

Abstract
Immunoglobulin E (IgE) and its high affinity receptor (FcεRI) are well known participants in the allergic response. The interaction of allergens with FcεRI-bound IgE antibodies is an essential step in mast cell/basophil activation and subsequent release of allergic mediators. It is known that the affinity of the interaction between an IgE antibody and an allergen may differ, raising the question of whether FcεRI can decipher such differences in interaction affinities. If so, do the cellular and physiological outcomes differ? Are the molecular mechanisms intiated by FcεRI similar under low or high affinity interactions? Could the resulting inflammatory response differ? Recent discoveries summarized herein begin to shed new light on these important questions. What we have learned from such studies is that IgE and FcεRI form a complex regulatory network influencing the inflammatory response in allergy and beyond.
Keywords: Antigen affinity, Autoimmunity, Basophils, FcεRI, IgE, Mast Cells, Systemic Lupus Erythematosus
Introduction
Differences in the affinity of IgE antibodies, for an allergen (a term used for allergy-provoking antigens), have long been recognized [1]. Recent studies, primarily done in mice, have shown that some IgE antibodies can be generated in peripheral tissues [2]; in contrast to IgG antibodies. Antibodies generated in peripheral tissues undergo direct class switching rather than a sequential class switch through an IgG intermediate and thus do not undergo an extensive process of affinity maturation through somatic hypermutation [3]. Such IgE antibodies are thought to be of lower affinity, whereas high affinity IgE antibodies are thought to be produced in germinal centers through a IgG1 intermediate that undergoes affinity maturation before sequential switching to the IgE subclass [4,5]. IgE antibody production in peripheral tissue may not be unusual, as it has been demonstrated that the majority of allergen-specific IgE in the blood of allergic patients is not derived from blood-derived B cells or plasma cells [6]. Recent work has also demonstrated that IgE antibodies can be found in various diseases with inflammatory components [7] arguing that dysregulation of IgE production occurs in many inflammatory diseases and may have (patho)physiological consequences. Such findings suggest that differences in the affinity of IgE antibody interactions with an antigen are likely to affect immunological responses. Thus, multiple questions arise: 1. How are differences in the affinity of IgE antibody and antigen interactions interpreted? Does the FcεRI sense these differences? 2. How do these differences influence the molecular signals generated upon FcεRI activation? 3. What are the consequences of these affinity differences at the cellular level? 4. Are there differences in the physiological outcome when the affinity of IgE and antigen interactions differ? 5. Does this suggest a role for IgE antibodies beyond allergic disease?
On mast cells and basophils, the FcεRI consist of an IgE-binding αchain [8], a β chain that serves to amplify signaling [9,10], and two homodimeric γ chains that are essential for the signaling capability of this receptor. The FcεRI's ability to interpret its engagement by IgE and antigen is encoded in immunoreceptor tyrosine-based activation motifs (ITAMs) found in the β and γ chains (fig.1). Phosphorylation of the FcεRI and subsequent phosphorylation of other molecules requires aggregation of FcεRI by IgE-antigen interactions. This allows the appropriate proximity of receptors with the associated Src family protein tyrosine kinase (SrcPTK), Lyn resulting in transphosphorylation [11,12]. The phosphorylation of tyrosine residues found in the ITAMs by Lyn results in the formation of novel docking sites for multiple signaling proteins that become activated and amplify signaling; the most notable example being that of Syk kinase [13,14]. Downstream of these events a plethora of molecular signals in the regulation of FcεRI-dependent effector responses have been uncovered. These events include phosphorylation of scaffold adaptors, like the linker for activation of T cells (LAT) 1 and subsequent association and phosphorylation of phopholipsae Cγ, molecules that serve to organize and promote calcium responses and degranulation in mast cells (fig. 1) [15]. The topic of FcεRI signaling has been extensively covered over the years [16,17] and will not be detailed herein. However, it has been proposed [18,19] that the occurrence of such downstream signaling events leading to a productive effector response requires that the FcεRI remain in an aggregated (or clustered) state for sufficient time to allow such signals to be successfully propagated downstream. This kinetic proofreading hypothesis [18] argues that weak or low affinity stimuli may not promote the appropriate residence time of a receptor in the cluster to allow for productive signaling. Thus, while not absolute, it implies that low affinity IgE antibody and antigen interactions may not yield a productive response. Yet, it is known that individuals with IgE antibodies of low affinity can develop allergies. At the same time, in some diseases the presence of high amounts of IgE [20] or of autoreactive IgE [21,22] does not appear to be associated with increased incidence of allergic disease.
Figure 1. Schematic model of FcεRI signaling generated by high or low affinity antigen and IgE antibody inteactions.
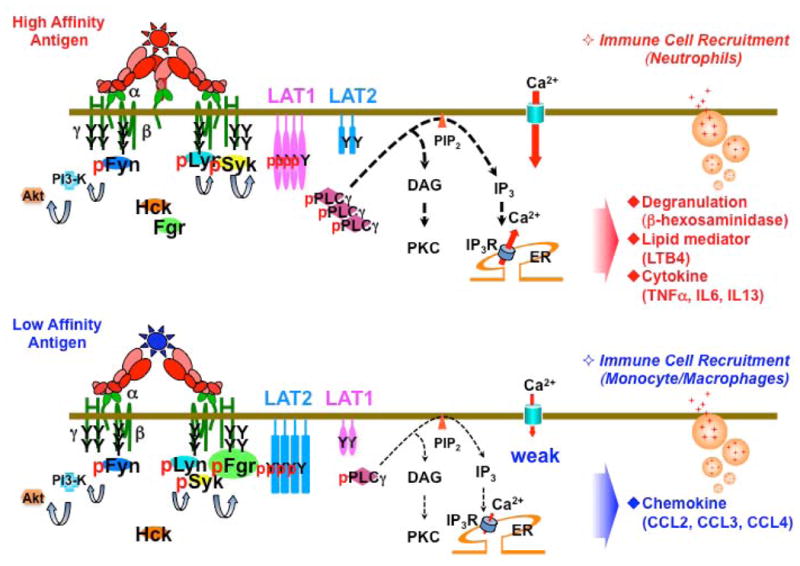
FcεRI is comprised of an IgE-binding α chain, a tretraspanning β chain and two disulfide linked γ chains. Aggregation of FcεRI with multivalent allergen (antigen) results in phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) by Lyn kinase, and leads to activation of Syk kinase through ITAM binding. In case of high affinity antigens (upper panel), activation of Syk results in the phosphorylation of the adapter protein linker for activation of T cells (LAT) 1 and phosphorylation of phopholipase Cg (PLCγ), which is required for generation of inositide trisphosphates (IP3) and normal calcium responses. The Lyn-Syk-LAT1 signals generated by high affinity allergens cooperate with Fyn kinase signals that positively regulate phosphatidylinositol 3-OH kinase (PI3-K) and PKB/Akt pathways. Both Lyn-Syk-LAT1 and Fyn-PI3-K-Akt pathways are required for the initiation of degranulation, lipid mediator release, and cytokine production (upper panel). While the Lyn-Syk-LAT1 signals are dominant under high affinity antigen engagement, low affinity antigens more effectively recruit another Src kinase, Fgr, which shows an enhanced colocalization with FcεRI clusters (lower panel). Furthermore low affinity antigens enhance phosphorylation of the LAT1 related adapter protein, LAT2, which is important for chemokine production and is recruited to the FcεRI clusters. In vivo, high or low affinity antigen stimulation of FcεRI leads to the respective recruitment of neutrophils or monocyte/macrophages and to the development of tissue inflammation. Thus, changes in the usage of both kinases and adapter proteins underlies how FcεRI is able to interpret high or low affinity IgE/antigen interactions, resulting in selective responses that regulate the inflammatory response.
In this commentary we focus on some of our recent studies that uncover previously unappreciated intracacies in the role of IgE and FcεRI in allergy and beyond. We believe such studies begin to explain how differences in the affinity of antibody and antigen interactions are interpreted at the cellular and physiological levels, and how such interactions may play a role in inflammation.
Binding of IgE to FcεRI: Properties and Functional Outcome
The binding of IgE to FcεRI is of very high affinity (KA ≥ 1010M-1). Yet, it is reversible with a half life of greater than six days [23]. The apparent lack of immediate functional cellular consequences (such as degranulation) upon IgE binding to mast cells or basophils led to the view that the binding of IgE is a passive or inert event and that the IgE-occupied FcεRI could be thought of as the “resting” receptor [24,25]. This view was further substantiated by studies [26] demonstrating that cells which had not seen IgE, and were subsequently stimulated with an aggregating antibody directed to FcεRI, had a similar degranulation response as cells sensitized with IgE and reacted with antibody to IgE [27,28]. Later studies, using chemically oligomerized IgE, demonstrated that degranulation could be elicited by an IgE aggregate as small as a dimer whereas monomers of IgE failed to provide a measurable response [26]. Such early studies made a strong argument for the essential role of FcεRI clustering in the induction of mast cell or basophil degranulation and release of allergic mediators.
However, in the past decade, a number of studies demonstrated that the binding of monomeric IgE to FcεRI on mouse mast cells induced both mast cell signals and various effector response [29-31]. A curiosity in such early studies, was that the observed response seemed to be associated with the particular IgE clone used in the study. It was later recognized that such clonal IgE molecules showed heterogeneity in their ability to induce cell responses. These IgE's were subsequently termed highly cytokinergic (HC) or poorly cytokinergic (PC) IgEs based on their respective ability to promote cytokine and other responses [32,33]. It is still unclear as to whether monomeric HC or PC IgE is relevant in humans. Some studies, using either human lung mast cells [34] or human cord blood-derived mast cells [35], reported that monomeric human IgE induced Ca2+ influx, dose-dependent histamine release, LTC4 production, and IL-8 synthesis as well as production of the chemokines I-309, GM-CSF, and MIP-1α. While these findings suggest that monomeric human IgE can promote cellular responses, there remains considerable uncertainty on whether these effects occur in the absence of FcεRI aggregation. This is because structural studies by James and colleagues [36] demonstrated that one type of mouse HC IgE (derived from the SPE-7 clone) changes its conformation upon binding to FcεRI inducing crossreactivity to an undefined cell surface antigen. In addition, HC IgEs are polyreactivity to cellular autoantigens and their functional effects can be disrupted by monovalent haptens [37]. This latter point makes a strong argument that clustering of FcεRI is a necessity for the functional responses elicited by these HC IgEs. Thus it seems that in principle, the effects of “monomeric” IgE are similar to those of IgE-antigen clustering of FcεRI but the aggregation induced by monomeric IgE may possibly differ in the extent (or size) of the receptor clusters and perhaps in their stability.
The Affinity of IgE and Antigen Interactions: Effect on FcεRI Signaling and Function
As mentioned above, it is well known that the affinity of the interaction between an IgE antibody and its cognate antigen can vary. The difference in affinity is likely to influence the size and/or stability of the FcεRI clusters formed. However, whether differences in the size or stability of FcεRI clusters can be recognized and interpreted by the cell, and whether such differences have associated functional consequences, has long remained unknown [38,39]. The aforementioned kinetic proofreading hypothesis [18] argues that weak or low affinity interactions would be non-productive, as they would fail to surpass the required thresholds and thus molecular signals would be abated. However, such models fail to explain how individuals who generate low affinity IgE antibodies can develop allergic disease when exposed to sufficient quantities of an allergen. In addition, kinetic proofreading models have focused on the dominant cellular responses and fail to consider that many other (patho)physiological meaningful cellular responses can occur. Early evidence of an exception to kinetic proofreading paradigm came from a study demonstrating that MCP-1 (CCL2) mRNA production appeared to be independent of the requirement for continued FcεRI clustering [40]. Additional evidence was provided by a later study demonstrating that the production of chemokines like CCL2 required very low FcεRI occupancy by IgE/antigen complexes thus suggesting that weak clustering (somewhat akin to low affinity conditions) of FcεRI might suffice in eliciting some responses [41].
These early studies led us to propose that FcεRI might effectively interpret low affinity IgE and antigen interactions but that the cellular responses might differ relative to those seen with a high affinity IgE and antigen interaction. To test this hypothesis, we utilized previously described [42] antigens (2-nitrophenyl (2NP) and dinitrophenyl (DNP) conjugated to an immunoglobulin Fab fragment as the carrier) that differed in their relative affinities by approximately three orders of magnitude. The IgE used was derived from the H1-DNPε 26.82 clone [43], which has a high affinity for DNP. To test whether downstream molecular signaling, that might lead to a productive cellular response, failed to occur upon low affinity FcεRI engagement we used concentrations of high or low affinity antigens that promoted similar FcεRI phosphorylation. Analysis of multiple mast cell responses demonstrated that low affinity engagement of FcεRI led to a productive response characterized by enhanced chemokine production (fig. 1) [44]. In contrast, degranulation and cytokine production was reduced under these conditions when compared to high affinity engagement of FcεRI. As might be expected, clustering of FcεRI by low affinity IgE and antigen interactions differed from that seen under high affinity conditions. Specifically, the dynamics and mobility of FcεRI clusters under low affinity conditions were markedly reduced relative to high affinity conditions with more FcεRI clusters found in the cells' periphery and with some forming a loose synpase-like structure at the cells' center. In comparison, high affinity engagement of FcεRI resulted in highly dynamic receptor clusters that rapidly moved towards the cell center forming a tight synapse-like structure. Phosphotyrosine was associated with FcεRI clusters under both high and low affinity conditions but the co-localization of phosphotyrosine-containing proteins with FcεRI clusters was more pronounced under low affinity conditions.
To explore whether these differences in FcεRI clustering might be interpreted into changes in the underlying molecular signaling, we investigated the molecular events generated under both conditions. Unexpectedly, a marked increase in association of FcεRI with the SrcPTK Fgr was observed under low affinity engagement of FcεRI (fig. 1) [44]. In contrast, no differences were observed in the association of Lyn with FcεRI under high or low affinity engagement. While it had previously been shown that Fgr could associate with FcεRI and might contribute to its function [45], it was unclear how this kinase might be used by FcεRI. Further examination revealed that the balance in phosphorylation of two related adaptor proteins, LAT1 and LAT2 [44], was altered. These molecules are key as organizers of signalsomes in the plasma membrane of mast cells as well as in other cells. Thus, changes in their phosphorylation status reflect changes in the molecular signals generated. We observed that under low affinity engagement of FcεRI, stronger phosphorylation of LAT2 occurred relative to LAT1 whereas under high affinity FcεRI engagement phosphorylation of LAT1 was favored (fig. 1). Since LAT1 is essential for mast cell calcium responses and contributes to mast cell degranulation [15], a reduction in its phosphorylation under low affinity FcεRI engagement is consistent with the reduced degranulation observed under these conditions. LAT2 appeared to be more important in driving phosphatidyl inositol 3-OH kinase (PI3K) activation and downstream signaling in this pathway. This apparently augments chemokine production and secretion. To test the importance of Fgr and LAT2 to the production of chemokines in mast cells, we utilized mast cells derived from genetically-altered mice deficient in these molecules and found a diminshed production of chemokines [44]. Thus, the findings argue that low affinity engagement of FcεRI is interpreted into functional consequences through alterations of molecular signals that promote selective responses. This would suggest that there may be many exceptions to the kinetic proofreading paradigm and that the affinity of the interaction between the IgE antibody and its cognate antigen when bound to FcεRI is likely to determine whether the receptor is under kinetic proofreading regulatory control.
(Patho)Physiological relevance of low affinity IgE/antigen interactions via FcεRI
While the aforementioned work demonstrated that low affinity IgE/antigen engagement of FcεRI alters cellular signalling and responses, the physiological consequences of this altered response remained of considerable interest. Specifically, one might ask the question: Do alterations in a mast cells' effector response result in an inflammatory response and what are the resulting (patho)physiological consequences? To address these questions we utilized the mouse model of passive cutaneous anaphylaxis, which is based on the elicitation of mast cell responses locally at the site of injection (ear). Such a model would allow us to measure (patho)physiological differences in the local tissue environment following low or high affinity engagement of FcεRI. Conduct of these experiments revealed that both low and high affinity challenges led to similar ear swelling responses, thickness of dermis, and numbers of immune cells infiltrating the tissues. However, vascular permeability was reduced upon low affinity challenge when compared to high affinity challenge [44] consistent with fewer cells showing a degranulated phenotype under low affinity challenge. These findings demonstrated that low affinity engagement of FcεRI in vivo results in an inflammatory response that has strong similarities to that of high affinity engagement but clearly differed in the release of preformed granule content. Exploration of the immune cell types infiltrating the local tissue revealed some important differences. Whereas neutrophils were the dominant cell type infiltrating the local tissue under high affinity engagement of FcεRI, monocyte/macrophages were more abundant in the local tissue when low affinity engagement of FcεRI occurred [44] (fig. 1). The physiological relevance of the difference in immune cell recruitment is unclear. One might speculate that since monocyte/macrophages are more effective producers of cytokines and chemokines than neutrophils, perhaps, low affinity engagement of FcεRI may require the former cells for amplification of the inflammatory response. Regardless, such findings demonstrate that low affinity engagement of FcεRI can promote immune cell recruitment and inflammation in tissues.
IgE and FcεRI; beyond allergic disease
The above findings argue that there may be circumstances whereby engagement of FcεRI may result in inflammation but not necessarily in allergic inflammation (where preformed granule allergic mediators are abundantly released). A postulate in this hypothesis might be that increased amounts of total IgE antibody (over normal circulating levels) may not be neccesary for such an inflammatory response. While the postulate does not exclude increased levels of total IgE, it argues that the presence of normal levels of IgE antibodies in the context of an appropriate antigen might suffice to elicit physiological responses. Given the recent reports of IgE dysregulation in some inflammatory diseases [7], it seems naïve to consider that the production of IgE in such circumstances does not contribute to the disease process. Thus, we set out to test whether IgE contributes in autoimmune inflammation, an inflammatory response generally linked to Th1 and Th17 responses. These studies were in part based on our previous observation of a role for autoreactive IgE in Systemic Lupus Erythematosus (SLE) [46]. This initial study demonstrated that autoreactive IgE functioned to amplify autoimmunity by FcεRI-dependent activation of basophils, which played a key role in plasma cell expansion and survival. This latter point was also supported by additional work demonstrating that activated basophils are highly effective in expansion and survival of plasma cells [47]. Importantly, an initial pilot study of human SLE subjects [46] also demonstrated that autoreactive IgE was associated with increased disease activity, the presence of lupus nephritis, but these subjects did not demonstrate increased allergic responses. These findings suggested that in SLE, the role of IgE and FcεRI was not associated with increased allergic responses. In addition, it should be noted that in human SLE subjects autoreactive antibodies of high and low affinity are prevalent. Thus, it is possible that IgE antibodies can elicit cellular responses independent of degranulation and the release of allergic mediators. In the following sections we will detail these studies and present new evidence for the role of IgE in promoting the immune response.
Prevalence of autoreactive IgE in SLE and its diease association
The overall relevance of our initial report [46] describing a role for autoreactive IgE in SLE onset and progression was questioned [48], based on previous studies demonstrating that approximately only 30% of human SLE subjects had detectable levels of autoreactive IgE [49]. This percentage was determined primarily on the detection of dsDNA IgE, which similar to dsDNA IgG showed a highly significant association with disease parameters. Thus, we conducted an expanded study to determine the overall prevalence of autoreactive IgE in SLE, what auto-antigens induced these antibodies, and what auto-antigen specificities were associated with disease parameters. Our study utilized approximately 200 human subjects in France and the United States [50]. Overall, the two cohorts studied did not differ markedly and individuals with known allergies or infections were excluded from the study. Screening of these individuals for IgE-reactive auto-antigens revealed that IgE antibodies were generated to at least seven autoantigens. Four of these were the well known SLE auto-antigens, dsDNA, Sm, Ro/SSA, and La/SSB. The three other auto-antigens were less well known, APEX nuclease 1 (APEX), N-methylpurine DNA-glycosolase (MPG), and CAP-Gly domain-containing linker protein family member 4 (CLIP4), but all are nuclear proteins. These latter three were found in many SLE subjects but were not detected in the majority of healthy controls. Assessment of the prevalence of these autoreactive IgE's in the studied SLE subjects demonstrated that approximately 65% of these subjects had autoreactive IgE to one or more of these seven auto-antigens. Additionally, when SLE subjects were stratified on the basis of active disease (using the SLE-disease activity index; SLEDAI), almost 83% of subjects with active disease had autoreactive IgE to one or more of these seven auto-antigens. Of particular interest, was the finding that the auto-antigens APEX, MPG, and CLIP4 seemed to selectively elicit IgE and not IgG antibody responses in SLE subjects [50]. All IgE autoantibodies showed a statistically significant association with increased disease activity, albeit dsDNA IgE showed the highest significance in association [50]. Autoreactive IgE's with specificity towards dsDNA, APEX, MPG, and CLIP4 also showed a statistically significant association with lupus nephritis and with hypocomplementemia, a serological marker of active disease. A multivariate analysis of the predictive value of dsDNA IgE and dsDNA IgG for SLE diagnosis demonstrated that the use of both of these parameters, along with age, in disease diagnosis enhanced the predictive value. Thus, these findings demonstrated that autoreactive IgE is highly prevalent in SLE and, like autoreactive IgG, is linked to disease activity and lupus nephritis. These findings strongly suggest that the presence of autoreactive IgE is playing a contributory role in autoimmune inflammation without manifesting as an enhanced allergic responses in the studied populations.
Uncovering the role of IgE in autoimmune inflammation
The aforementioned study [50] led us to develop mouse models in which one might study the role of IgE in autoimmune disease in the absence of a dominant Th2 environment. Previous work [46] using Lyn kinase-deficient mice (that develop spontaneous lupus-like disease) suggested that IgE was an important contributor to disease development. However, in the early life of these mice there is a strong Th2 skewing with the development of an atopic-like allergic disease. Thus, one could not exclude that this phenotype might be contributory in exacerbating the lupus-like disease seen in later life. To remove this confounding factor, we chose a well-described model [51] of spontaneous lupus-like disease, the FcγRII-deficient mice in the C57BL/6 background. We also studied the role of IgE in FcγRIIB-/- mice carrying the chromosomal translocation of Y-linked autoimmune acceleration (Yaa) [51], which accelerates the development of the lupus-like phenotype due to a duplication of the Tlr7 gene in male mice only and is demonstated in a more aggressive disease [52,53]. The FcγRIIB locus encodes an inhibitory receptor that controls the activation of the many cell types [54] and polymorphisms in this gene were previously associated with lupus [55]. Neither of these models demonstrated enhanced Th2 reponses, increased levels of total IgE, nor spontaneous allergic-like phenotype, despite the reported higher susceptibility of FcγRIIB-/- mice to type I hypersensitivity due to increased effector cell responses [56]. These mice developed increased levels of autoreactive IgE providing a model that could be crossed to the well characterized [57] Igh7-/- mice (IgE-deficient in C57BL/6 background) to study the role of IgE autoantibody production in the inflammation and lupus-like phenotype seen in FcγRIIB-/- and FcγRIIB-/-.Yaa (BD and JR, submitted manuscript).
Study of the life span of IgE-deficient FcγRIIB-/- and FcγRIIB-/-.Yaa mice provided some unexpected results. FcγRIIB-/- and FcγRIIB-/-.Yaa mice have a median survival of 7.3 and 4.8 months, respectively. However, when they are incapable of producing IgE antibodies the median survival for FcγRIIB-/- mice was undetermined, as they survived well beyond a 12 month period, and FcγRIIB-/-.Yaa mice survived a median of 11 months (Table 1). This prolongation of life, when IgE antibodies are absent (in particular in the highly aggressive FcγRIIB-/-.Yaa mouse model), demonstrated that the production of IgE plays a role in disease onset and progression. Concommitant with the increased survival, analysis of circulating autoantibodies of the IgG subclass as well as plasma cell numbers in the spleen and lymph nodes demonstrated a marked reduction in both (Table 1). Nonetheless, it is clear that the FcγRIIB-/-.Yaa mice still developed disease in late life, implying that IgE is functioning to amplify autoimmune inflammation but is not essential to the development of disease in this highly aggressive model. To further understand the role of IgE, we investigated the impact of IgE-deficiency on immune cell recruitment to the secondary lymphoid organs in both FcγRIIB-/- and FcγRIIB-/-.Yaa mice (submitted manuscript). Remarkably, the absence of IgE caused a marked diminution of immune cells (of all types) to the secondary lymphoid organs, demonstrating a previously unknown role for IgE in immune cell recruitment during inflammation. These findings, together with our previous demonstration of a role for basophils in SLE [46], raise the question of whether the basophil is contributing to the autoimmune inflammation seen in FcγRIIB-/- and FcγRIIB-/-.Yaa mice. While we cannot conclusively answer this question with the current study, our findings showed that the basophil numbers in the secondary lymphoid organs of IgE-deficient FcγRIIB-/- and FcγRIIB-/-.Yaa mice did not differ from IgE-sufficient FcγRIIB-/- and FcγRIIB-/-.Yaa mice. However, whereas the basophils in the secondary lymphoid organs of FcγRIIB-/- and FcγRIIB-/-.Yaa mice were activated those in the secondary lymphoid organs of IgE-deficient FcγRIIB-/- and FcγRIIB-/-.Yaa mice were not. Thus, this finding suggests a role for autoreactive IgE-FcεRI activated basophils, residing in the secondary lymphoid tissues, in facilitating the recrutiment of other immune cells into these tissues. A more robust model in which the presence or absence of basophils in these tissues can be manipulated should provide more definitive evidence.
Table 1.
Phenotypic differences in the lupus-like disease model of FcγRIIB-/- and FcγRIIB-/-.Yaa mice that express the IgE locus or not.
| lgE+ | IgE- | ||||
|---|---|---|---|---|---|
|
| |||||

|

|
||||
|
| |||||
| Kidney | ACR | 0.5 | 1.3 | 0.1 | 0.5 |
| C3 | ++ | ++ | - | - | |
| IgG | + | ++ | - | - | |
| Nephritis score | 6 | 10 | - | 1 | |
|
| |||||
| Serum | dsDNA IgG | ++ | + | + | - |
| RNP IgG | + | ++ | - | - | |
| Histones IgG | ++ | + | - | - | |
|
| |||||
| SLO | Size | Hypertrophy | Hypertrophy | Normal | Normal |
| CD138+ cells | ↑ | ↑↑ | Normal | ↑ | |
| CD19+ B220+ cells | ↑ | ↑↑ | Normal | Normal | |
| CD11b+ CD11c+ cells | ↑ | ↑↑ | Normal | Normal | |
| Ly6Clo-int cells | ↑↑ | ↑↑ | Normal | Normal | |
| Ly6Chi cells | ↑↑ | ↑↑ | Normal | Normal | |
| Neutrophils cells | ↑↑ | ↑↑ | Normal | Normal | |
| CD200R+ basophils | ↑ | ↑↑ | Normal | Normal | |
|
| |||||
| Average Life spam (months) | 7.3 | 4.8 | >12 | 11 | |
ACR: albumin/ creatinine ratio: C3: complement 3: CD: cluster of differentiation; RNF: nbonucleoproteins
Perspectives
A role for IgE in allergic and autoimmune inflammation in both human and mouse is evident from the aforementioned studies. We speculate that the low affinity IgE and antigen interactions, which elicit cellular responses in the absence of the release of preformed allergic mediators, may be an essential component to the development of IgE-dependent inflammation with no apparent allergic manifestation. This is consistent with the accumulating evidence of IgE production independently of germinal center maturation [3], not being derived from circulating B cells or plasma cells [6], but instead produced at the local site of inflammation [5]. Nonetheless, the majority of allergen-specific IgE in the blood of allergic subjects is thought to be locally produced [6] and thus assumed to be of “low affinity”, yet these subjects develop allergic symptoms. Our findings show that, in contrast to most allergic subjects, the total circulating IgE levels in SLE subjects is normal. This would suggest that occupancy of FcεRI by antigen-specific IgE is likely to be significantly greater in allergic subjects when compared to SLE subjects. This may be an important factor in eliciting the release of preformed mediators from mast cells or basophils, since we previously demonstrated that antigen-specific IgE occupancy of FcεRI must exceed 10% for degranulation to occur whereas chemokine production is elicited at this level of receptor occupancy [41]. It may also be possible that the tissue microenvironment in SLE is inhibitory for mast cell or basophil degranulation. Regardless, the lower levels of antigen-specific IgE in SLE subjects bodes well for strategies in which IgE binding to FcεRI may be employed as a means to intervene in IgE-dependent non-allergic inflammation, and clinical studies have been initiated to explore the effectiveness of anti-IgE therapy (omalizumab) in the treatment of SLE. While a clear mechanistic understanding of the role of IgE in inflammation remains to be elucidated, the uncovering of the contribution of IgE and antigen interactions in promoting non-allergic inflammation extends the current view of the IgE-FcεRI axis beyond allergic disease.
Acknowledgments
The authors wish to acknowledge the many co-authors and contributors to the work summarized herein. We particularly wish to acknowledge Dr. Nicolas Charles and the clinical teams at NIAMS-NIH and at Hôpital Bichat, Assistance Publique-Hôpitaux de Paris, Université Paris Diderot, for their invaluable contributions in the study of human SLE subjects.
Footnotes
The work described herein was primarily supported by the Intramural Research Program of the National Insititute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health.
Contributor Information
Barbara Dema, Email: barbara-isabel.dema-jimenez@inserm.fr.
Ryo Suzuki, Email: suzukir@phar.nagoya-cu.ac.jp.
Juan Rivera, Email: juan_rivera@nih.gov, SciKnow259@comcast.net.
References
- 1.Jackola DR, Pierson-Mullany LK, Liebeler CL, Blumenthal MN, Rosenberg A. Variable binding affinities for allergen suggest a ‘selective competition’ among immunoglobulins in atopic and non-atopic humans. Mol Immunol. 2002;39:367–377. doi: 10.1016/s0161-5890(02)00108-6. [DOI] [PubMed] [Google Scholar]
- 2.Takhar P, Smurthwaite L, Coker HA, Fear DJ, Banfield GK, Carr VA, Durham SR, Gould HJ. Allergen drives class switching to ige in the nasal mucosa in allergic rhinitis. Journal of immunology. 2005;174:5024–5032. doi: 10.4049/jimmunol.174.8.5024. [DOI] [PubMed] [Google Scholar]
- 3.Erazo A, Kutchukhidze N, Leung M, Christ AP, Urban JF, Jr, Curotto de Lafaille MA, Lafaille JJ. Unique maturation program of the ige response in vivo. Immunity. 2007;26:191–203. doi: 10.1016/j.immuni.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong H, Dolpady J, Wabl M, Curotto de Lafaille MA, Lafaille JJ. Sequential class switching is required for the generation of high affinity ige antibodies. J Exp Med. 2012;209:353–364. doi: 10.1084/jem.20111941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiong H, Curotto de Lafaille MA, Lafaille JJ. What is unique about the ige response? Adv Immunol. 2012;116:113–141. doi: 10.1016/B978-0-12-394300-2.00004-1. [DOI] [PubMed] [Google Scholar]
- 6.Eckl-Dorna J, Pree I, Reisinger J, Marth K, Chen KW, Vrtala S, Spitzauer S, Valenta R, Niederberger V. The majority of allergen-specific ige in the blood of allergic patients does not originate from blood-derived b cells or plasma cells. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2012;42:1347–1355. doi: 10.1111/j.1365-2222.2012.04030.x. [DOI] [PubMed] [Google Scholar]
- 7.Heimall J, Freeman A, Holland SM. Pathogenesis of hyper ige syndrome. Clin Rev Allergy Immunol. 2010;38:32–38. doi: 10.1007/s12016-009-8134-1. [DOI] [PubMed] [Google Scholar]
- 8.Metzger H, Kinet JP, Blank U, Miller L, Ra C. The receptor with high affinity for ige. Ciba Found Symp. 1989;147:93–101. discussion 101-113. [PubMed] [Google Scholar]
- 9.Lin S, Cicala C, Scharenberg AM, Kinet JP. The fcerib subunit functions as an amplifier of fcerig-mediated cell activation signals. Cell. 1996;85:985–995. doi: 10.1016/s0092-8674(00)81300-8. [DOI] [PubMed] [Google Scholar]
- 10.On M, Billingsley JM, Jouvin MH, Kinet JP. Molecular dissection of the fcrb signaling amplifier. J Biol Chem. 2004;279:45782–45790. doi: 10.1074/jbc.M404890200. [DOI] [PubMed] [Google Scholar]
- 11.Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the high-affinity receptor for ige is phosphorylated upon aggregation. Proc Natl Acad Sci U S A. 1994;91:11246–11250. doi: 10.1073/pnas.91.23.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonakis BM, Chen H, Haleem-Smith H, Metzger H. The unique domain as the site on lyn kinase for its constitutive association with the high affinity receptor for ige. J Biol Chem. 1997;272:24072–24080. doi: 10.1074/jbc.272.38.24072. [DOI] [PubMed] [Google Scholar]
- 13.Paolini R, Serra A, Kinet JP. Persistence of tyrosine-phosphorylated fceri in deactivated cells. J Biol Chem. 1996;271:15987–15992. doi: 10.1074/jbc.271.27.15987. [DOI] [PubMed] [Google Scholar]
- 14.Yamashita T, Suzuki R, Backlund PS, Yamashita Y, Yergey AL, Rivera J. Differential dephosphorylation of the fcrg immunoreceptor tyrosine-based activation motif tyrosines with dissimilar potential for activating syk. J Biol Chem. 2008;283:28584–28594. doi: 10.1074/jbc.M802679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saitoh S, Arudchandran R, Manetz TS, Zhang W, Sommers CL, Love PE, Rivera J, Samelson LE. Lat is essential for fceri-mediated mast cell activation. Immunity. 2000;12:525–535. doi: 10.1016/s1074-7613(00)80204-6. [DOI] [PubMed] [Google Scholar]
- 16.Rivera J, Gilfillan AM. Molecular regulation of mast cell activation. J Allergy Clin Immunol. 2006;117:1214–1225. doi: 10.1016/j.jaci.2006.04.015. quiz 1226. [DOI] [PubMed] [Google Scholar]
- 17.Galli SJ, Tsai M. Ige and mast cells in allergic disease. Nat Med. 2012;18:693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKeithan TW. Kinetic proofreading in t-cell receptor signal transduction. Proc Natl Acad Sci U S A. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hlavacek WS, Redondo A, Metzger H, Wofsy C, Goldstein B. Kinetic proofreading models for cell signaling predict ways to escape kinetic proofreading. Proc Natl Acad Sci U S A. 2001;98:7295–7300. doi: 10.1073/pnas.121172298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson EE, Irons JS, Patterson R, Roberts M. Serum ige concentration in atopic dermatitis. Relationship to severity of disease and presence of atopic respiratory disease. J Allergy Clin Immunol. 1974;54:94–99. doi: 10.1016/0091-6749(74)90037-2. [DOI] [PubMed] [Google Scholar]
- 21.Atta AM, Sousa CP, Carvalho EM, Sousa-Atta ML. Immunoglobulin e and systemic lupus erythematosus. Braz J Med Biol Res. 2004;37:1497–1501. doi: 10.1590/s0100-879x2004001000008. [DOI] [PubMed] [Google Scholar]
- 22.Budde IK, de Heer PG, Natter S, Mahler V, van der Zee JS, Valenta R, Aalberse RC. Studies on the association between immunoglobulin e autoreactivity and immunoglobulin e-dependent histamine-releasing factors. Immunology. 2002;107:243–251. doi: 10.1046/j.1365-2567.2002.01475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furuichi K, Rivera J, Isersky C. The receptor for immunoglobulin e on rat basophilic leukemia cells: Effect of ligand binding on receptor expression. Proc Natl Acad Sci U S A. 1985;82:1522–1525. doi: 10.1073/pnas.82.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metzger H. The cellular receptor for ige Receptors and recognition P Cuatrecasas and m F Greaves. London: Chapman and Hall; 1977. pp. 75–102. [Google Scholar]
- 25.Metzger H. The ige-mast cell system as a paradigm for the study of antibody mechanisms. Immunol Rev. 1978;41:186–199. doi: 10.1111/j.1600-065x.1978.tb01465.x. [DOI] [PubMed] [Google Scholar]
- 26.Fewtrell C, Metzger H. Larger oligomers of ige are more effective than dimers in stimulating rat basophilic leukemia cells. Journal of immunology. 1980;125:701–710. [PubMed] [Google Scholar]
- 27.Ishizaka T, Chang TH, Taggart M, Ishizaka K. Histamine release from rat mast cells by antibodies against rat basophilic leukemia cell membrane. Journal of immunology. 1977;119:1589–1596. [PubMed] [Google Scholar]
- 28.Isersky C, Taurog JD, Poy G, Metzger H. Triggering of cultured neoplastic mast cells by antibodies to the receptor for ige. Journal of immunology. 1978;121:549–558. [PubMed] [Google Scholar]
- 29.Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, Krystal G. Monomeric ige stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. 2001;14:801–811. doi: 10.1016/s1074-7613(01)00159-5. [DOI] [PubMed] [Google Scholar]
- 30.Kashiwakura J, Otani IM, Kawakami T. Monomeric ige and mast cell development, survival and function. Adv Exp Med Biol. 2011;716:29–46. doi: 10.1007/978-1-4419-9533-9_3. [DOI] [PubMed] [Google Scholar]
- 31.Asai K, Kitaura J, Kawakami Y, Yamagata N, Tsai M, Carbone DP, Liu FT, Galli SJ, Kawakami T. Regulation of mast cell survival by ige. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 32.Kitaura J, Eto K, Kinoshita T, Kawakami Y, Leitges M, Lowell CA, Kawakami T. Regulation of highly cytokinergic ige-induced mast cell adhesion by src, syk, tec, and protein kinase c family kinases. Journal of immunology. 2005;174:4495–4504. doi: 10.4049/jimmunol.174.8.4495. [DOI] [PubMed] [Google Scholar]
- 33.Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, Kawakami Y, Liu FT, Lowell CA, Barisas BG, Galli SJ, Kawakami T. Evidence that ige molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the fceri. Proc Natl Acad Sci U S A. 2003;100:12911–12916. doi: 10.1073/pnas.1735525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cruse G, Kaur D, Yang W, Duffy SM, Brightling CE, Bradding P. Activation of human lung mast cells by monomeric immunoglobulin e. Eur Respir J. 2005;25:858–863. doi: 10.1183/09031936.05.00091704. [DOI] [PubMed] [Google Scholar]
- 35.Gilchrest H, Cheewatrakoolpong B, Billah M, Egan RW, Anthes JC, Greenfeder S. Human cord blood-derived mast cells synthesize and release i-309 in response to ige. Life Sci. 2003;73:2571–2581. doi: 10.1016/s0024-3205(03)00607-6. [DOI] [PubMed] [Google Scholar]
- 36.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 37.Kashiwakura J, Okayama Y, Furue M, Kabashima K, Shimada S, Ra C, Siraganian RP, Kawakami Y, Kawakami T. Most highly cytokinergic iges have polyreactivity to autoantigens. Allergy Asthma Immunol Res. 2012;4:332–340. doi: 10.4168/aair.2012.4.6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacGlashan DW, Jr, Peters SP, Warner J, Lichtenstein LM. Characteristics of human basophil sulfidopeptide leukotriene release: Releasability defined as the ability of the basophil to respond to dimeric cross-links. Journal of immunology. 1986;136:2231–2239. [PubMed] [Google Scholar]
- 39.MacGlashan DW, Jr, Schleimer RP, Lichtenstein LM. Qualitative differences between dimeric and trimeric stimulation of human basophils. Journal of immunology. 1983;130:4–6. [PubMed] [Google Scholar]
- 40.Liu ZJ, Haleem-Smith H, Chen H, Metzger H. Unexpected signals in a system subject to kinetic proofreading. Proc Natl Acad Sci U S A. 2001;98:7289–7294. doi: 10.1073/pnas.121171998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Espinosa C, Odom S, Olivera A, Hobson JP, Martinez ME, Oliveira-Dos-Santos A, Barra L, Spiegel S, Penninger JM, Rivera J. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity ige receptor on mast cells. J Exp Med. 2003;197:1453–1465. doi: 10.1084/jem.20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torigoe C, Inman JK, Metzger H. An unusual mechanism for ligand antagonism. Science. 1998;281:568–572. doi: 10.1126/science.281.5376.568. [DOI] [PubMed] [Google Scholar]
- 43.Liu FT, Bohn JW, Ferry EL, Yamamoto H, Molinaro CA, Sherman LA, Klinman NR, Katz DH. Monoclonal dinitrophenyl-specific murine ige antibody: Preparation, isolation, and characterization. Journal of immunology. 1980;124:2728–2737. [PubMed] [Google Scholar]
- 44.Suzuki R, Leach S, Liu W, Ralston E, Scheffel J, Zhang W, Lowell CA, Rivera J. Molecular editing of cellular responses by the high-affinity receptor for ige. Science. 2014;343:1021–1025. doi: 10.1126/science.1246976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, Kim JW, Kim do K, Kim HS, Park HJ, Park DK, Kim AR, Kim B, Beaven MA, Park KL, Kim YM, Choi WS. The src family kinase fgr is critical for activation of mast cells and ige-mediated anaphylaxis in mice. Journal of immunology. 2011;187:1807–1815. doi: 10.4049/jimmunol.1100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charles N, Hardwick D, Daugas E, Illei GG, Rivera J. Basophils and the t helper 2 environment can promote the development of lupus nephritis. Nat Med. 2010;16:701–707. doi: 10.1038/nm.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez Gomez M, Talke Y, Goebel N, Hermann F, Reich B, Mack M. Basophils support the survival of plasma cells in mice. Journal of immunology. 2010;185:7180–7185. doi: 10.4049/jimmunol.1002319. [DOI] [PubMed] [Google Scholar]
- 48.Bosch X, Lozano F, Cervera R, Ramos-Casals M, Min B. Basophils, ige, and autoantibody-mediated kidney disease. Journal of immunology. 2011;186:6083–6090. doi: 10.4049/jimmunol.1002648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Atta AM, Santiago MB, Guerra FG, Pereira MM, Sousa Atta ML. Autoimmune response of ige antibodies to cellular self-antigens in systemic lupus erythematosus. Int Arch Allergy Immunol. 2010;152:401–406. doi: 10.1159/000288293. [DOI] [PubMed] [Google Scholar]
- 50.Dema B, Pellefigues C, Hasni S, Gault N, Jiang C, Ricks TK, Bonelli MM, Scheffel J, Sacre K, Jablonski M, Gobert D, Papo T, Daugas E, Illei G, Charles N, Rivera J. Autoreactive ige is prevalent in systemic lupus erythematosus and is associated with increased disease activity and nephritis. PLoS One. 2014;9:e90424. doi: 10.1371/journal.pone.0090424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in fcgammariib(-/-) mice. J Exp Med. 2002;195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive b cell responses to rna-related antigens due to tlr7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 53.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malbec O, Fridman WH, Daeron M. Negative regulation of hematopoietic cell activation and proliferation by fc gamma riib. Curr Top Microbiol Immunol. 1999;244:13–27. [PubMed] [Google Scholar]
- 55.Li X, Ptacek TS, Brown EE, Edberg JC. Fcgamma receptors: Structure, function and role as genetic risk factors in sle. Genes Immun. 2009;10:380–389. doi: 10.1038/gene.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in fc gamma rii-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 57.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in ige-deficient mice. Nature. 1994;370:367–370. doi: 10.1038/370367a0. [DOI] [PubMed] [Google Scholar]