

Osteoglophonic dysplasia or osteoglophonic dwarfism (OGD, OMIM#166250) is a rare skeletal disorder caused by mutations in fibroblast growth factor receptor 1 (FGFR1)(1,2). It is inherited as an autosomal dominant trait. OGD is characterized by gross stunting of stature associated with profound craniofacial abnormalities (3,4). We presented a 12-year old Chinese girl diagnosed with OGD. Her parents and other siblings are otherwise normal. This patient has short stature i.e. her height was 102 centimetres, which was well below the 3rd percentile for her age and corresponded to the 50th percentile for a 4 year-old girl. She was dysmorphic as she had mandibular prognathism, midfacial hypoplasia, hypertelorism, right eye hypertropia, an everted nose with a depressed nasal bridge. [Figure 1] She also had brachydactyly with a single palmar crease and there was fusion of the proximal interphalangeal joints in all four limbs. Her skeletal survey showed rhizomelic shortening of the humerus and femur bilaterally, short metacarpals with fusion of the proximal and middle phalanx, enlargement of the metaphysis of the distal femur bilaterally and anterior beaking of her lumbar vertebral body. Her skull was of abnormal configuration with presence of craniosynostosis, small midface and prognathic mandible. Another striking feature was she presented with giant cell lesions of her right maxilla and left mandible.
Figure 1.

Facial features showing mandibular prognathism, midfacial hypoplasia and everted nose with a depressed nasal bridge
Blood investigations revealed normal serum calcium and phosphate (2.22 mmol /L, 1.33 mmol/L respectively), normal alkaline phosphatase at 167 U/L [normal 130-560 U/L], normal parathyroid hormone level, a 7.57 pmol/L and fibroblast growth factor 23 (FGF23) at 74 pg/ml (this value was at the upper end of normal for adults as to date, there is no relevant data for children). Mutation analysis of the FGFR1 gene revealed a heterozygous mutation c.1141C>T in exon 10 [based on the FGFR1 transcript variant 1 mRNA (NM_023110)]. [Figure 2] This mutation results in a cysteine-to-arginine change (Cys381Arg) in the FGFR1 transmembrane domain (1).
Figure 2.
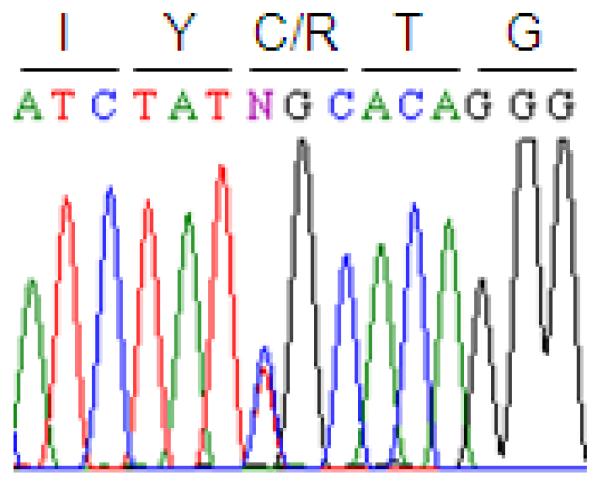
Heterozygous mutation c.1141C>T of FGFR1 in exon 10
The FGFRs are part of a tyrosine kinase receptor family; they comprise an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular tyrosine kinase region (2). FGFR1 and FGFR2 mutations cause syndromes involving craniosynostosis whereas FGFR3 mutations are associated with dwarfing syndromes such as achondroplasia and hypochondroplastic dwarfism (1). OGD is caused by activating mutations in a highly conserved domain of FGFR1 and shares characteristics with both the craniosynostosis and dwarfing syndromes (1). In our patient, molecular analysis of FGFR1 revealed a missense mutation (c.1141C>T) in exon 10 resulting in a substitution of cysteine 381 to arginine. This substitution creates an unpaired cysteine residue and may disrupt disulfide bonding required for proper dimerization of FGFR1. To date, mutations have been reported in only 6 other OGD patients (1,2). Our patient’s mutation has been reported in two of these 6 patients, which indicates that Cys381Arg may be a ‘common’ mutation in this rare disorder (1,2). One of the two reported patients was the index case who was first described by Beighton (3); the other one was patient 4, described in 2005 by White et al (1). Both these patients showed similar radiological features to ours, including metaphyseal lucencies affecting the long bones with anterior beaking of the vertebral bodies. The other similarity was their serum phosphate and FGF23 levels were at normal levels. However, our patient displayed pathological features of giant cell lesions in her maxilla and mandible. This lesion was also shown in one of the six previously reported OGD cases but, with different type of mutation (N330I).
Acknowledgement
The authors would like to express their appreciation to Ms. Sharifah Azween Syed Omar for her assistance in facilitating the process of sample collection and transportation. Portions of this work were supported by NIH grant AR42228 (to MJE).
REFERENCES
- 1.White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR 1 in bone elongation. Am J Hum Genet. 2005;76:361–67. doi: 10.1086/427956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrow EG, Davis SI, Mooney SD, et al. Extended mutational analyses of FGFR1 in osteoglophonic dysplasia. Am J Medical Genet. 2006;140A:537–539. doi: 10.1002/ajmg.a.31106. [DOI] [PubMed] [Google Scholar]
- 3.Beighton P, Cremin BJ, Kozlowski K. Osteoglophonic dwarfism. Pediatr Radiol. 1980;10:46–50. doi: 10.1007/BF01644343. [DOI] [PubMed] [Google Scholar]
- 4.Roberts TS, Stephen L, Beighton P. Osteoglophonic dysplasia: dental and orthodontic implications. Orthod Craniofacial Res. 2006;9:153–156. doi: 10.1111/j.1601-6343.2006.00369.x. [DOI] [PubMed] [Google Scholar]