

Abstract
Recently, several laboratories have reported thousands of circular RNAs (circRNAs) in animals. Numerous circRNAs are highly stable and have specific spatiotemporal expression patterns. Even though a function for circRNAs is unknown, these features make circRNAs an interesting class of RNAs as possible biomarkers and for further research. We developed a database and website, “circBase,” where merged and unified data sets of circRNAs and the evidence supporting their expression can be accessed, downloaded, and browsed within the genomic context. circBase also provides scripts to identify known and novel circRNAs in sequencing data. The database is freely accessible through the web server at http://www.circbase.org/.
Keywords: gene expression, circular RNA, database
INTRODUCTION
In recent years, several laboratories have reported thousands of circular RNAs (termed circRNAs) expressed in animal cells (Salzman et al. 2012, 2013; Jeck et al. 2013; Memczak et al. 2013). These single-stranded RNA molecules correspond to circular isoforms, often produced from exons in which the 5′ and 3′ ends are covalently closed to form a “head-to-tail” splice junction (or “backsplice”). It has been shown that in humans, circRNAs are the predominant isoform of exon-scrambling events and that circularization is a widespread and general feature of gene expression (Salzman et al. 2012). Two recent articles provided evidence that the human circRNAs (CDR1as/ciRS-7) can have regulatory function by acting as a miRNA sponge (Hansen et al. 2013; Memczak et al. 2013). However, it is still unknown if circRNAs have biological function since loss-of-function experiments are missing and, in many cases, are difficult to carry out due to the overlap between circular and linear isoforms. Nevertheless, numerous circRNAs seem well expressed (Jeck et al. 2013; Memczak et al. 2013; Salzman et al. 2013), often in a tissue-specific and developmental stage–specific manner (Memczak et al. 2013). Moreover, circRNAs are also unusually stable RNA molecules, presumably because their lack of ends prevents them from regulation by conventional RNA degradation pathways. Thus, circRNAs can be diagnostic of gene expression patterns and may constitute an interesting, novel class of biomarkers.
Thousands of circRNAs in humans, the mouse, and other animals have been reported by computational analysis of sequencing data, and the numbers are likely to grow. For an overview of detection methods used, see articles by Hoffmann et al. (2014) and Jeck and Sharpless (2014). To meet the need for an integrated database that enables the study, comparison, or download of circRNAs, we developed “circBase,” with the following objectives: (1) For each circRNA, the evidence for its existence and expression should be summarized and accessible; (2) circRNAs should be presented within the genomic context and together with available expression or regulatory data; (3) a search engine should allow flexible queries, for example, by sequence, by gene, by genomic location, etc.; (4) it should be possible to intersect and download data sets in multiple ways and formats; and (5) users should be able to upload tracks with their own circRNA expression and intersect with the available data. In essence, we intend to serve the community by pooling together and making accessible published data sets from different laboratories. In its current implementation (version 1.0), circBase provides a solution to these objectives by (1) a simple searching interface that is convenient for users focusing on a small number of genes or loci; (2) various methods of bulk data retrieval; (3) merging, unifying, and annotating published data sets; and (4) linking circRNAs to the UCSC genome browser (Kent et al. 2002) and doRiNA (Anders et al. 2012), a database for post-transcriptional regulatory elements. Additionally, we (Memczak et al. 2013) have described a method for detecting circRNAs by computational analysis from RNA sequencing data. This method does not depend on preexisting genome annotation (e.g., known transcripts or splice sites). The latest version of software needed to perform this analysis is also available on circBase.
The database currently hosts data from various Homo sapiens, Mus musculus, Caenorhabditis elegans, and Latimeria samples. We are expecting the range of organisms and samples to extend in the future. circBase will not cover viroids, which are already collected in other resources (Rocheleau and Pelchat 2006; Hamilton et al. 2011).
RESULTS AND DISCUSSION
Searching circBase
There are three main ways to query circBase: (1) Simple search, (2) List search, and (3) Table browser.
Simple search, available from the server homepage, is intended for simple queries by identifiers, genomic location, sequence, gene description, or Gene Ontology term identifiers. Apart from internal identifiers, we provide support for RefSeq transcript identifiers, HGNC gene symbols, and names of particular circRNAs from the literature (e.g., CDR1as). We cannot exclude the possibility of different investigators using the same name for multiple circRNAs. Therefore, only circBase internal identifiers, which are based on the genomic location of a head-to-tail splice site, uniquely identify database records. In the search field, genomic locations can be given in a UCSC compatible format (e.g., chrX:134,619,072–134,756,686) or with whitespace-separated chromosome, start and end coordinates. When searching by genomic coordinates, the list of results will contain all circRNAs that overlap the specified genomic region. Search by gene description terms (e.g., “apoptosis”) will return circRNAs that are produced from transcripts of genes matching the description. Description terms are derived from official full gene names in NCBI Entrez Gene database (Maglott et al. 2011). Users can also query the database by DNA or RNA sequence, in which case the number of exact sequence matches per circRNA will be shown. This search option is limited to sequences >6 nucleotides (nt), and only exact matches of queried sequence, or its reverse complement, will be reported. Searches can be limited to a particular animal species. For the nonexact sequence search, users may use Blat (Kent 2002), available from the main menu. circBase hosts Blat references of human, mouse, and C. elegans circRNAs. circRNAs are cut opposite to the head-to-tail splice junction to keep it intact and allow looking for continuous matches that overlap the junction.
List search allows the intersection of a large number of search terms with database contents. Upon selecting the organism and assembly, the user can paste or upload a list of any identifier type supported by circBase. It is also possible to submit a list of genomic regions (for example in BED format) and retrieve all overlapping circRNAs in the database.
The Table browser can be used for conditional data retrieval (Fig. 1A). After selecting the organism and experiment of interest, the user can further refine the selection by a number of options, such as presence in a particular sample, range of genomic or spliced sequence lengths, number of reads supporting the head-to-tail splice junction, and many more. This interface was developed to enable simple generation of complex queries such as “Retrieve all the circRNAs from experiment A that are present in HEK293 cells, intergenic, between 1000 and 2000 bp long and do not overlap repeat sequences.”
FIGURE 1.

(A) circBase table browser. Table browser enables the user to design queries for conditional data retrieval. In this example, the user will retrieve all circRNAs from a CD34+ sample, 100–500 nt in length, that are transcribed from coding regions and do not overlap repeat sequences. (B) circBase results page. The user is presented with basic information about each circRNA that matched the search query. Columns, from left to right, contain information on the following: (1) the organism data came from; (2) genomic position; (3) the strand circRNA is transcribed from; (4) a unique identifier; (5) genomic length; (6) length of an in silico predicted spliced form; (7) list of samples the circRNA was observed in; (8) number of reads overlapping the circular junction; (9) repeat sequences overlapping the 3′ and 5′ ends that give rise to a head-to-tail splice junction; (10) circRNA annotation (annotation terms are described in documentation available on the circBase website); (11) an overlapping transcript; (12) gene symbol of the overlapping gene; and (13) a list of studies in which the particular circRNA was detected. (C) Reads mapped to a head-to-tail splice junction. Alignments of reads that support a head-to-tail splice junction can be retrieved from a single record page. Nucleotides that match the genomic reference are printed as dots, while mismatched nucleotides are represented as letters. (D) circBase single record page. In addition to data available from a results page, the user can explore more information about a particular circRNA on a single record page. (E) UCSC genome browser on doRiNA, a database for post-transcriptional regulatory elements. A region surrounding the CDR1as circRNA is shown. In addition to the basic genome browser tracks, the user can explore RNA binding protein PAR-CLIP tracks, ribosome profiling data, miRNA target prediction tracks, and much more.
All search results are returned as a hyperlinked table (Fig. 1B). The table contains basic information on every circRNA that matched the search criteria, while additional information can be accessed by following the related hyperlink. Clicking the genomic position links to the local copy (Anders et al. 2012) of UCSC genome browser, while gene symbols and best transcripts are linked to the NCBI Entrez Gene (Maglott et al. 2011) and Reference Sequence (RefSeq) (Pruitt et al. 2007) databases, respectively (with the exception of C. elegans where they are linked to WormBase) (Chen et al. 2005). Data set and circRNA IDs link to pages with additional information. Apart from data visible in a results table, circRNA single-record pages (Fig. 1D) contain evidence for the occurrence of a particular circRNA, such as prediction score, read counts, alignment of reads to a head-to-tail splice junction (Fig. 1C), and the literature-based evidence for circRNAs that was verified experimentally. Genomic positions are linked to our local UCSC Genome browser, which is a part of the doRiNA database. Therefore, in addition to information retrieved from circBase, the user can explore RNA binding protein target sites, predicted miRNA binding sites and ribosome profiling tracks (Fig. 1E).
Data export
Results can be exported using options in the “Export Results” ribbon below the main menu. Results tables are available in comma-separated values (csv), tab-delimited text (tsv), and Excel Workbook (xlsx) formats, while nucleotide sequence of particular circRNAs can be downloaded as a compressed fasta file. Upon clicking the “fasta” option from the results page, the user can select between genomic or inferred spliced sequence, define a number of upstream and downstream flanking nucleotides, or download an arbitrary number of nucleotides around the 5′ and 3′ splice sites.
Additional flat file export options are available from the “downloads” page.
Conclusions and future work
circBase facilitates circRNA research by (1) providing users with a single repository that collects, unifies, and annotates circRNA data in standardized formats; (2) extending public data sets with additional basic analysis; (3) integrating this data with external resources, such as the UCSC Genome Browser and NCBI databases; and (4) enabling users to interact with the database contents through a simple interface. While we developed circBase, a special emphasis was put on building a simple and self-explanatory interface, as well as writing thorough user documentation that covers all the advanced options that may not be used routinely. We intend to regularly update the database with newly published data. Direct data submission by the users is currently not supported, but the users are encouraged to contact us with requests for adding their data to circBase.
Availability
The database is freely accessible through the web server at http://www.circbase.org/.
MATERIALS AND METHODS
Implementation and contents
Our web server user interface is implemented using HTML, CSS, and JavaScript. Data are stored in a MySQL database, which is interfaced to the front end by a series of server-side Perl CGI scripts. A web browser fully compatible with HTML5 and CSS3 is required for optimal performance. We have thoroughly tested the web server in Google Chrome 31.0, Mozilla Firefox 25.0, and Internet Explorer 10.0.
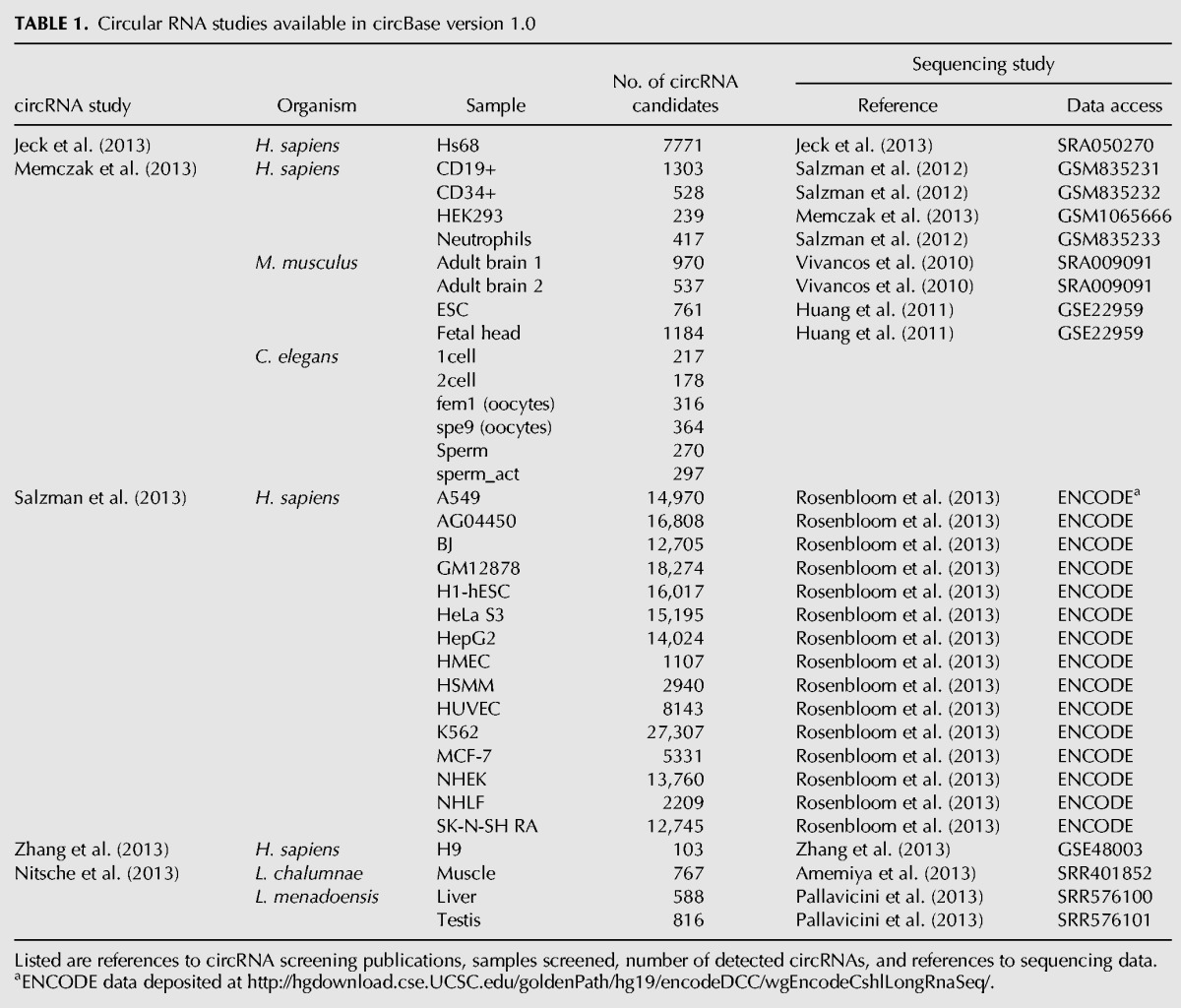
To the best of our knowledge, circBase contains data from all studies of large-scale circRNA identification published to date (Table 1; Jeck et al. 2013; Memczak et al. 2013; Nitsche et al. 2013; Salzman et al. 2013; Zhang et al. 2013). In addition to the information made available by the investigators, we have annotated all circRNA transcripts, predicted their putative spliced forms, and, where applicable, provided alignments of reads spanning head-to-tail junctions. Transcript reannotation was necessary to standardize the content and level the amount of information contained across some publications (Jeck et al. 2013; Salzman et al. 2013). Unique identifiers are assigned to circRNAs and will provide fixed references for future circBase releases.
TABLE 1.
Circular RNA studies available in circBase version 1.0
ACKNOWLEDGMENTS
We thank all members of the N. Rajewsky laboratory for helpful discussions and comments on circBase interface and contents. We thank Christine Kocks and Benedikt Obermayer for their valuable suggestions on improving the manuscript. Marvin Jens is thanked for providing the software for transcript annotation and splice site predictions. P.G. was partially funded by the DFG Graduate School “Computational Systems Biology” (CSB-GRK 1772) and DEEP–German Epigenome Programme. P.P. was funded by the MDC-NYU PhD Exchange Programme.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.043687.113.
REFERENCES
- Amemiya CT, Alfoldi J, Lee AP, Fan S, Philippe H, Maccallum I, Braasch I, Manousaki T, Schneider I, Rohner N, et al. 2013. The African coelacanth genome provides insights into tetrapod evolution. Nature 496: 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C. 2012. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res 40: D180–D186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Harris TW, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Bradnam K, Canaran P, Chan J, Chen CK, et al. 2005. WormBase: a comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res 33: D383–D389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Neeno-Eckwall EC, Adhikari BN, Perna NT, Tisserat N, Leach JE, Levesque CA, Buell CR. 2011. The Comprehensive Phytopathogen Genomics Resource: a web-based resource for data-mining plant pathogen genomes. Database (Oxford) 2011: bar053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. 2013. Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388 [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt L, Teupser D, Hackermueller J, et al. 2014. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol 15: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, Marks H, Klampfl T, Kralovics R, Stunnenberg HG, et al. 2011. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One 6: e27288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sharpless NE. 2014. Detecting and characterizing circular RNAs. Nat Biotechnol 32: 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT: the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. 2011. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 39: D52–D57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338 [DOI] [PubMed] [Google Scholar]
- Nitsche A, Doose G, Tafer H, Robinson M, Saha NR, Gerdol M, Canapa A, Hoffmann S, Amemiya CT, Stadler PF. 2013. Atypical RNAs in the coelacanth transcriptome. J Exp Zool B Mol Dev Evol 10.1002/jez.b.22542 [DOI] [PubMed] [Google Scholar]
- Pallavicini A, Canapa A, Barucca M, Alf Ldi J, Biscotti MA, Buonocore F, De Moro G, Di Palma F, Fausto AM, Forconi M, et al. 2013. Analysis of the transcriptome of the Indonesian coelacanth Latimeria menadoensis. BMC Genomics 14: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35: D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau L, Pelchat M. 2006. The Subviral RNA Database: a toolbox for viroids, the hepatitis delta virus and satellite RNAs research. BMC Microbiol 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. 2012. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. 2013. Cell-type specific features of circular RNA expression. PLoS Genet 9: e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivancos AP, Guell M, Dohm JC, Serrano L, Himmelbauer H. 2010. Strand-specific deep sequencing of the transcriptome. Genome Res 20: 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. 2013. Circular intronic long noncoding RNAs. Mol Cell 51: 792–806 [DOI] [PubMed] [Google Scholar]