

Abstract
Bcl-3 is an atypical member of the IκB family and modulates gene expression via interaction with p50/NF-κB1 or p52/NF-κB2 homodimers. We report here that Bcl-3 is required in dendritic cells (DCs) to assure effective priming of CD4 and CD8 T cells. Lack of Bcl-3 in bone marrow-derived DCs (BMDCs) blunted their ability to expand and promote effector functions of T cells upon antigen/adjuvant challenge in vitro and after adoptive transfers in vivo. Importantly, the critical role of Bcl-3 for priming of T cells was exposed upon antigen/adjuvant challenge of mice specifically ablated of Bcl-3 in DCs. Furthermore, Bcl-3 in endogenous DCs was necessary for contact hypersensitivity responses. Bcl-3 modestly aided maturation of DCs, but most consequentially, Bcl-3 promoted their survival, partially inhibiting expression of several anti-apoptotic genes. Loss of Bcl-3 accelerated apoptosis of BMDCs during antigen presentation to T cells and DC survival was markedly impaired in the context of inflammatory conditions in mice specifically lacking Bcl-3 in these cells. Conversely, selective over-expression of Bcl-3 in DCs extended their lifespan in vitro and in vivo, correlating with increased capacity to prime T cells. These results expose a previously unidentified function for Bcl-3 in DCs survival and the generation of adaptive immunity.
Introduction
Dendritic cells (DCs) are the most potent antigen presenting cells and are crucial for the initiation of adaptive immune responses. The immunogenicity of DCs is determined by their ability to capture, process and present antigens, their production of cytokines and other soluble mediators and their expression of co-stimulatory molecules, but also by their longevity. Enhanced survival or expansion of DCs can result in autoimmunity (1-3). Upon activation, the lifetime of DCs may need to be strictly regulated to maintain a balanced and functional immune response (4-6). However how DCs manage to carefully control their own survival, particularly during priming of T cells is largely unknown.
NF-κB is a master regulator of inflammation and several NF-κB subunits have been described to control DC functions (7-11). The dimeric NF-κB transcription factors are composed of five, variously combined polypeptides that comprise the Rel/NF-κB family (RelA [p65], RelB, c-Rel, p50 [NF-κB1], and p52 [NF-κB2]). Both p50 and p52 lack transactivation domains and the abundant p50 homodimers have been implicated in inhibition of NF-κB-dependent gene transcription (12, 13). NF-kB activity is regulated by the IκB family proteins, which include the classical members IκBα, IκBβ and IκBε the p105/NF-κB1 and p100/NF-κB2 precursors, and the atypical members IκBζ, IκBNS and Bcl-3. The atypical members modulate transcriptional activities of NF-κB complexes in the nucleus.
Bcl-3 exclusively binds homodimers of p50 or p52 and may convert these homodimers into transactivating complexes owing to transactivation domains present within Bcl-3, yet Bcl-3 may also enhance their inhibitory function. The exact outcome may depend on the particular target gene and cellular context, which also involves not well-understood post-translation modifications of Bcl-3 (12, 14). The specific cellular functions and mechanisms of action of Bcl-3 in biologic contexts remain poorly understood. Nevertheless, much evidence points to profound roles of Bcl-3 in vivo. Its gene is a partner in recurring chromosomal translocations, especially in B cell leukemias (15, 16), and its expression is elevated in various solid tumors (17). Bcl-3 is critical for both innate and adaptive immune responses to pathogens and contributes to immune system development (18-22).
Here we have explored, for the first time, the role of Bcl-3 in DCs. We discovered that Bcl-3 was required for efficient priming of CD4 and cross-priming of CD8 T cells in vitro and in vivo. Mice specifically ablated for Bcl-3 in CD11c+ cells failed to generate a proper antigen-specific CD4 T cell response and failed to develop a normal CD8-depedent contact hypersensitivity reaction. Mechanistically, Bcl-3 contributed to expression of co-stimulatory factors on DCs and modestly reduced expression of some T cell inhibitory factors, but most notably, promoted survival of DCs to allow for optimal priming of T cells. Conversely, transgenic mice overexpressing Bcl-3 in CD11c+ cells prolonged the lifespan of DCs in vitro and in vivo and enhanced T cell priming. Our findings reveal an unexpected and critical role for Bcl-3 in DCs to assure adequate survival and efficient priming of T cells.
Materials and Methods
Mice
All mice used were on C57BL/6 backgrounds. OT-I, OT-II, and LPR mice were from Taconic Farms and IL-10−/−, Itgax-cre (CD11c-cre) from Jackson Laboratories; Bcl-3−/−) (18) and Bcl-3 transgenic mice have been described (23); Bcl-3flx/flx mice are described in Supplementary Figure 2. All mice were housed in NIAID Institute facilities and all experiments were done with approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Flow cytometry
Samples were stained at 4 °C with Fc Block present (2.4G2; BD Biosciences) in flow cytometry buffer (PBS 2% FBS). Antibodies used: allophycocyanin-conjugated anti-CD4 (RM4-5), allophycocyanin anti-CD11c (HL3), allophycocyanin anti-NK1.1 (PK136), allophycocyanin anti-CD8 (53-6.7), FITC–anti-CD3ε (145-2C11), PE–anti-CD86 (B7-2), FITC anti-CD54 (3E2), PE anti-CD40 (3/23), PercP anti-CD45.2 (104), PercP anti-Vα2 TCR (B20.1), phycoerythrin-cyanine7 anti-CD11c (HL-3) (all from BD Biosciences); phycoerythrin-cyanine7 anti-IFNγ (XMG1.2), phycoerythrin-cyanine5 anti-MHC-II (M5/114.15.2), PE anti-MHC-II (M5/114.15.2), allophycocyanin anti-CD207 (eBioRMUL.2), PE anti-FasL (MFL3), eFluor-450 anti-CD11b (M1/70), PE anti-PD-L1 (MIH5), PE anti-CD103 (2E7) (all from eBioscience); allophycocyanin anti-CD8 (53-6.7), allophycocyanin anti-CD49b (DX5), allophycocyanin anti-CD25 (PC61), allophycocyanin-cy7 MHC-II (M5/114.15.2), FITC–anti-CD80 (16-10A1), FITC anti-MHC-I (34-1-2s) (all from Biolegend). For PI/AnnexinV analysis the AnnexinV e-Fluor-450 apoptosis detection kit was used (eBioscience). Caspase-3 activation was measured with NucView 488 Caspase-3 Assay Kit (Biotium, Hayward, CA). Dead cells were excluded with Aqua Live/Dead fixable kit (Invitrogen). Stained cells were analyzed on a FACS CANTO and data analyzed with FlowJo software (BD Biosciences).
In vitro priming of T cells
BMDCs were generated with GM-CSF for 7-9 days(24). DC yield was monitored with flow cytometry after anti-CD11b, anti-CD11c staining. BMDCs were stimulated with ultrapure LPS (Escherichia coli 0111:B4; List Biological Laboratories). Surface markers were assessed with flow cytometry 24h after LPS (100ng/ml). Cytokines present in cell supernatants of BMDCs (105/well) after 16h with LPS were measured with cytometric bead analysis (CBA, BD Biosciences, CBA Mouse inflammation kit). For standard antigen-presentation experiments BMDCs (5×104/well of 96-well plate) were incubated for 3h with chicken ovalbumin (OVA; Calbiochem) (typically 100μg/ml), stimulated with LPS (100 ng/ml) overnight, washed and co-cultured with 2.5×105 CD4+ OT-II transgenic T cells/well for 72h. OT-II T cells were purified with the CD4+ T-cell isolation kit (Miltenyi Biotec), and labeled with CFSE (Molecular Probes). T cell proliferation was measured with flow cytometry and recorded as Proliferation Indices (number of divisions/number of dividing cells) and Division Indices (average number of divisions/cell in original population). In some co-culture experiments OT-II (not labeled) were analyzed for CD25 or supernatants for IL-2 or IFNγ with CBA (Th1-Th2 mouse kit; BD). Also, BMDCs were stimulated with LPS overnight and then pulsed with H2b-restricted OVA peptide 323– 339 (AnaSpec, CA) and washed prior to addition of T cells. For cross-priming, 5×104/well BMDCs were stimulated with LPS overnight, pulsed with OVA (100μg/ml) for 3h and co-cultured with 2.5×105/well OT-I CD8+ T cells for 72h. OT-I cells were purified with the CD8+ T-cell isolation kit (Miltenyi Biotec), OT-I cells were analyzed as described above for OT-II.
In vivo T cell priming
CD45.1 mice were injected i.v. with 5×106 CFSE-labeled CD45.2 OT-II cells, previously isolated by negative or positive selection (CD4+ T-cell isolation kits, Miltenyi Biotec). One day later, mice were injected intraperitoneally (i.p.) with 1×106 OVA-loaded (100μg/ml) and LPS (100ng/ml)-stimulated BMDCs (described above). 3 days thereafter, splenocytes were isolated and OT-II proliferation (CFSE dilution) measured with flow cytometry, gated on CD45.2+, CD4+ cells. CD45.2 WT and Bcl-3−/− mice were injected i.v. with 5×106 CFSE-labeled CD45.1 OT-I T cells, previously isolated by negative selection (CD8+ T-cell isolation kit, Miltenyi Biotec). One day later, mice were injected intradermally (i.d.) with 1μg OVA. Cells were isolated from spleens and draining lymphnodes 3 days later and OT-I proliferation (CFSE dilution) assessed with flow cytometry, gated on CD8+, CD45.1+ cells. Bcl-3-Δ-DC (and control) mice were injected with 5×106 CFSE-labeled OT-II, and one day later with 1μg OVA and 30μg LPS subcutaneously (s.c.). 3 days thereafter cells from draining lymphnodes were analyzed with flow cytometry for OT-II proliferation (CFSE dilution) after gating on CD4+, Vα2TCR+ cells.
Contact hypersensitivity and skin DC isolation
Mice were sensitized on shaved bellies with 25μl of 100μg/ml oxazolone (Sigma, 1:5 olive oil:acetone solution) for 2 consecutive days. 5 days later, both sides of ears were challenged with of 5μl of 10μg/ml of oxazolone or solvent control. Ear thickness was measured for at time of challenge with oxazolone on ears and over the course of the next 4 consecutive days.
To extract skin DCs from mouse ears, ears were split in two parts (dorsal and ventral) and incubated for 30’ at 37°C in PBS containing 2.5 mg/ml dispase II (Roche) to allow separation of dermal and epidermal sheets. The separated epidermal and dermal sheets were incubated for 1 h at 37°C with a solution of RPMI containing 2.5mg/ml LiberaseTM (Roche) to obtain homogeneous cell suspensions.
DC apoptosis
BMDCs (3×105/well of 24-well plate) were incubated for 3h with 100μg/ml OVA, stimulated with LPS (100 ng/ml) o.n., washed and co-cultured with 1.5×106 OT-II cells/well (24-well plate) for 72h. Apoptosis of BMDCs was analyzed with flow cytometry after staining for CD11c, Annexin-V/7-AAD or caspase-3 and gating on CD11c+. WT and Bcl-3−/− mice were injected i.v. with 30μg of LPS or PBS. After 48 hours, splenocytes were isolated, stained for CD11c and MHC-II assessed with flow cytometry. Absolute numbers of CD11c+MHC-II+ in the spleen were determined using countbright absolute counting beads (Invitrogen). In situ apoptosis of splenic DC was determined with TUNEL assay (Histoserv, Inc) and quantitated by counting in 0.63mm2 areas.
Western analysis
BMDCs were stimulated for various times with LPS (100ng/ml) and nuclear and cytoplasmic fractions were prepared by Nuclear Extraction-Protein Extraction Reagent Kit (Pierce). Proteins from cell lysates were separated by standard SDS-PAGE and analyzed by immunoblotting with antibodies specific for p65 (CT, Millipore), p50 (sc-114,), actin (sc-47778) and LaminB (sc-6216) (all Santa Cruz).
PCR array
3×106 WT and Bcl3−/− BMDCs were stimulated for 24h with LPS (100ng/ml) in 6-well plates. Cells were lysed in TRIZOL and apoptotic gene expression was analyzed with RT2 Profiler PCR Array (SABiosciences, Qiagen).
Statistical analysis
Data were recorded as the mean ±SEM. Differences between groups were analyzed by unpaired, two-tailed Student's t-tests. p≤ 0.05 was considered significant (Prism; GraphPad Software). For multiple comparisons, data were analyzed by the one-way ANOVA followed by Bonferroni's multiple-comparisons test. The number of independent data points (n) for each experiment is stated in figure legends.
Results
Bcl-3 promotes dendritic cell-mediated priming of CD4 T cells
Bone marrow-derived DCs (BMDC) from wild-type (WT) and Bcl-3 deficient (Bcl-3−/−) animals were loaded with ovalbumin, stimulated with LPS and co-cultured with CFSE-labeled OT-II CD4 T cells for 3 days (lack of Bcl-3 did not affect generation of BMDCs; Supplemental Fig. 1A). Bcl-3−/− BMDCs were much less effective in inducing proliferation of T cells than WT BMDCs (Fig. 1A). This was not due to a defect in either uptake or processing of antigen (Supplemental Fig. 1B,C) and T cell proliferation was not rescued upon loading with Ova peptide (Fig. 1B). The defect in priming was not specific to TLR4, since CD4 proliferation was also impaired upon stimulation with CpG or poly I:C (Fig. 1C). We also observed diminished induction of CD25, IL-2 and IFNγ when T cells were primed by Bcl-3−/− BMDCs (Fig. 1D).
Figure 1.

Bcl-3 is required for efficient BMDC-mediated CD4 priming in vitro and in vivo. (A) WT and Bcl3−/− BMDCs were loaded with different doses of ovalbumin (OVA) and stimulated with LPS (100ng/ml) overnight (o.n.), then co-cultured with CFSE-labeled OT-II T cells for 72h. T cells were analyzed by flow cytometry after staining and gating for CD4. Representative FACS plots and proliferation and division indices (FloJo) are shown; mean ± SEM; n=5/group. (B) WT and Bcl-3−/− BMDCs were stimulated with LPS o.n., pulsed for 3h with different doses of OVA-peptide, and co-cultured for 72h with CFSE-labeled OT-II T cells. T cells were analyzed by flow cytometry after staining and gating for CD4 and data presented as in (A) (n=4/group). (C) WT and Bcl3−/− BMDCs were loaded with 100μg/ml OVA and stimulated with LPS (100ng/ml), CpG (10μg/ml) or poly I:C (100μg/ml) o.n., then co-cultured with CFSE-labeled OT-II T cells for 72h. T cells were analyzed by flow cytometry after staining and gating for CD4. Division index (FloJo) is shown; mean ± SEM; n=3/group. (D) WT and Bcl3−/− BMDCs were treated as in (A) (100μg/ml OVA) and incubated with OT-II T cells for 72h. T cells were analyzed for CD25 after staining for CD4 and CD25 and gating on CD4. A representative mean florescent intensity (MFI) plot and summary analysis are shown in left two panels. The shaded area represents naïve cells (controls). Cell culture supernatants were analyzed for IL-2 and IFNγ with CBA, shown in right two panels. Mean ± SEM; n=3/group. (E) 5×106 CFSE-labeled CD45.2 OT-II T cells were injected i.v. into CD45.1 congenic mice. 24h later animals were injected i.p. with 1×106 OVA-loaded (100μg/ml) and LPS stimulated WT and Bcl-3−/− BMDCs. Splenocytes were isolated 72h later, stained and gated for CD45.2 and CD4 and analyzed by flow cytometry. Data shown as in (A); n=10 mice/group based on 2 experiments. (F) LPS-stimulated and CFSE-labeled (WT) and cell trace violet-labeled (Bcl-3−/−) BMDCs were co-injected (5×106/each group) into WT mice i.p.. 18h later spleens were harvested and cells stained and gated for CD11c. Absolute numbers of CFSE+CD11c+ and violet+CD11c+ were determined and data are shown as mean ±SEM; n=4 mice/group; an additional experiment yielded similar data. *P<0.05, **P<0.01 and ***P<0.0001.
To investigate priming in vivo, CD45.1 mice were injected with CFSE labeled CD45.2 OT-II cells, then with LPS-stimulated and ovalbumin-pulsed WT or Bcl-3−/− BMDCs and proliferation of splenic CD45.2 CD4 OT-II cells was monitored three days later. Compared to WT, Bcl-3−/− BMDCs induced significantly less OT-II proliferation in vivo (Fig. 1E). Of note, Bcl-3−/− BMDC migrated to the spleen as efficiently as WT BMDCs (Fig. 1F). Therefore, expression of Bcl-3 in BMDCs is required for efficient priming of CD4 T cells in vitro and in vivo.
Bcl-3 promotes DC-mediated cross-priming of CD8 T cells
To investigate cross-priming of CD8 T cells, we cultured LPS-stimulated and ovalbumin-pulsed WT or Bcl-3−/− BMDCs together with CFSE-labeled OT-I CD8 T cells. Both proliferation of T cells (Fig. 2A) and levels of IL-2 (Fig. 2B) were significantly reduced when BMDCs lacked Bcl-3. To assess cross-priming in vivo, WT and Bcl-3−/− mice were injected with CFSE-labeled CD8 OT-I cells, ovalbumin was administered one day later and proliferation of OT-I CD8 T cells was monitored in spleen and draining lymph node three days thereafter (Fig. 2C). Cross-priming in Bcl-3−/− compared to WT mice resulted in reduced proliferation of T cells in both spleen and lymph node. Therefore, efficient cross-priming of CD8 T cells requires Bcl-3 expression in DCs in vitro and in vivo (see also below).
Figure 2.

Bcl-3 is required for efficient DC-mediated cross-priming in vitro and in vivo. (A) WT and Bcl3−/− BMDCs were stimulated with LPS (100ng/ml) o.n., pulsed with OVA (100 μg/ml) for 3h, and co-cultured with CFSE-labeled OT-I T cells for 72h. T cells were analyzed by flow cytometry after staining and gating for CD8. Representative FACS plots and proliferation and division indices (FloJo) are shown. Mean ± SEM; n=5/group. (B) WT and Bcl3−/− BMDCs were treated as in (A) and IL-2 production in supernatants was analyzed with CBA. Mean ± SEM; n=5/group. (C) 5×106 CFSE-labeled CD45.1 OTI T cells were injected i.v. into CD45.2 WT and Bcl3−/− mice. 24h later animals were injected i.d. with OVA (1μg). Cells were isolated from spleens and draining lymphnodes 72h later, stained and gated for CD45.1 and CD8 and analyzed by flow cytometry. Data shown as in (A), with n=10 mice/group based on 2 experiments. *P<0.05, **P<0.01.
Ablation of Bcl-3 in CD11c+ cells impairs priming of T cells in mice
To investigate the role of Bcl-3 in endogenous antigen-presenting DCs we made use of mice in which DCs lacked Bcl-3. We generated conditional Bcl-3 ‘knock-out’ mice (Bcl-3flx/flx) and deleted Bcl-3 in DCs with CD11c-driven Cre (CD11c-Cre; Bcl-3flxffx; hereafter referred to as Bcl-3-Δ-DC) (Supplemental Fig. 2A,B). We confirmed that BMDCs generated from Bcl-3-Δ-DC mice were defective in priming CD4 T cells in vitro (Supplemental Fig. 2C) and that loss of Bcl-3 in CD11c+ cells did not alter the total numbers of DCs, CD4, CD8, or B cells in spleens (Supplemental Fig. 2D).
CFSE-labeled OT-II CD4 cells were injected into Bcl-3flx/flx (‘WT’) and Bcl-3-Δ-DC mice, followed by challenge with ovalbumin and LPS, and proliferation of OT-II cells in draining lymph nodes was measured three days thereafter. OT-II proliferation was significantly reduced in Bcl-3-Δ-DC compared to WT mice (Fig. 3A). Bcl-3 was thus critical within endogenous CD11c+ cells to properly prime CD4 T cells. To assess migration of DCs from skin to lymph node, we painted shaved bellies of WT and Bcl-3-Δ-DC mice with, injected LPS s.c. and enumerated FITC-labeled CD11c+ cells in draining lymph nodes 18 hours later (Fig. 3B). Loss of Bcl-3 did not appear to affect migration.
Figure 3.

Selective ablation of Bcl-3 in CD11c+ cells impairs CD4 and CD8 responses in mice. (A) 5×106 CFSE-labeled OT-II T cells were injected i.v. into WT and Bcl-3-Δ-DC mice. 24h later animals were injected s.c. with OVA (μg) and LPS (30μg). Cells were isolated from draining lymphnodes 72h later, stained and gated for Vα2 TCR and CD4, and analyzed by flow cytometry. Representative FACS plots and proliferation and division indices shown. Mean ±SEM; n=8 mice/group based on 2 experiments. (B) Shaved abdomens of WT and Bcl-3-Δ-DC mice were painted with FITC solution and injected s.c. with LPS (30μg). Inguinal lymph nodes were obtained after 18 hours, stained for CD11c and MHC-II and absolute numbers of CD11c+MHC-II+-FITC+DCs were obtained using countbright absolute counting beads. Data shown as mean ±SEM; n=4 mice/group. (C) WT and Bcl-3-Δ-DC mice were sensitized with Oxazolone applied to shaved abdominal skin on two consecutive days. 5 days later mice were challenged on ears and ear swelling was measured blindly at indicated time points post challenge, presented as mean of increase ±SEM in thickness over basal level of solvent-only treated ears; n=5 mice/group; an additional experiment yielded similar data. *P<0.05.
To further explore the role of Bcl-3 in DCs in cross-priming in vivo, we employed a CD8-dependent contact hypersensitivity (CHS) model. Bcl-3-Δ-DC and WT mice were sensitized ventrally to the hapten oxazolone, re-challenged on ears 5 days later, and ear thickness was measured as a read-out of inflammation at time of challenge and over the course of the next 4 consecutive days. Bcl-3-Δ-DC mice had notably reduced ear thickening compared to WT mice (Fig. 3C). To exclude the possibility that the observed reduction in CHS was not due to the lack of one or more DC subtypes in the skin, we analyzed the skin DC subsets in ears of WT and Bcl-3−/− (25, 26). We did not find any significant differences in the skin DC populations between WT and Bcl-3−/− mice (Supplemental Fig. 2E). These findings implicate Bcl-3 in endogenous dendritic cells for both priming of CD4 and cross-priming of CD8 T cells.
Bcl-3 contributes to BMDC maturation
Efficient priming requires engagement of T cells with antigen-bound MHC-II, with costimulatory ligands on DCs and stimulation by DC-produced cytokines (27). Lack of Bcl-3 in BMDCs partially reduced LPS-induced increases in expression of the costimulatory proteins CD80 and CD86 (Fig. 4A). LPS-induced levels of MHC-II were also somewhat lower, while those of PD-L1 were enhanced. However, addition of PD-L1 blocking antibodies to co-cultures failed to improve priming (not shown). Expression levels of MHC-I, CD40 and ICAM-1 were not noticeably different in the absence of Bcl-3 (Fig. 4A).
Figure 4.

Bcl-3 contributes to BMDC maturation but is dispensable for inflammatory cytokine production. (A) WT and Bcl-3−/− BMDCs were left unstimulated or stimulated with LPS (100ng/ml). 24h later BMDCs were stained for markers indicated, and analyzed by flow cytometry, gated on CD11c+CD11b+ cells. Representative MFI plots in top rows, summaries for differentially expressed markers in bottom row. Isotype control staining is represented by shaded area. Data presented as mean ±SEM; n=10/group. (B) WT and Bcl-3−/− BMDCs (105) were stimulated with LPS for 18h in 96-well plates, and indicated cytokines present in supernatants were measured with CBA. Data shown as mean ±SEM; n=3-4/group. (C) WT, Bcl3−/− and Bcl3−/−/IL-10−/− BMDCs were loaded with OVA (100μg/ml) and stimulated with LPS (100ng/ml) o.n., and co-cultured with CFSE-labeled OT-II T cells for 72h. Cells were stained and gated for CD4, analyzed with flow cytometry and data shown as the mean ±SEM; n=2/group. (D) WT and Bcl-3−/− BMDCs were treated and seeded together with CFSE-labeled OT-II T cells as in (C) in bottom chambers of a transwell plate. LPS stimulated WT and Bcl-3−/− BMDCs were seeded in upper chambers, allowing for every combination of BMDCs in the two chambers as indicated. After 72h, cells in bottom chamber were analyzed as in (C). Data shown as mean ±SEM; n=4/group based on 2 experiments. *P<0.05, **P<0.01.
WT and Bcl-3−/− BMDCs were stimulated with LPS overnight to measure protein levels of IL-6, IL-12p70, TNF-α and IL-10. Levels of IL-10 were modestly increased in Bcl-3−/− BMDCs, noted previously (28), while production of IL-6, TNF-αand IL-12p70 were normal (Fig. 4B). To assess the relevance of increased IL-10 production, BMDCs from WT, Bcl-3−/− and [Bcl-3−/−IL-10−/−] mice (DKO) were used in standard co-cultures; DKO BMDCs failed to improve defective T cell proliferation (Fig. 4C). Furthermore, IL-10 blocking antibodies in co-cultures failed to improve priming by Bcl-3−/− BMDCs (Supplemental Fig. 3A).
To determine whether soluble factor(s) differentially secreted between LPS-stimulated WT and Bcl-3−/− BMDCs caused defective T cell priming, we seeded ovalbumin-loaded and LPS-stimulated WT or Bcl-3−/− BMDCs together OT-II T cells in the bottom chambers, and LPS-stimulated WT or Bcl-3−/− BMDCs in the upper chambers of a transwell plate, allowing for all combinations of BMDCs. Only the presence or absence of Bcl-3 in BMDCs in the lower chamber mattered for priming of T cells (Fig. 4D). This suggests soluble factors were not primarily responsible for defective priming.
Bcl-3 promotes survival of DCs in vitro and in vivo
LPS-activated WT and Bcl-3−/− BMDCs were transcriptionally profiled with PCR arrays. Expression of several pro-apoptotic genes was increased in LPS-stimulated Bcl-3−/−, compared to WT BMDCs (especially caspases 4 and 12, NF-κB1, and RIPK1, but also including FasL and Bax) (Fig. 5A). We then investigated whether Bcl-3 might promote survival in DCs, especially since the life-expectancy of DCs may be critical for efficient priming of T cells (1, 2). We therefore assessed the survival of BMDCs in standard 3 days co-culture experiments with Annexin-V and 7-AAD staining (Fig. 5B,C) or caspase-3 activation (Fig. 5D). Bcl-3−/− BMDCs survived significantly less well than WT BMDCs, evident by day two, and they exhibited increased caspase-3 activation, indicating a role for Bcl-3 in preventing premature apoptosis. Of note, the absence of Bcl-3 in BMDCs did not affect the cytoplasmic and nuclear levels of p50 and p65 during a time course of stimulation with LPS (Supplemental Fig. 3B). This was expected, as Bcl-3 is not known to affect the activation/nuclear translocation of NF-κB; instead, it affects the transcriptional activity of these complexes.
Figure 5.

Bcl-3 promotes survival of DCs. (A) WT and Bcl-3−/− BMDCs were stimulated with LPS (100ng/ml) for 24h and analyzed with RT2 profiler apoptosis superarray. Mean of fold-change for n=5 mice/group. (B) WT and Bcl3−/− BMDCs were treated and co-cultured with OT-II as in Figure 1A (standard conditions). Cells were stained for CD11c, Annexin V and 7-AAD and analyzed by flow cytometry after gating on CD11c. Representative FACS plot in top panels and % live and apoptotic BMDCs in bottom panels; mean ±SEM, 4 experiments with n=12 mice/group. (C) WT and Bcl3−/− BMDCs were treated and co-cultured with OT-II (24, 48 or 72h) and BMDCs analyzed as in (B). Mean of % live cells ±SEM, n=3/group (additional experiment yielded similar results). (D) WT and Bcl3−/− BMDCs were treated and co-cultured with OT-II cells as in (B) and CD11c-gated BMDCs assayed for caspase-3 activity. Mean ±SEM, n=3 mice/group (additional experiment yielded similar results). (E) WT and Bcl-3−/− and (F) WT and Bcl-3-Δ-DC mice were injected with LPS (30μg) or PBS i.v., splenocytes isolated 48h later and stained for CD11c and MHC-II. Absolute DC numbers shown as mean ±SEM; n=5-8 mice/group, based on 2 experiments. (G) WT and Bcl-3-Δ-DC mice were treated as in (E,F) and spleen sections stained for TUNEL+ cells. Representative sections shown in left panels and data summarized in right panel; mean of counts/section area ±SEM; n=4 mice/group with blinded analysis of 5 random sections/mouse. *P<0.05, **P<0.01 and ***P<0.0001.
To test survival of DCs in vivo, we injected WT, Bcl-3−/− and Bcl-3-Δ-DC mice i.v. with LPS and monitored numbers of CD11chighMHC-II+ splenic DCs at days 0 and 2. DCs undergo rapid activation-induced cell death under these conditions (29, 30). We observed an even more pronounced reduction in splenic DCs lacking Bcl-3 (Fig. 5E,F). Splenic sections from LPS-treated Bcl-3-Δ-DC and WT mice were stained for TUNEL+ (apoptotic) cells; their numbers were significantly higher in Bcl-3-Δ-DC than WT mice (Fig. 5G). These data implicate Bcl-3 in survival of DCs in vitro and in vivo, suggesting a mechanism by which Bcl-3 promoted T cell priming.
Surface staining for FasL confirmed increased, albeit still low expression levels in Bcl-3−/− BMDCs (Supplemental Fig. 3C). Because FasL may impair priming due to Fas-mediated apoptosis of T cells and/or BMDCs (1, 2, 31), we added the Fas-Fc soluble inhibitor to co-cultures. However, limited Fas engagement on naïve T cells during activation may promote proliferation, while only extensive engagement may favor apoptosis (32, 33). Consistent with this, Fas-Fc significantly reduced proliferation of T cells primed by WT BMDCs, and further reduced poor proliferation when primed by Bcl-3−/− BMDCs; however, the difference between WT and Bcl-3−/− BMDCs was no longer as apparent in the presence of Fas-Fc (Supplemental Fig. 3D). To assess apoptosis of co-cultured T cells, induced via Fas or otherwise, we measured caspase-3 activation and stained for live/dead cells (Supplemental Fig. 3E,F). We observed a limited increase in apoptosis of OT-II cells when primed by Bcl-3−/− compared to WT BMDCs. We furthermore tested Fas-deficient CD4 OT-II T cells (from OT-II/LPR crosses). These cells proliferated less well than Fas-sufficient OT-IIs, consistent with the noted positive role of Fas. However, the difference in proliferation of these cells when primed by WT compared to Bcl-3−/− BMDCs was no longer as marked now (Supplemental Fig. 3G). Therefore increased expression of FasL on BMDCs appears to have moderately increased T cell apoptosis, but unlikely to be primarily responsible for impaired priming by Bcl-3−/− BMDCs.
Overexpression of Bcl-3 in DCs promotes survival and T cell priming
BMDCs were generated from Bcl-3 transgenic (Tg) mice that express the transgene in DCs only (CD11c-Cre-mediated removal of loxP-flanked Stop cassette) (23). When WT and Bcl-3 Tg BMDCs were used in standard co-culture experiments. the Tg BMDCs caused a significant increase in T cell proliferation above that of WT BMDCs (Fig. 6A). Importantly, Tg BMDCs also exhibited significantly improved survival in these co-cultures (Fig. 6B). To assess survival of DCs in vivo, we injected WT and Tg mice i.v. with LPS and monitored CD11chighMHC-II+ splenic DCs at days 0 and 2. As discussed, LPS caused a drastic reduction of DCs in WT mice after 2 days, while significantly more DCs remained at that time in mice expressing the Bcl-3 Tg in DCs (Fig. 6C). Thus survival of DCs was extended by increased Bcl-3 levels and correlated with improved T cell priming.
Figure 6.
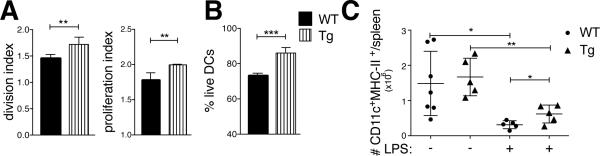
Bcl-3 overexpression promotes T cell priming and DC survival. (A) WT and Tg BMDCs were treated and co-cultured with CFSE-labeled OT-II cells and T cells analyzed as in Figure 1A (standard conditions and analysis). Data shown as mean ±SEM; n=3/group. (B) WT and Tg BMDCs were treated and co-cultured as in (A), then cells stained for CD11c, Annexin V and 7-AAD and analyzed by flow cytometry after gating on CD11c. Data shown as mean of % live DCs ±SEM; n=3/group. (C) WT and Tg mice were injected with LPS (30μg) or PBS i.v.; splenocytes were isolated 48h later and stained and gated for CD11c and MHC-II. Data shown as mean of absolute numbers ±SEM; n=5 mice/group. *P<0.05, **P<0.01 and ***P<0.0001
Discussion
The present study explores, for the first time, the function of the NF-κB regulator Bcl-3 in antigen presenting DCs. We discovered that Bcl-3 plays a surprisingly critical role in DC-mediated priming of CD4 and cross-priming of CD8 T cells. We demonstrate this with BMDCs, and importantly, with antigen-challenged mice specifically ablated of Bcl-3 in DCs. Thus, Bcl-3 was essential within the dendritic cell population normally present in animals to initiate an appropriate adaptive immune response. Bcl-3 contributed in several ways; it promoted expression of some co-activators associated with DC maturation, and delimited expression of some potentially negative mediators of T cell activation. However these contributions were relatively minor and Bcl-3's most consequential function was to prevent the premature demise of DCs. Loss of Bcl-3 shortened the lifetime of activated DCs, both in vitro and in vivo. By contrast, overexpression of Bcl-3 increased their lifetime in vitro and in vivo, correlating with increased priming. Our findings suggest that activated, Bcl-3-deficient DCs failed to survive long enough to assure efficient continuous priming of T cells, thereby compromising the development of an adequate adaptive immune response in vivo.
DC maturation signals invariably activate NF-κB, and prior studies have suggested roles for various Rel subunits in this process, including expression of cytokines (8-10, 34). Bcl-3 interacts with homodimers of p50/NF-κB1 and p52/NF-κB2 (14). NF-κB2 and NF-κB1 have been suggested to act as negative regulators of DC functions, based on studies with BMDCs and/or mice lacking these proteins. Ablation of NF-κB2 increased expression of some co-stimulatory molecules, postulated to be due to loss of the RelB inhibitor p100/NF-κB2 (35). Ablation of NF-κB1 increased expression of some inflammatory cytokines, especially TNFα(7), but also of some maturation markers, and enhanced IFNγ production by co-cultured T cells (36). It was furthermore suggested that loss of inhibitory p50 homodimers might be primarily responsible for these changes. However these interpretations are confounded by the fact that NF-κB1 and NF-κB2 encode the IκB-like precursors p105 and p100 as well as the processed p50 and p52 subunits, which form NF-κB heterodimers in addition to homodimers. The different forms have distinct activities and it is unclear which are critical for DC-mediated priming. Prior studies have suggested that Bcl-3 enforces inhibitory activities of p50 homodimers (37), and based on the above-cited studies, Bcl-3 would then be expected to inhibit DC functions. However, contrary to this notion, we found that Bcl-3 was indispensible for efficient antigen-specific priming of both CD4 and CD8 T cells, as demonstrated both in vitro and in mice conditionally ablated for Bcl-3 in antigen-presenting dendritic cells, including a CD8 T cell-dependent contact hypersensitivity model. Rather than enforcing inhibitory functions of p50 homodimers, Bcl-3 may reverse these functions, at least for some genes, which is conceivable given that Bcl-3 contains transactivation domains. In addition, Bcl-3 may execute important functions via p52 homodimers.
To understand how Bcl-3 may aid DCs in effective priming of T cells, we investigated antigen uptake, processing, maturation and cytokine expression. LPS-induced expression of MHC II and the co-stimulators CD80 and CD86 was modestly reduced in Bcl-3-deficient BMDCs, and expression of the potential inhibitors PDL-1, FasL and IL-10 was somewhat increased. However, blocking or eliminating these inhibitors failed to overcome defective T cell priming by Bcl-3-deficient BMDCs, and in the case of FasL, appeared to result in an only moderate reversal. This argues against significant roles for these inhibitors, although in vivo contributions cannot be ruled out. By contrast, loss of Bcl-3 markedly increased apoptosis in DCs, which we demonstrated in BMDCs in vitro as well as in DCs in vivo. Prior reports have suggested that the lifespan of DCs significantly affects the magnitude of immune responses to antigens and is critical for the balance between tolerance and inflammation (1, 4-6). Extending the lifetime of DCs, such as via over-expression of anti-apoptotic or elimination of apoptotic proteins enhanced antigen-specific T cell responses and/or led to autoimmunity and inflammation; conversely, elimination of the anti-apoptotic regulator Bcl-xL shortened DC survival in vivo and blunted an antigen-specific T cell response (38). Therefore the observed premature apoptosis of Bcl-3-deficient DCs may be the primary reason for impaired priming of T cells. In line with this interpretation, overexpression of Bcl-3 increased survival of DCs in vitro and in vivo and enhanced priming of T cells. The Bcl-3-mediated survival of activated and antigen-loaded DCs may increase their chances to encounter cognate T cells and, in addition, may aid activation of these cells via prolonged interaction.
How may Bcl-3 assure adequate survival of DCs? Transcriptional profiling indicated elevated expression of several pro-apoptotic genes in Bcl-3-deficient BMDCs, including Bax, caspases 12 and 4, RipK1 and NF-kB1. Loss of NF-κB1 has been suggested to prolong the lifespan of DCs (36) and it is conceivable that Bcl-3 may act in part by limiting expression of this protein, not just by modulating its functions. RPK1 is involved in apoptosis and necroptosis, and pretreatment of BMDCs with the inhibitor of necroptosis Nec-1 did not rescue T cell priming (data not shown) (39). Bcl-3 appears to modulate the expression of multiple proteins and it may be the combination of these changes that impact the physiology of DCs.
The ability of DCs to prime T cells has also recently been linked to a major shift in metabolic programming from oxidative phosphorylation to aerobic glycolysis (40). A similar switch occurs in highly proliferative and stressed tumor cells and activated lymphocytes. It may be in the context of metabolic shifts and stress conditions that Bcl-3 is required to carefully control the adequate survival of activated DCs, thereby assuring an effective adaptive immune response.
Supplementary Material
Acknowledgements
We greatly appreciate the constructive inputs provided by all members of the Siebenlist laboratory and the Laboratory of Molecular Immunology.
This research was supported by the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases and the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health.
Abbreviations
- CBA
cytometric bead array
- BMDC
bone marrow-derived dendritic cells
References
- 1.Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 2.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, Fink PJ, Chervonsky AV. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujikado N, Saijo S, Yonezawa T, Shimamori K, Ishii A, Sugai S, Kotaki H, Sudo K, Nose M, Iwakura Y. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nature medicine. 2008;14:176–180. doi: 10.1038/nm1697. [DOI] [PubMed] [Google Scholar]
- 4.Chen M, Wang J. Regulation of Immune Responses by Spontaneous and T cell-mediated Dendritic Cell Death. Journal of clinical & cellular immunology. 2011:S3. doi: 10.4172/2155-9899.S3-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kushwah R, Hu J. Dendritic cell apoptosis: regulation of tolerance versus immunity. Journal of immunology. 2010;185:795–802. doi: 10.4049/jimmunol.1000325. [DOI] [PubMed] [Google Scholar]
- 6.Granucci F, Zanoni I. The dendritic cell life cycle. Cell Cycle. 2009;8:3816–3821. doi: 10.4161/cc.8.23.9998. [DOI] [PubMed] [Google Scholar]
- 7.Dissanayake D, Hall H, Berg-Brown N, Elford AR, Hamilton SR, Murakami K, Deluca LS, Gommerman JL, Ohashi PS. Nuclear factor-kappaB1 controls the functional maturation of dendritic cells and prevents the activation of autoreactive T cells. Nature medicine. 2011;17:1663–1667. doi: 10.1038/nm.2556. [DOI] [PubMed] [Google Scholar]
- 8.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 9.Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, Yu T, Fagerlund R, Asagiri M, Zuniga EI, Hoffmann A. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-kappaB pathways. Nature immunology. 2012;13:1162–1170. doi: 10.1038/ni.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, Beg AA. Distinct roles of different NF-kappa B subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. Journal of immunology. 2007;178:6777–6788. doi: 10.4049/jimmunol.178.11.6777. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, D'Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8alpha-dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity. 1998;9:839–847. doi: 10.1016/s1074-7613(00)80649-4. [DOI] [PubMed] [Google Scholar]
- 12.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Claudio E, Brown K, Siebenlist U. NF-kappaB guides the survival and differentiation of developing lymphocytes. Cell Death Differ. 2006;13:697–701. doi: 10.1038/sj.cdd.4401894. [DOI] [PubMed] [Google Scholar]
- 14.Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- 15.Ohno H, Takimoto G, McKeithan TW. The candidate proto oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 16.Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, Jundt F, Anagnostopoulos I, Bommert K, Lichter P, Stein H, Scheidereit C, Dorken B. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106:4287–4293. doi: 10.1182/blood-2004-09-3620. [DOI] [PubMed] [Google Scholar]
- 17.Maldonado V, Melendez-Zajgla J. Role of Bcl-3 in solid tumors. Mol Cancer. 2011;10:152. doi: 10.1186/1476-4598-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, Love P, Sher A, Siebenlist U. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 19.Kreisel D, Sugimoto S, Tietjens J, Zhu J, Yamamoto S, Krupnick AS, Carmody RJ, Gelman AE. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest. 2011;121:265–276. doi: 10.1172/JCI42596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pene F, Paun A, Sonder SU, Rikhi N, Wang H, Claudio E, Siebenlist U. The IkappaB family member Bcl-3 coordinates the pulmonary defense against Klebsiella pneumoniae infection. Journal of immunology. 2011;186:2412–2421. doi: 10.4049/jimmunol.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruan Q, Zheng SJ, Palmer S, Carmody RJ, Chen YH. Roles of Bcl-3 in the pathogenesis of murine type 1 diabetes. Diabetes. 2010;59:2549–2557. doi: 10.2337/db10-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Wang H, Claudio E, Brown K, Siebenlist U. A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity. 2007;27:438–452. doi: 10.1016/j.immuni.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Paun A, Claudio E, Wang H, Siebenlist U. The tumor promoter and NF-kappaB modulator Bcl-3 regulates splenic B cell development. Journal of immunology. 2013;191:5984–5992. doi: 10.4049/jimmunol.1300611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tassi I, Cella M, Castro I, Gilfillan S, Khan WN, Colonna M. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. European journal of immunology. 2009;39:1369–1378. doi: 10.1002/eji.200839313. [DOI] [PubMed] [Google Scholar]
- 25.Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, de Bovis B, Devilard E, Viret C, Azukizawa H, Kissenpfennig A, Malissen B. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. The Journal of experimental medicine. 2010;207:189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annual review of immunology. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 28.Riemann M, Endres R, Liptay S, Pfeffer K, Schmid RM. The IkappaB protein Bcl-3 negatively regulates transcription of the IL-10 gene in macrophages. Journal of immunology. 2005;175:3560–3568. doi: 10.4049/jimmunol.175.6.3560. [DOI] [PubMed] [Google Scholar]
- 29.De Smedt T, Pajak B, Klaus GG, Noelle RJ, Urbain J, Leo O, Moser M. Antigen-specific T lymphocytes regulate lipopolysaccharide-induced apoptosis of dendritic cells in vivo. Journal of immunology. 1998;161:4476–4479. [PubMed] [Google Scholar]
- 30.Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, Rocchetti M, Mingozzi F, Foti M, Chirico G, Costa B, Zaza A, Ricciardi-Castagnoli P, Granucci F. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460:264–268. doi: 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- 31.Ramaswamy M, Cruz AC, Cleland SY, Deng M, Price S, Rao VK, Siegel RM. Specific elimination of effector memory CD4+ T cells due to enhanced Fas signaling complex formation and association with lipid raft microdomains. Cell Death Differ. 2011;18:712–720. doi: 10.1038/cdd.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desbarats J, Wade T, Wade WF, Newell MK. Dichotomy between naive and memory CD4(+) T cell responses to Fas engagement. Proc Natl Acad Sci U S A. 1999;96:8104–8109. doi: 10.1073/pnas.96.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maksimow M, Santanen M, Jalkanen S, Hanninen A. Responding naive T cells differ in their sensitivity to Fas engagement: early death of many T cells is compensated by costimulation of surviving T cells. Blood. 2003;101:4022–4028. doi: 10.1182/blood-2002-06-1904. [DOI] [PubMed] [Google Scholar]
- 34.O'Keeffe M, Grumont RJ, Hochrein H, Fuchsberger M, Gugasyan R, Vremec D, Shortman K, Gerondakis S. Distinct roles for the NF-kappaB1 and c-Rel transcription factors in the differentiation and survival of plasmacytoid and conventional dendritic cells activated by TLR-9 signals. Blood. 2005;106:3457–3464. doi: 10.1182/blood-2004-12-4965. [DOI] [PubMed] [Google Scholar]
- 35.Speirs K, Lieberman L, Caamano J, Hunter CA, Scott P. Cutting edge: NF-kappa B2 is a negative regulator of dendritic cell function. Journal of immunology. 2004;172:752–756. doi: 10.4049/jimmunol.172.2.752. [DOI] [PubMed] [Google Scholar]
- 36.Larghi P, Porta C, Riboldi E, Totaro MG, Carraro L, Orabona C, Sica A. The p50 subunit of NF-kappaB orchestrates dendritic cell lifespan and activation of adaptive immunity. PLoS One. 2012;7:e45279. doi: 10.1371/journal.pone.0045279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 38.Hon H, Rucker EB, 3rd, Hennighausen L, Jacob J. bcl-xL is critical for dendritic cell survival in vivo. Journal of immunology. 2004;173:4425–4432. doi: 10.4049/jimmunol.173.7.4425. [DOI] [PubMed] [Google Scholar]
- 39.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Everts B, Pearce EJ. Metabolic Control of Dendritic Cell Activation and Function: Recent Advances and Clinical Implications. Frontiers in immunology. 2014;5:203. doi: 10.3389/fimmu.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.