

Abstract
The tumor suppressor protein Par-4, which is secreted by normal cells, selectively induces apoptosis in cancer cells. We identified a 3-arylquinoline derivative, designated Arylquin 1, as a potent Par-4 secretagogue in cell cultures and mice. Mechanistically, Arylquin 1 binds to vimentin, displaces Par-4 from vimentin for secretion and triggers the efficient paracrine apoptosis of diverse cancer cells. Thus, targeting vimentin with Par-4 secretagogues efficiently induces paracrine apoptosis of tumor cells.
Keywords: 3-Arylquinolines, Par-4, Apoptosis, Secretagogues
Lung cancer is the most frequently diagnosed cancer and the leading cause of cancer-related deaths in the world 1. Lung tumor cells with p53 mutations or deletions often develop resistance to chemotherapy and radiation therapy leading ultimately to the demise of the patients 2-4. Rather interestingly, such p53-deficient cancer cells are susceptible to apoptosis by the pro-apoptotic tumor suppressor called Prostate apoptosis response-4 (Par-4) 5, 6, 7. Par-4 induces apoptosis in diverse cancer cells but not in normal cells 7. Par-4 is ubiquitously expressed in normal cells and tissues, but it is inactivated, down-regulated, or mutated in several types of cancers 7, 8, 9. Both intracellular and secreted Par-4 play a role in apoptosis induction by caspase-dependent mechanisms 7. Par-4 is secreted in cell culture-conditioned medium (CM) or systemically in mice from normal cells, and extracellular Par-4 binds to its receptor GRP78 on the cancer cell surface and induces apoptosis 5, 6. In contrast, normal cells express low to undetectable levels of basal or inducible cell surface GRP78 and are resistant to apoptosis by extracellular Par-4 5, 6.
Because the baseline levels of Par-4 secreted by normal cells are generally inadequate to cause massive apoptosis in cancer cell cultures, secretogogues that bolster the release of Par-4 constitute an important therapeutic advance. Nutlin-3a, originally developed as an MDM2 inhibitor 10, stimulated Par-4 secretion at micromolar levels in mouse embryonic fibroblast (MEF) cells 11. The presence of halogen substituents on an aromatic ring, two aromatic rings separated by a two-atom spacer (i.e., 1,2-diphenylethane subunit) and a nitrogen-containing heterocycle (i.e., an imidazole subunit) in Nutlin-3a enabled us to screen an in-house library that possessed similar features, namely halogenated aromatic rings separated by two-atom spacers (i.e.,1,2-diphenylethene or stilbene subunits) and nitrogen-containing heterocycles. Specifically, we focused on halogenated 3-arylquinolines, 3-arylquinolones, and 3-arylthioquinolones, which possessed stilbene subunits and nitrogen- containing heterocyclic rings imbedded within their structures. A stilbene subunit imbedded within a 3-arylquinoline, for example, is highlighted in Supplementary Results, Supplementary Note. We screened representative members of each of these heterocyclic families on a compound-by-compound basis for the secretion of Par-4 protein from normal mouse fibroblasts under conditions that were not toxic to the cells. Initial expectations were that these heterocycles would serve as Nutlin-3a surrogates and inhibit MDM2, but studies reported herein established a completely different mechanism of action (Supplementary Figure 1), reflecting that the structural dissimilarities between these heterocycles and Nutlin-3a outweighed the similarities that led to their initial selection for screening.
Within this library of compounds, the fluorinated 3-arylquinolines proved particularly promising in promoting Par-4 secretion. Structure-activity studies defined that 3-arylquinolines, such as Arylquin 1 (1) (Figure 1a and Supplementary Figure 2), was most active as the leading member of a new class of “small-molecule” Par-4 secretagogues. Arylquin 1 produced a dose-dependent secretion in MEF cells (Figure 1b). Arylquin 1 also induced robust secretion of Par-4 in normal/immortalized human cells, but failed to induce the secretion of Par-4 in a panel of lung tumor cells (Supplementary Figure 3). By contrast, prostate cancer cells showed induction of Par-4 secretion with Arylquin 1 relative to vehicle control (Supplementary Figure 3). Consistent with previous studies 5, Brefeldin A, which blocked anterograde endoplasmic reticulum-Golgi traffic, inhibited basal as well as Arylquin 1-inducible Par-4 secretion (Supplementary Figure 3). These findings indicated that Arylquin 1 regulated Par-4 secretion via the classical secretory pathway.
Figure 1. Arylquin 1 displaces Par-4 bound to vimentin to induce Par-4 secretion.
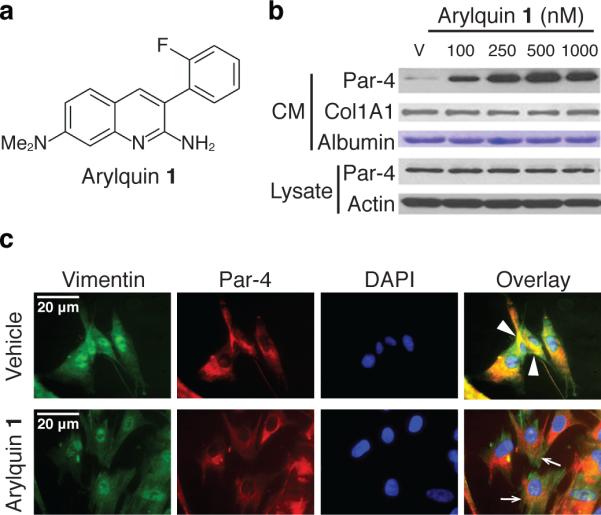
a. Chemical structure of Arylquin 1. Arylquin 1 contains 2-amino and 7-(N,N-dimethyl)amino substituents on the quinoline ring and an ortho-fluorine on the C-3 aryl group.
b. Arylquin 1 induces dose-dependent secretion of Par-4. MEF cells were treated with the indicated concentrations of Arylquin 1 or vehicle (V), and Par-4 in the conditioned medium (CM) or whole-cell lysate was quantified by Western blot analysis. Albumin or collagen 1A1 in the CM, or intracellular β-actin in the lysate served as a loading control. Uncut gels for Figure 1b can be found in Supplementary Figure 17.
c. Par-4 co-localizes with vimentin and is displaced from vimentin by Arylquin 1 treatment of cells. HEL cells, treated with vehicle or Arylquin 1 (500 nM) for 24 h, were subjected to ICC for Par-4 (red fluorescence) and vimentin (green fluorescence). Cells were stained with DAPI to reveal their nuclei (cyan fluorescence). Colocalization of Par-4 and vimentin in the Overlay images shown in vehicle panel is indicated by arrowheads (yellow fluorescence), and dissociation of Par-4 and vimentin (loss of yellow fluorescence, but retention of red and green fluorescence) is indicated by arrows in the Arylquin 1 panel.
To identify the molecular target responsible for the observed Par-4 secretory activity, we synthesized a biotinylated Arylquin 9 (9) (Supplementary Note). Biotinylated Arylquin 9 was confirmed experimentally to retain Par-4 secretory properties (Supplementary Figure 4) and was then used in pull-down experiments for potential protein targets in mouse fibroblasts (MEFs) and human fibroblasts (HEL). We identified vimentin, a cytoskeletal intermediate filament protein 12, as its principal target (Supplementary Figures 5 and 6). The binding of Par-4 to vimentin was experimentally confirmed by co-immunoprecipitation experiments: the Par-4 antibody co-immunoprecipitated endogenous vimentin, and the vimentin antibody coimmunoprecipitated endogenous Par-4 (Supplementary Figure 7). Immunocytochemical analysis confirmed that Par-4 co-localized with vimentin (Figure 1c). On the other hand, cells treated with Arylquin 1 showed neither Par-4 co-immunoprecipitation (Supplementary Figure 7) nor co-localization (Figure 1c) with vimentin, indicating that Arylquin 1 displaced Par-4 from vimentin. This action of Arylquin 1 was not associated with inhibition of vimentin expression (Supplementary Figure 8), suggesting that Arylquin 1 may cause conformational changes in vimentin to inhibit its ability to bind and sequester Par-4 or compete for a hydrophobic binding region on vimentin crucial for Par-4 binding. The differential regulation of Par-4 secretion in normal and various cancer cells by Arylquin 1 (noted above in Supplementary Figure 3) may reflect distinct posttranslational modification patterns of Par-4 and/or vimentin; studies are underway to address the underlying mechanism.
Computer modeling using molecular dynamics simulations led to a minimum-energy structure in which Arylquin 1 binds tetrameric vimentin in a hydrophobic pocket that lies between a pair of head-to-tail α-helical dimers (Supplementary Figure 9). The spatial arrangement of functional groups within Arylquin 1 was ideally suited to stabilize binding to vimentin (Supplementary Figure 9). Additional modeling revealed that Arylquin 1 and its analogs examined bind vimentin in the same orientation but with different binding energies (Supplementary Figure 10-12). The relative values of the calculated binding energies are qualitatively consistent with experimental trends: Arylquin 1, Arylquin 6 (6) and Arylquin 8 (8) with the largest binding energies promoted the highest levels of Par-4 secretion (Supplementary Figure 2 and Supplementary Table 1). The fluorine group in Arylquin 1 was indispensable for activity, and the removal of the fluorine was accompanied by reduced binding (Supplementary Table 1) and concomitant loss of Par-4 secretory activity (Supplementary Figure 2).
Because targeting vimentin may induce apoptosis, we tested normal cells and diverse cancer cells for apoptosis by Arylquin 1. Arylquin 1 induced the dose-dependent apoptosis in cancer cells but not in normal cells (Figure 2a and Supplementary Figure 13). Importantly, 500 nM amounts of Arylquin 1, which triggered secretion of Par-4 from normal cells but not lung cancer cells, did not directly induce apoptosis in normal or cancer cells. By contrast, 500 nM amounts of Arylquin 1 induced apoptosis of PC-3 cells and its derivative PC-3MM2, which are sensitive to apoptosis by Par-4, but not in LNCaP or DU145 cells, which are resistant to apoptosis by Par-4 5,6.
Figure 2. Arylquin 1 induces paracrine apoptosis in cancer cells.

a. Arylquin 1 induces apoptosis. The indicated amounts of Arylquin 1 or vehicle (V) were added to the cells to test for apoptosis.
b. Arylquin 1 induces paracrine apoptosis. Cancer cells were co-cultured with MEFs and treated with Arylquin 1 (500 nM) or vehicle and tested for apoptosis.
c. Arylquin 1 induces systemic Par-4 pro-apoptotic activity. (Left Panel) Serum from mice injected with Arylquin 1 (Aq) or corn oil vehicle (V), was examined by Western blot analysis. (Right Panels) Aliquots of serum from these mice were either directly added to the growth medium of cells in culture, or incubated with the indicated antibody, and then added to the growth medium of PC-3 MM2 cells to test for apoptosis.
Panels a-c. Apoptotic cells were scored after 24 h and data shown represent mean values from three independent experiments ± s.d. Asterisks (**) or (*) indicate statistical significance (P < 0.0001) or (P < 0.001), respectively, by the Student t test. Uncut gels for Figure 2 can be found in Supplementary Figure 18.
We next tested co-cultures of normal cells with cancer cells for the apoptotic effect of Arylquin 1 at 500 nM, as this low concentration induced the secretion of Par-4 from normal cells yet did not induce apoptosis in normal or cancer cells. Arylquin 1 treatment of the co-cultures containing Par-4+/+ MEFs and cancer cells resulted in apoptosis of the cancer cells, relative to vehicle-treatment (Figure 2b). Only the cancer cells, but not normal HEL cells, underwent apoptosis in such co-culture experiments. By contrast, Arylquin 1 treatment of the co-cultures containing Par-4−/− MEFs and cancer cells did not induce apoptosis. Paracrine apoptosis induced in the cancer cells by Par-4, which was secreted from Par-4+/+ MEFs but not Par-4−/− MEFs in response to Arylquin 1 treatment (Supplementary Figure 14), was mediated via cell surface GRP78 (Supplementary Figure 15). Moreover, vimentin-deficient cells 13,14 showed robust increase in secretion of pro-apoptotic Par-4 activity in the CM relative to counterpart wild type cells, and Arylquin 1 did not further induce Par-4 secretion in these cells (Supplementary Figure 16). Based on these findings, we infer that: (a) vimentin sequestered Par-4 and prevented its secretion, and (b) Arylquin 1 bound to vimentin and thereby altered the vimentin-Par-4 association to facilitate Par-4 secretion.
To determine the physiological significance of these findings, we injected immunocompetent mice with Arylquin 1 or vehicle and examined their serum for circulating levels of Par-4. Arylquin 1 produced 5-fold higher Par-4 secretion relative to vehicle control in serum (Figure 2c). Serum from the Arylquin 1 treated mice, but not vehicle-treated mice, produced significantly higher (P <0.001) ex vivo apoptosis of cancer cell cultures (Figure 2c). The pro-apoptotic activity in the serum was neutralized by the Par-4 antibody. These findings implied that systemic Par-4 levels were elevated in response to Arylquin 1 treatment and that these levels were effective in producing apoptosis of cancer cells.
In summary, the present study identified a novel secretagogue, Arylquin 1 that produced a dose-dependent secretion of Par-4 at nanomolar concentrations from both normal fibroblasts and epithelial cells. Vimentin was the primary target of Arylquin 1, as determined using a biotinylated analog of Arylquin 1. Vimentin represents a particularly important therapeutic target because of its elevation in diverse tumors and its causal role in EMT and metastasis 12. Importantly, this chemical genetics approach led to the identification of vimentin as a novel binding partner of Par-4 and indicated that Arylquin 1 exhibited its function by binding to vimentin and releasing vimentin-bound Par-4 for secretion. At low concentrations, Arylquin 1 by itself did not kill normal cells and most cancer cells, but instead, it caused robust secretion of Par-4 from normal cells and triggered apoptosis in cancer cells only when they were used in co-culture experiments with normal cells. These findings, which implicated Par-4 secreted from normal cells in the apoptotic death of cancer cells, were corroborated by the observation that Arylquin 1 treatment of cancer cells co-cultured with Par-4-null normal cells failed to induce apoptosis of the cancer cells. Thus, Arylquin 1 induced paracrine apoptosis in cancer cells via Par-4 secreted by normal cells. Because Par-4 produced apoptosis in diverse tumors and because there were no previously reported compounds that acted at nanomolar concentrations to produce the levels of Par-4 secretion discovered in this study, these findings have potential, translational significance.
Methods Online
Chemistry
Nutlin-3a, an inhibitor of MDM2 that is reported to bind directly to MDM2, release, stabilize and activate p53 10, was acquired from Cayman Chemical Company. Brefeldin A, N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone(zVAD-fmk) and other chemicals were purchased from Sigma Aldrich or Fisher Scientific or were synthesized according to literature procedures. The synthesis of Arylquin 1, which utilized 4-(N,N-dimethylamino)-2-aminobenzaldehyde in a Friedländer condensation with 2-fluorophenylacetontrile 15, and other heterocyclic families is described in Supplementary Note. The condensation of 2-amino-4-(N,N-dimethylamino)benzaldehyde with 2-(2-fluorophenyl)acetyl chloride secured 7-(dimethylamino)-3-(2-fluorophenyl)quinolin-2(1H)-one, and treatment with Lawesson's reagent 16 provided 7-(dimethylamino)-3-(2-fluorophenyl)quinoline-2(1H)-thione. S-alkylation of this intermediate with (+)-biotinyl-iodoacetamidyl-3,6-dioxaoctanediamine led to biotinylated Arylquin 9 (Supplementary Note). Solvents were used from commercial vendors without further purification unless otherwise noted. Nuclear magnetic resonance spectra were determined on a Varian instrument (1H, 400MHz; 13C, 100Mz). High resolution electrospray ionization (ESI) mass spectra were recorded on a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The FT resolution was set at 100,000 (at 400 m/z). Samples were introduced through direct infusion using a syringe pump with a flow rate of 5 µL/min. MALDI mass spectra were obtained on a Bruker Utraflexstreme time-of-flight mass spectrometer (Billerica, MA), using DHB (2,5-dihydroxybenzoic acid) matrix. Purity of compounds was established by combustion analyses by Atlantic Microlabs, Inc., Norcross, GA. Compounds were chromatographed on preparative layer Merck silica gel F254 unless otherwise indicated.
Cells and plasmids
Human lung cancer cells H1299, HOP92, A549, H460, mouse lung cancer cells LLC1, human prostate cancer cells LNCaP, DU145, PC-3, and primary human lung fibroblast cells HEL and epithelial cells HBEC and BEAS-2B were from ATCC, MD; normal human prostate epithelial cells PrE and human prostate stromal cells PrS were from Lonza Inc., Allendale, NJ. KP7B cells were from Tyler Jacks, MIT, MA. PC-3 derivatives PC-3 MM2 were from Sue-Hwa Lin, M.D. Anderson Cancer Center, Houston, Texas. Par-4+/+ and Par-4−/− MEFs were derived from wild type and Par-4-null C57BL/6 mice generated by Taconic 11. Vimentin-null (Vim−/−) and wild type MEFs, as well as vimentin-expressing (Vim+) and vimentin-deficient (Vim-) SW13 cells were from Anthony Brown (Ohio State University).
Antibodies and siRNA duplexes
Par-4 (R334), Col1A1 (H-197), Vimentin (H-84) for Western blot; Vimentin (RV202) for ICC and immunoprecipitation; GRP78 (N20), Col1A1 (H-197), p53 (DO-1) and pan-cytokeratin (C11) antibodies were from Santa Cruz Biotechnology, Inc. Active caspase 3 antibody (Asp175) (5A1E) and p53 antibody (1C12) were from Cell Signaling. The β-actin antibody was from Sigma Chemical Corp.
Pull down experiments
To identify the target protein for compound Arylquin 1, pull-down experiments were performed as described previously 17. MEFs or HEL cells (grown to confluence in 15 cm plates) were lysed in 50 ml lysis buffer (40 mM Hepes, pH 7.8; 140 mM NaCl; 10 mM NaF; 10% Glycerol, 1 mM EDTA; 1% Triton 100), and the lysates were pre-cleared at 4oC for 1 h with 100 μL streptavidin beads (Novagen, Strep-Tactin Superflow Agarose). Binding reactions were performed by incubating the pre-cleared cell lysates with 50 μL beads ± 25 μg of biotinylated Arylquin 1 at 4°C for 2 h. The beads were then washed four times with buffer (40 mM Hepes, pH 7.8; 140 mM NaCl; 10 mM NaF; 10% Glycerol, 1 mM EDTA), and bound protein was eluted with 50 μL of 2.5 mM Biotin/PBS. Eluted proteins were resolved by SDS-PAGE and Coomassie blue staining.
Co-immunoprecipitation and Western blot analysis
Protein extracted from cell lysates was filtered, pre-cleared with 25 μL (bed volume) of protein G-Sepharose beads and immunoprecipitated with 1 μg of respective antibodies. The eluted proteins were resolved by SDS-PAGE, and subjected to Western blot analysis as described 18.
Apoptosis assays and detection of cell surface GRP78
Apoptotic nuclei were identified by immunocytochemical (ICC) analysis for active caspase-3, and nuclei were revealed by 4, 6-diamidino-2-phenylindole (DAPI) staining 5,6. A total of three independent experiments were performed; and approximately 500 cells were scored in each experiment for apoptosis under a fluorescent microscope. Cell surface GRP78 expression on the cancer cell surface was quantified by FACS analysis in the Flow Cytometry Shared Resource Facility, Markey Cancer Center as previously described 5.
Animal experiments
C57BL/6 mice were injected via the intraperitoneal route with Arylquin 1 (10 mg/kg body weight) or corn oil vehicle, and whole-blood samples were collected 24 h later. Serum was separated from the blood samples, heated at 56°C to inactivate complement. Aliquots of the mouse serum samples were added to the growth medium (final 20% mouse serum) of normal and cancer cells in culture and tested for induction of ex vivo apoptosis in cancer cells. All animal procedures were performed with University of Kentucky IACUC approval.
Computational modeling
Molecular modeling of vimentin binding with Arylquin 1 and the analogs was performed by using the previously reported computational protocol 19,20. Briefly, each ligand was docked into the binding cavity and the resulting poses were refined by molecular dynamics (MD)-simulations. The most favorable binding mode (with the lowest binding free energy), which was identified in the docking procedure, was subjected to an MD simulation for 1 ns at 298 K and used in binding free energy calculations.
Computational methods
Each ligand was docked into the binding cavity of the vimentin structure 20 using the SABRE program 21. The docked vimentin-ligand structure was used as an initial structure for MD simulation in water. The general procedure for carrying out the MD simulations in water was essentially the same as that used in our previously reported computational studies 22,23. Briefly, the MD simulations were performed using the Sander module from Amber12 24. The vimentin-ligand binding complex was neutralized by adding counter ions and was solvated in an orthorhombic box of TIP3P water molecules with a minimum solute-wall distance of 10 Å. The solvated systems were energy-minimized and carefully equilibrated. These systems were gradually heated from T = 10oK to T = 298.15oK in 50 ps before the production MD run. The MD simulations were performed with a periodic boundary condition in the NPT ensemble at T = 298.15o K using the Berendsen temperature coupling 25 and constant pressure (P = 1 atm) with isotropic molecule-based scaling. A time step of 2 fs was used, with a cutoff of 12 Å for the non-bonded interactions, and the SHAKE algorithm was employed to keep all covalent bonds involving hydrogen atoms rigid 26. Long-range interactions were handled using the particle mesh Ewald (PME) algorithm 27. During the energy minimization and MD simulation, only the ligand and residue side chains in the binding pocket were permitted to move. A residue-based cutoff of 12 Å was utilized for non-covalent interactions. The production MD simulation was then carried out for 1 ns and we made sure that the MD trajectory was stable. During the simulation, the atomic coordinates of the system were collected every 1 ps. The last 50 snapshots of the simulated structure of the MD trajectory were used to carry out the molecular mechanics with generalized Born and surface area solvation (MM/GBSA) binding energy calculations. The MM/GBSA binding energy calculations were performed by using the MM/GBSA method implemented in the Amber12 24. Our MM/GBSA calculation for each snapshot was carried out in the same way as we did for other protein-ligand systems 28. The entropic contribution (-T△S) was neglected in the binding free energy calculations due to several reasons: (a) the calculation of the entropic contribution (-T△S) to the binding free energy would require some additional approximations; (b) we only needed to have a rough estimate of the relative binding free energies in this study; and (c) the -T△S values for all of the ligands examined in this study are expected to be close to each other and, thus, are not expected to change the order of the calculated binding energies.
Statistical analysis
All experiments were performed in triplicate to verify the reproducibility of the findings. The results show a mean of at least 3 experiments ± Standard Deviation (s.d.). Statistical analyses were carried out with Statistical Analysis System software (SAS Institute, Cary, NC) and P values were calculated using the Student t test. The effect of interaction between two different treatments was analyzed using a two-way ANOVA model with data normality and equality of variance assumptions.
Supplementary Material
Acknowledgments
This work was supported by National Center for Research Resources in a grant entitled “COBRE Center for Biomedical Research Excellence” grant P20 RR020171 (to L. Hersh, PI and DW as core director), NIH/NCI R01 CA60872 and R21 CA179283 (to VMR) and University of Kentucky CCTS Drug Discovery Pilot Grant (to VMR, DW and CL).
Footnotes
Author Contributions
R.B., contribution to experimental biological studies; V.M.S., contribution to arylquin organic syntheses; N.H, contribution to experimental biological studies; W.Z. contribution to experimental biological studies; W.J.L., contribution to NMR studies characterizing arylquins; A.H, contribution to computational studies; C.G.Z., contribution to computational studies; D.S.W, contribution to design of arylquin syntheses; C.L., contribution to biological studies of biotinylated arylquin; V.M.R., contribution to design of Par-4 biological studies.
Competing Financial Interests
Vivek M. Rangnekar, David S. Watt and Chunming Liu hold 30% equity each in a limited liability corporation, Curonc LLC, founded for the purpose of advancing translational research involving arylquins, and the University of Kentucky has filed a preliminary patent application for these compounds in which these authors are co-inventors.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Viallet J, Minna JD. Am. J. Respir. Cell Mol. Biol. 1990;2:225–232. doi: 10.1165/ajrcmb/2.3.225. [DOI] [PubMed] [Google Scholar]
- 3.Luo SY, Lam DCL. Translational Respiratory Med. 2013;1:6–8. doi: 10.1186/2213-0802-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen F, Wang W, El-Deiry WS. Biochem Pharmacol. 2010;80:724–730. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Burikhanov R, et al. Cell. 2009;138:377–388. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burikhanov R, et al. Cancer Res. 2013;73:1011–1019. doi: 10.1158/0008-5472.CAN-12-3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebbar N, Wang C, Rangnekar VM. J Cell Phys. 2012;227:3715–3721. doi: 10.1002/jcp.24098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrestha-Bhattarai T, Hebbar N, Rangnekar VM. Cancer Cell. 2013;24:3–5. doi: 10.1016/j.ccr.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez JV, et al. Cancer Cell. 2013;24:30–44. doi: 10.1016/j.ccr.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vassilev LT, et al. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 11.Burikhanov R, et al. Cell Reports. 2014;6:271–277. doi: 10.1016/j.celrep.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satelli A, Shulin L. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckes B, et al. J Cell Sci. 2000;113:2455–2462. doi: 10.1242/jcs.113.13.2455. [DOI] [PubMed] [Google Scholar]
- 14.Gillard BK, Harrell RG, Marcus DM. Glycobiology. 1996;6:33–42. doi: 10.1093/glycob/6.1.33. [DOI] [PubMed] [Google Scholar]
- 15.Amberg W, et al. PCT Int Appl WO2007022946 A1 20070301. 2007 [Google Scholar]
- 16.Lecher HZ, Greenwood RA, Whitehouse KC, Chao TH. J Am Chem Soc. 1956;78:5018–5022. [Google Scholar]
- 17.Zhang W, et al. ACS Chem Biol. 2013;8:796–803. doi: 10.1021/cb3005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goswami A, et al. Cancer Res. 2008;68:6190–6198. doi: 10.1158/0008-5472.CAN-08-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bargagna-Mohan P, et al. J Biol Chem. 2010;285:7657–7669. doi: 10.1074/jbc.M109.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bargagna-Mohan P, et al. Chem Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamza A, et al. J Chem Inform Model. 2014;54:1166–1173. doi: 10.1021/ci500025r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamza A, Zhan C-G. J Phys Chem B. 2009;113:2896–2908. doi: 10.1021/jp8082612. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, et al. Mol Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 24.Case DA, et al. AMBER 12. University of California; San Francisco, CA: 2012. [Google Scholar]
- 25.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 26.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 27.Darden T, York D, Pedersen L. J. Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 28.Hamza A, et al. J Phys Chem B. 2010;114:5605–16. doi: 10.1021/jp100668y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.