPs evoked in the LA by photostimulation of auditory inputs was decreased to 15% of their control values measured prior to drug administration (Fig. 4A). This is consistent with our in vitro slice results (Fig. 2A) and confirms that EFPs evoked in the LA in response to photostimulation are mediated by glutamatergic excitatory synapses (Sah et al. 2003). When the same animal was retested 20 h later, the amplitudes of light-evoked EFP had recovered up to ∼80% of the values measured prior to drug treatment, indicating that the effect of kynurenic acid was reversible.
Figure 4.

Optogenetic CS was mediated by glutamatergic synaptic transmission in the LA. (A) EFP in the LA was tested before and after the microinfusion of kynurenic acid. EFP amplitude was dramatically decreased 10 min after kynurenic acid treatment (2 mM, 1 μL) and gradually recovered after 20 h. (B) Experimental procedures of optogenetic fear conditioning with drug infusion. (C) The effect of kynurenic acid on fear conditioning. The mice injected with kynurenic acid into the LA (n = 6) showed significantly lower freezing than vehicle group mice (n = 5). (D) The kynurenic acid group mice showed normal learning following retraining after vehicle injection. Data are expressed as mean ± SEM. (*) P < 0.05, (**) P < 0.01.
These results allowed us to next use kynurenic acid to determine the role of excitatory synaptic transmission in the behavioral response to photostimulation. For this purpose, either kynurenic acid or control vehicle solution was locally infused into the LA immediately prior to training and mice were then fear conditioned with the optogenetic CS as described earlier. When tested 24 h later (Fig. 4B, Retention 1), the mice injected with kynurenic acid showed significantly less freezing upon presentation of the CS alone compared with the control mice (Fig. 4C; P = 0.043, unpaired t-test). However, when the kynurenic acid group of mice was retrained 2 d after infusion of vehicle (Fig. 4B, Retention 2), they showed normal learning in response to photostimulation and exhibited robust CS-induced freezing during the retention test (Fig. 4D; two-way repeated-measures ANOVA, Drug × CS interaction: F(1,4) = 18.82, P = 0.0123. Bonferroni post hoc test between pre-CS and CS: P = 0.104 for Retention 1, P = 0.002 for Retention 2). These results establish that fear conditioning was mediated by photostimulation of presynaptic glutamatergic auditory inputs to the LA. Taken together, our results revealed that activation of auditory projections to neurons can serve as a CS that, when paired with unconditioned footshock, can produce fear conditioning and form a long-term associative memory.
Discussion
A large amount of evidence supports the notion that the LA is a key site for encoding and storing the associative memory for Pavlovian fear conditioning. This hypothesis predicts that activating only the synaptic inputs that deliver CS information to LA neurons should be sufficient both to form fear memory and to allow its subsequent retrieval. However, this prediction has never been tested directly. Here we have used selective photostimulation of auditory presynaptic inputs in the LA as a CS for fear conditioning, thereby presenting a CS directly to the LA rather than through upstream sensory brain regions such as the thalamus and the cortex. After learning, the same photostimulus alone could activate retrieval of the fear memory. Thus, upon photostimulation, mice exhibited strong freezing that corresponded to the normal fear response formed by natural auditory cues. This artificial fear memory trace was formed by the associative activation of auditory inputs and the US pathway; fear memory did not occur if the CS photostimulus was presented alone or was temporally uncoupled from the US.
The electrical stimulation methods that have been used in many previous studies to activate brain regions suffer from a lack of specificity. For example, although it has been reported that strong stimulation of the MGm/PIN causes an unconditioned freezing response (Cruikshank et al. 1992; Kwon and Choi 2009), it is possible that this response was caused by activation of axonal projections, including nociceptive inputs that pass through this region (Lanuza et al. 2004). In contrast, the ChR2-mediated photostimulation technique that we have used allowed us to activate auditory inputs in a highly specific manner. Indeed, we observed that photostimulation alone did not cause any freezing behavior, indicating that we were selectively activating the auditory CS inputs rather than mixed and undefined inputs from thalamic and cortical projections.
In our experiments, we simultaneously stimulated both thalamic and cortical auditory pathways. In the auditory fear conditioning circuit, auditory information reaches the LA through both of these pathways and both are thought to be important for encoding fear conditioning. It has been suggested that the thalamic pathway delivers a rapid but simple pattern of auditory information, whereas the cortical pathway encodes slow but detailed information (LeDoux 1996). In addition, some studies have suggested that the interaction between thalamic and cortical inputs may be advantageous for inducing plastic synaptic changes within the LA (Shin et al. 2006; Cho et al. 2012). Therefore, by simultaneously stimulating both thalamic and cortical projections, we attempted to closely mimic the processes that deliver natural sensory information. Nevertheless, it is possible that activating a single auditory pathway, either thalamic or cortical, could be sufficient to form and support long-term fear memory. Further experiments will be needed to test this possibility.
Although our optogenetic strategy specifically targeted auditory inputs projecting to the LA, it is possible that the fear memory trace is represented in regions in addition to LA. Specifically, other subdivisions of the amygdala, such as BA and CE, and still other brain regions downstream from LA, such as the periaqueductal gray matter, are involved in memory representation (Wilensky et al. 2006). For example, recent studies show that fear learning induces long-term synaptic potentiation in the CE (Ciocchi et al. 2010). It is very unlikely that direct activation of BA or CE through the optical stimulation of axons projecting to the BA or CE mediated fear memory formation in our experiments. Our analysis showed highly restricted expression of ChR2 in the LA, whereas the BA showed very weak, if any, ChR2-expressing auditory projections (see Fig. 1D,E). Moreover, although some anatomical studies have reported that CE also directly receives sensory projections (Turner and Herkenham 1991; McDonald 1998), these would not have been activated in our experiments (Fig. 1E). In addition, photostimulation produced no fear memory during local block of glutamatergic synaptic transmission in the LA. Thus, we conclude that a fear memory trace can be formed in the LA by the association of photostimulated LA-projecting auditory inputs along with the US. However, the LA serves as an interface for the amygdala, by receiving initial sensory inputs, and has strong interconnections to the downstream BA and CE (Sah et al. 2003; Ehrlich et al. 2009). It is, therefore, possible that there are learning-related changes occurring downstream from LA, but in our experiments these would secondarily result from activation of LA neurons. More generally, our findings do not mean that circuit components other than those we manipulated have no role for memory formation. Rather, broader neural circuits, encompassing sensory perception to behavioral expression, may be necessary to shape different aspects of the memory such as its strength, maintenance, specificity, and other features.
In conclusion, we demonstrated for the first time that the activity of synaptic inputs to the LA is sufficient to serve as a CS to produce long-term associative memory for fear conditioning. Our study strongly supports the idea that activation of sensory input to LA synapses may be a crucial and sufficient step to form the memory for association of the CS and the US during natural fear conditioning and that this specific brain site is the place where the memory is formed, providing important new insights into the role of specific circuit elements within the broader neural circuitry for associative memory formation.
Materials and Methods
Mice
129/C57Bl/6 hybrid background mice (2–3 mo old, 23–35 g) were group-housed on a 12-h light/dark cycle at a constant temperature of 22 ± 1°C with 40%–60% humidity. Food and water were available ad libitum throughout the experiment. All procedures used were consistent with the animal ethics guidelines of the institutions where the experiments were performed (KAIST and KIST).
Virus production
For ChR2 gene delivery, we produced recombinant adeno-associated virus vectors. DNA plasmids coding AAV-hSyn-ChR2-Venus or AAV-hSyn-EGFP were amplified and purified using a Maxiprep kit (Qiagen). The purified plasmids were mixed into a CaCl2 solution with the DNA plasmid coding AAV2/1 and pAΔF6 and cotransfected into HEK293T cell using calcium phosphate precipitation. Cells were harvested 72 h after transfection and the relevant virus was purified in an ultracentrifuge on an iodixanol gradient. The viral concentration was determined by quantitative PCR, and the final titer was 0.5–1 × 1012 vg/mL.
Brain surgery
Mice were anesthetized by intraperitoneal injection of pentobarbital (83 mg/kg) and placed in a stereotaxic frame. A glass micropipette containing an AAV virus solution was positioned on the targeted AuV and MGm brain region (AuV: AP = −2.7 mm, ML = +4.4 mm, DV = −3.2 mm, MGm: AP = −3.2 mm, ML = +1.9 mm, DV = −3.5 mm from bregma). AAV solutions were unilaterally injected at a rate of 0.1 μL/min for 8 min (total, 0.8 μL/region). Micropipettes were left in place for an additional 10 min to ensure diffusion. The pipette was removed, head skin was clipped, and mice were then allowed to recover on a heat pad and moved back to their home cage. Experiments were conducted at least 4 wk later, to allow sufficient time for expression of ChR2 in axons projecting to the LA. Three weeks after AAV injection, mice used for behavioral experiments were implanted with a guide cannula for optical fiber insertion. The guide cannula was positioned 1 mm above the LA (AP = −1.8 mm, ML = ±3.4 mm, DV = −3.0 mm from bregma) and fixed with dental cement.
To quantify ChR2-Venus expression, mice were perfused and fixed with 4% PFA, and the brain was then sliced into 40-μm thick sections. Brain sections corresponding to −1.7, −1.9, and −2.1 mm posterior from bregma were then selected for analysis. The amygdala was subdivided into dorsal/ventral LA, BA, and CE (Fig. 1D; Franklin and Paxinos 2008). Under a fluorescent microscope, fluorescence intensity values were measured in the four subdivisions using NIS imaging software (Nikon). The adjacent piriform cortex of the same sections, which did not express ChR2-Venus, was selected for measurement of background fluorescence. Image acquisition conditions were adjusted to keep the intensity value of the background fluorescence constant across all slices. The mean intensity value for each region was calculated by averaging the three sections from a single mouse, and mean fluorescence intensity values were averaged for each region to determine whether ChR2-Venus expression was successful or was a failure for each mouse.
Acute brain slice recordings
Coronal brain slices (1.6 mm posterior from bregma) were obtained from mice 1 mo after AAV injection into MGm and AuV and were prepared using conventional methods (Pettit and Augustine 2000; Nakajima et al. 2004; Wang et al. 2007). In brief, a Vibratome (VT-1200, Leica) was used to slice brains into 350-μm thick coronal sections in a solution containing high sucrose. This was composed of (in mM): 87 sodium chloride, 75 sucrose, 25 NaHCO3, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, and 25 d(+)-glucose. pH was maintained at 7.4 by gassing the solution with 95% O2/5% CO2. Slices were then transferred to an incubation chamber filled with oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d(+)-glucose, 2.5 CaCl2, 1 MgCl2, 3 sodium pyruvate, and 1 ascorbic acid. Slices were incubated at 36°C for 30 min and at least 30 min at room temperature prior to use.
Whole-cell patch-clamp recordings were performed at 32°C in an upright microscope (FV1000MPE, Olympus) in a recording chamber perfused with 95% O2/5% CO2 aerated ACSF. To characterize photostimulation of AuV or MGm neurons (Fig. 2A, top), coronal sections 3.0 mm posterior from the bregma were used and recordings were made from somata of neurons within these regions. For analysis of LA responses to synaptic photostimulation (Fig. 2A, bottom), coronal sections from bregma −1.6 mm were used; these do not contain somata of AuV and MGm but do contain ChR2-expressing axonal fibers coming from the infected site and projecting to LA excitatory neurons (Fig. 1D). Occasionally, kynurenic acid (2 mM, Sigma) was added to the ACSF to block excitatory synaptic transmission. Pyramidal neurons in MGm, AuV, or LA were identified with infrared differential interference contrast (IR-DIC) optics in combination with a digital video camera (MCE-B013-U, Mightex). Whole-cell patch-clamp recordings were made from these neurons using glass pipettes (5–12 MΩ) filled with internal solution containing (in mM): 130 K-gluconate, 2 NaCl, 4 MgCl2, 20 HEPES, 4 Na2ATP, 0.4 Na3GTP, 0.5 EGTA, and 10 Na2 phosphocreatine, pH adjusted to 7.25 with 1 M KOH. Osmolality of this solution was 290–295 mOsm. Current measurements were made under voltage clamp, at a holding potential of −60 mV. Electrical responses were detected with an amplifier (Multiclamp 700B, Molecular Devices), digitized 20 kHz via an A–D converter (Digidata 1440A, Molecular Devices), acquired using pClamp software (Molecular Devices), and analyzed using Clampfit software (Molecular Devices).
A mercury arc lamp (USH-1030L, Olympus,) filtered by a band-pass filter (470–495 nm) was used to activate ChR2. Photostimuli were applied through a 25× (1.05 NA) water-immersion objective lens and the entire width of the microscope field (∼500 μm diameter) was illuminated. During synaptic photostimulation, this light spot surrounded an LA neuron and was used to photostimulate ChR2-expressing axonal fibers projecting from AuV and MGm. Pulse duration was controlled by an electronic shutter (Uniblitz VS25, Vincent). For direct photostimulation of AC and MG neurons, light pulses of 0.7-mW/mm2 luminance and 10-msec duration were applied at 5 Hz for 2 sec. For evoking synaptic responses in the LA, slightly brighter single light pulses (1.2-mW/mm2, 10-msec duration) were delivered every 30 sec.
In vivo recording
Photostimulation of neurons in ChR2-expressing mice was tested by recording local field potentials. Mice were unilaterally injected with AAV-ChR2 or AAV-GFP into MGm and AuV. Four weeks after virus injection, mice were anesthetized with pentobarbital (83 mg/kg, i.p.) and placed in a stereotaxic frame. An optrode, which consisted of a tungsten electrode (5 MΩ) and an optical fiber, was positioned at the point where the LA receives axonal projections from the virus-injected sites. Electrode placement was confirmed via postmortem histology. Recordings were band-pass filtered between 10 and 300 Hz using a DAM80 differential amplifier (World Precision Instruments), and digitized at 10 kHz. Signals were processed via custom-made programs written in Labview (National Instruments). Photostimuli were delivered through an optical fiber connected to a 473-nm diode laser (Crystalaser). Light-evoked local field potentials were acquired and analyzed off-line. Five waveforms induced by photostimuli delivered at 1 min intervals were averaged and the baseline-to-negative peak amplitude was calculated from these average signals. For tests of axonal back-propagation, an additional optrode was placed into the MGm or AuV and EFPs were measured in MGm, AuV, and/or LA.
Fear conditioning with the optogenetic CS
After 7 d recovery from cannula implantation, mice were handled and habituated with optical fiber insertion. The optical fiber length was adjusted so that its tip was positioned at the LA and its back end was connected to a 473-nm diode laser (Crystalaser) via a rotary joint. Before training, light power at the fiber tip (200-μm core) was calibrated to 9.6 mW/mm2 by measurement with a light power meter (PM100D, Thorlabs). To prevent possible light leakage from the optical fiber serving as a visual cue during fear conditioning, we minimized light power and the fiber was double coated with a black jacket and with white paint.
During training, the optical fiber was positioned in the target LA through a guide cannula, and placed in a training chamber with video camera monitoring. Two minutes later, mice received six pairings of the optogenetic CS (20 sec duration) followed by a footshock that served as a US (0.4-mA shock for 2 sec). The optogenetic CS consisted of 10-msec light pulses that were repeated at 10 Hz, with 2 sec duration stimulus trains separated by 1 sec interval. Intertrial intervals were randomly given at an average interval of 2 min (Fig. 3A). Mice were kept in the chamber for an additional 30 sec after delivery of the last shock to monitor post-shock behavior and then removed from the chamber. In control experiments, the group receiving the CS alone received the same behavioral training procedure described above for the ChR2-paired group, except for elimination of the footshock during training. Mice in the ChR2-unpaired group were given explicitly unpaired presentations of the CS and the US, with an interstimulus interval longer than 1 min. Retention tests were conducted 24 h after training. For this purpose, mice were placed in a context-shifted test chamber. To clearly distinguish the CS-induced fear responses from general fear responses, the baseline freezing level was recorded after a mouse initiated movement with its hind paw; this was done because mice normally show continuous movement after initiating such a motion. After the 2-min baseline recording, 20 sec of the optical CS was presented three times, with a 10-sec interval, as used for fear conditioning (Fig. 3A). Mice were removed from the test chamber after additional 60 sec following termination of photostimulation. The freezing behavior measured during the 2 min from onset of photostimulation was used as an index of conditioned fear memory for data analysis.
In some experiments, mice were unilaterally infused with 1 μL of kynurenic acid (2 mM dissolved in ACSF) into the LA at a rate of 0.2 μL/min prior to starting training. After allowing 2 min for drug diffusion, these mice were then fear conditioned with the optogenetic CS and the next day they were subjected to the retention test as described earlier. Two days after the first retention test, mice received the same treatment except for infusion of ACSF instead of kynurenic acid.
Statistical analysis
Comparison of ChR2-Venus expression was performed by one-way ANOVA. Freezing behaviors were analyzed by Student's unpaired t-test or two-way repeated-measures ANOVA followed by Bonferroni post hoc tests for multiple comparisons. Prism (GraphPad software) was used for all statistical analysis in this study.
Supplementary Material
Acknowledgments
We thank members of our laboratories for critical discussions and helpful comments and Dr Bong-Kiun Kaang for DNA plasmids. J.-T.K. is supported by the project of Global Ph.D. Fellowship from the National Research Foundation of Korea. This work was supported by a National Research Foundation of Korea grant funded by the Korean government (2011-0013173); KAIST Future Systems Healthcare Project from the Ministry of Science, ICT and Future Planning; and Intelligent Synthetic Biology Center of Global Frontier Project funded by the Ministry of Science, ICT & Future Planning (2011-0031955) to J.-H.H. and the World Class Institute (WCI) program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (MEST) (WCI 2009-003); as well as a CRP grant from the National Research Foundation of Singapore to G.J.A.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.035816.114.
References
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. 2001. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8: 229–242 [DOI] [PubMed] [Google Scholar]
- Cahill L, Weinberger NM, Roozendaal B, McGaugh JL. 1999. Is the amygdala a locus of “conditioned fear”? Some questions and caveats. Neuron 23: 227–228 [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI. 2010. Targeted optogenetic stimulation and recording of neurons in vivo using cell-type-specific expression of Channelrhodopsin-2. Nat Protoc 5: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Bayazitov IT, Meloni EG, Myers KM, Carlezon WA Jr, Zakharenko SS, Bolshakov VY. 2012. Coactivation of thalamic and cortical pathways induces input timing-dependent plasticity in amygdala. Nat Neurosci 15: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, et al. 2010. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468: 277–282 [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Edeline JM, Weinberger NM. 1992. Stimulation at a site of auditory-somatosensory convergence in the medial geniculate nucleus is an effective unconditioned stimulus for fear conditioning. Behav Neurosci 106: 471–483 [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. 2009. Amygdala inhibitory circuits and the control of fear memory. Neuron 62: 757–771 [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. 2005. The neuroscience of mammalian associative learning. Annu Rev Psychol 56: 207–234 [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. 2008. The mouse brain in stereotaxic coordinates. Elsevier/Academic Press, London/New York [Google Scholar]
- Gdalyahu A, Tring E, Polack PO, Gruver R, Golshani P, Fanselow MS, Silva AJ, Trachtenberg JT. 2012. Associative fear learning enhances sparse network coding in primary sensory cortex. Neuron 75: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerren RA, Weinberger NM. 1983. Long term potentiation in the magnocellular medial geniculate nucleus of the anesthetized cat. Brain Res 265: 138–142 [DOI] [PubMed] [Google Scholar]
- Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, LeDoux JE. 2010. Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci 107: 12692–12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, LeDoux JE. 2011. Molecular mechanisms of fear learning and memory. Cell 147: 509–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Wolff SB, Luthi A, LeDoux JE. 2012. Controlling the elements: an optogenetic approach to understanding the neural circuits of fear. Biol Psychiatry 71: 1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Jung MW. 2006. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev 30: 188–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JT, Choi JS. 2009. Cornering the fear engram: long-term synaptic changes in the lateral nucleus of the amygdala after fear conditioning. J Neurosci 29: 9700–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JT, Jhang J, Kim HS, Lee S, Han JH. 2012. Brain region-specific activity patterns after recent or remote memory retrieval of auditory conditioned fear. Learn Mem 19: 487–494 [DOI] [PubMed] [Google Scholar]
- Lanuza E, Nader K, Ledoux JE. 2004. Unconditioned stimulus pathways to the amygdala: effects of posterior thalamic and cortical lesions on fear conditioning. Neuroscience 125: 305–315 [DOI] [PubMed] [Google Scholar]
- LeDoux JE. 1996. The emotional brain: the mysterious underpinnings of emotional life. Simon & Schuster, New York [Google Scholar]
- LeDoux JE, Farb C, Ruggiero DA. 1990. Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J Neurosci 10: 1043–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Luthi A. 2011. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480: 331–335 [DOI] [PubMed] [Google Scholar]
- Maren S. 2001. Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24: 897–931 [DOI] [PubMed] [Google Scholar]
- McDonald AJ. 1998. Cortical pathways to the mammalian amygdala. Prog Neurobiol 55: 257–332 [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. 1997. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611 [DOI] [PubMed] [Google Scholar]
- Nakajima R, Nakamura T, Ogawa M, Miyakawa H, Kudo Y. 2004. Novel method for quantification of brain cell swelling in rat hippocampal slices. J Neurosci Res 76: 723–733 [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. 2010. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90: 419–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Riedner BA, Gafford GM, Helmstetter FJ. 2006. The formation of auditory fear memory requires the synthesis of protein and mRNA in the auditory thalamus. Neuroscience 141: 1163–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Augustine GJ. 2000. Distribution of functional glutamate and GABA receptors on hippocampal pyramidal cells and interneurons. J Neurophysiol 84: 28–38 [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. 1997. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607 [DOI] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE. 1992. Equipotentiality of thalamo-amygdala and thalamo-cortico-amygdala circuits in auditory fear conditioning. J Neurosci 12: 4501–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE. 1993. Information cascade from primary auditory cortex to the amygdala: corticocortical and corticoamygdaloid projections of temporal cortex in the rat. Cereb Cortex 3: 515–532 [DOI] [PubMed] [Google Scholar]
- Romanski LM, Clugnet MC, Bordi F, LeDoux JE. 1993. Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav Neurosci 107: 444–450 [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. 2003. The amygdaloid complex: anatomy and physiology. Physiol Rev 83: 803–834 [DOI] [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E, Bolshakov VY. 2006. Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron 52: 883–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BH, Herkenham M. 1991. Thalamoamygdaloid projections in the rat: a test of the amygdala's role in sensory processing. J Comp Neurol 313: 295–325 [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. 2011. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature 471: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Peca J, Matsuzaki M, Matsuzaki K, Noguchi J, Qiu L, Wang D, Zhang F, Boyden E, Deisseroth K, et al. 2007. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci 104: 8143–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger NM. 1998. Physiological memory in primary auditory cortex: characteristics and mechanisms. Neurobiol Learn Mem 70: 226–251 [DOI] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. 2006. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci 26: 12387–12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.