

Abstract
Hepatocellular cancer (HCC) is a highly treatment refractory cancer and is also highly resistant to adverse cellular stress. While cell behavior can be modulated by non-coding RNAs (ncRNAs) within extracellular vesicles (EVs), the contributions of long non-coding RNAs (lncRNAs) are largely unknown. To this end, the involvement and functional roles of lncRNAs contained within EVs during chemotherapeutic stress in human HCC were determined. Expression profiling identified a subset of lncRNAs that were enriched in tumor cell derived vesicles released from two different cell lines. Of these, lincRNA-VLDLR (linc-VLDLR) was significantly up-regulated in malignant hepatocytes. Exposure of HCC cells to diverse anti-cancer agents such as sorafenib, camptothecin, and doxorubicin increased linc-VLDLR expression in cells as well as within EVs released from these cells. Incubation with EVs reduced chemotherapy-induced cell death and also increased linc-VLDLR expression in recipient cells. RNAi-mediated knockdown of linc-VLDLR decreased cell viability and abrogated cell cycle progression. Moreover, knockdown of VLDLR reduced expression of ABCG2 (ATP-binding cassette, sub-family G member 2), whereas over-expression of this protein reduced the effects of VLDLR knockdown on sorafenib-induced cell death. Here, linc-VLDLR is identified as an extracellular vesicle enriched lncRNA that contributes to cellular stress responses.
Implications
These findings provide new insight into the role of extracellular vesicles and demonstrate the capacity of lncRNAs to mediate chemotherapeutic stress response in HCC.
Keywords: Exosomes, non-coding RNA, hepatocellular cancer, drug transporters, intercellular signaling
Introduction
Hepatocellular cancer (HCC) is one of the most prevalent cancers worldwide, with an annual incidence of around 750,000 new cases.(1) This tumor is characterized by the alteration of multiple signaling pathways that modulate tumor behavior, local spread and a tendency for multifocal tumor development.(2) HCC is highly resistant to conventional therapies. Tumor progression is increased by the ability of HCC cells to resist adverse environmental stress such as hypoxia, radiation, and chemotherapy. Acquired resistance to adverse environmental conditions enhances tumor propagation, malignant progression and resistance to therapy, and is a central issue in both the pathophysiology and the therapy of HCC.(3, 4) Understanding the primary mechanisms or acquiring resistance to cellular stress will enable us to develop more effective treatments for HCC.
The major focus of attention in genetic regulation of HCC development, progression and behavior has been on protein-coding genes, and more recently microRNAs. In contrast, the contribution of long non-coding RNA to hepatocarcinogenesis has only recently become appreciated. Long non-coding RNAs (lncRNAs) are defined as non-coding RNAs more than 200 nucleotides in length.(5-8) Like miRNA, these lncRNA can impact on regulation of gene expression and have an impact on many different cellular processes. In contrast to miRNA, however, they have complex RNA structures and can function through a diverse and broad range of mechanism. Our previous studies suggested that TUC338, a lncRNA that contains an ultra-conserved element, is significantly increased in human cirrhosis and HCC and can promote cell growth of HCC cells.(9) On the other hand, maternally expressed gene 3 is strikingly down-regulated in HCC relative to expression in non-malignant hepatocytes and plays crucial role as a tumor suppressor.(10) Although these and other lncRNA such as MALAT-1, HULC and H19 have been implicated in human HCC, the functional contribution of these and other lncRNA genes is mostly unknown.
We have recently shown that HCC cells can release extracellular vesicles (EV) such as exosomes, which are membrane-derived vesicles that originate from endosomal multivesicular bodies and have a size range of 40–100nm when released into the interstitial fluid. These vesicles contain protein, lipids and RNA derived from their donor cell cytoplasm(11) and can be taken up by other cells. EVs are considered to be related to cell–cell communication and can transfer their content to modulate cellular activities in recipient cells.(12, 13) These vesicles have been reported to be secreted into the medium from a variety of normal or tumor cells in culture. We previously documented that HCC cell derived EVs contain miRNAs that can modulate transformed cell behavior in recipient cells.(14) Similar to miRNAs and mRNAs, HCC cell derived EVs could contain and transfer long non-coding RNAs. We postulated that this intercellular signaling could mediate resistance to chemotherapeutic stress in HCC cells. Our studies identified involvement of the lncRNA linc-VLDLR in modulation of chemotherapeutic responses by tumor cell extracellular vesicles. These findings provide several new insights into mechanisms of resistance to chemotherapy in HCC, and the contribution of extracellular lncRNA mediated signaling in tumor cell responses to adverse environmental.
Materials and Methods
Cell Lines, Culture, and Reagents
Non-malignant human hepatocytes were obtained from Sciencell (Carlsbad, CA) and cultured as recommended by the supplier. HCC cell lines HepG2, Hep3B, PLC/PRF-5 and Huh-7 were obtained from the American Type Culture Collection (Manassas, VA). HepG2.ST were obtained from HepG2 cells by spontaneous transformation.(15) Human cholangiocarcinoma cell line MzChA-1 were obtained as described (16). All cell lines were authenticated. All of the HCC cells were cultured in DMEM high glucose medium (HyClone Laboratories, Logan, UT), containing 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Life Technologies, Grand Island, NY), at 37°C with 5% CO2. Mz-ChA-1 cells were cultured in CMRL Medium 1066 (Life Technologies) with 10% FBS, 1% L-glutamine, and 1% antibiotic-antimycotic mix. For all studies with extracellular vesicles (EV), EV depleted medium was prepared by centrifuging cell-culture medium at 100,000g overnight to spin down any pre-existing EV content. Camptothecin and doxorubicin was obtained from Sigma-Aldrich (St. Louis, MO), and sorafenib was obtained from Selleck (Houston, TX). Compounds were dissolved in 100% DMSO and diluted with culture media to the desired concentration with a final DMSO concentration of 0.1%. DMSO 0.1% (v/v) was used as a solvent control.
Isolation of extracellular vesicles
HCC cells (1×106) were plated in 11mL of EV-depleted medium (DMEM, 10% FBS, 1% Anti-Anti) on collagen-coated 10-cm dishes. After 3-4 days, the medium was collected and sequential centrifugation was performed. The medium was first centrifuged at 300g for 10 minutes, then at 2,000g for 20 minutes in 4°C to remove cells and cell debris. The supernatant was then centrifuged at 10,000g for 70 minutes at 4°C. The supernatant was further ultracentrifuged at 100,000g for 70 minutes at 4°C to pellet EV, which were then washed by resuspending in phosphate-buffered saline (PBS) and ultracentrifuged at 100,000g for 70 minutes in 4°C. The final pellet, comprised of an extracellular vesicle (EV) preparation that contained a homogenous population of extracellular vesicles, and was used for isolation of extracellular RNA (exRNA) or other studies, or resuspended with 50-100 μl of PBS and stored at -80°C. The protein yield was measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific Inc., Rockford, IL). The number of EV was examined by NanoSight (NanoSight Ltd, Amesbury, UK).
RNA Extraction and Analysis
Total RNA was extracted from HCC cells using Trizol (Life Technologies) and extracellular RNA (exRNA) was isolated using ExoQuick-TC (System Biosciences, Mountain View, CA). HCC cells (1 × 106) were plated in 11mL of EV-depleted medium on collagen-coated 10-cm dishes. After 3-4 days, the medium was collected and sequentially centrifuged at 3000xg for 15min to remove cells and cell debris. The supernatant was transferred to a sterile vessel and combined with 2mL ExoQuick-TC. After an overnight precipitation at 4°C, exRNA was extracted using SeraMir™ Exosome RNA Amplification Kit (System Biosciences) according to the manufacturers’ instructions. RNA concentration was measured using NanoDrop ND-2000 (Nano-Drop Technologies, Wilmington, DE).
Real-time PCR Analysis
RNA was treated with RNase-free DNase I (Qiagen, Valencia, CA). One μg of RNA was reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (BIO-RAD Laboratories, Inc., Hercules, CA), and Real-time quantitative RT-PCR (qRT-PCR) was performed using a Mx3000p System (Stratagene, La Jolla, CA) to detect mRNA and ncRNA expression with SYBR green I (SYBR® Advantage® qPCR Premix, Clontech Laboratories, Inc., Mountain View, CA). The following PCR primers were used: linc-VLDLR, forward: 5’-AGCAGTCACATTCATCGCAC-3’, reverse: 5’-GAGGAATAGGTGCGAACTGC-3’, ATP-binding cassette half-transporter (ABCC1), forward: 5’- GAGAGTTCCAAGGTGGATGC-3’, reverse: 5’-AGGGCCCAAAGGTCTTGTAT-3’, ATP-binding cassette, sub-family G member 2 (ABCG2), forward: 5’-TTCGGCTTGCAACAACTATG-3’, reverse: 5’- TCCAGACACACCACGGATAA-3’, RNU6B, forward: 5’-CTCGCTTCGGCAGCACA-3’, reverse: 5’-AACGCTTCACGAATTTGCGT-3’.
Long non-codingRNA (lncRNA) profiling
Expression profiling of 90 human lncRNA was performed using the LncProfiler™ qPCR Array Kit (System Biosciences) according to the manufacturers’ instructions. RNA from EV or donor cells (n = 3 per each cell line) were treated with DNase I and 2 μg of DNase-treated RNA was reverse transcribed. Real-time PCR was performed (2X Maxima® SYBR Green with Rox, Fermentas, Glen Burnie, MD) and the cycle number at which the reaction crossed a threshold (CT) was determined for each gene. Raw CT values were normalized using a median CT value (ΔCT = CTlncRNA – CTmedian). For each lncRNA, the relative amount of each lncRNA between two sample sets A and B was described using the equation 2-ΔΔCT where ΔΔCT =ΔCTA- ΔCTB.
Transfection of siRNAs
siRNA against linc-VLDLR was designed using sidesign center (Dharmacon, Lafayette, CO). Two independent siRNAs against linc-VLDLR (VLDLR siRNA-1; GCACAACACCCAAAGACAT and VLDLR siRNA-2; CACAACACCCAAA-GACATA) or non-targeting (NT) control siRNA (siGENOME Non-Targeting siRNA) were purchased from Dharmacon (Lafayette, CO). HepG2 cells were transfected with 50 nM siRNA to linc-VLDLR or NT control using Lipofectamine 2000 (Life Technologies) for 48 hours before further experiments.
Western blotting
Total protein was extracted from cultured cells using cOmplete Lysis-M, EDTA-free and cOmplete Mini, EDTA-free, Protease Inhibitor Cocktail Tablet (Roche Applied Science, Indianapolis, IN). Equivalent amounts of protein samples were mixed with NuPAGE® LDS Sample Buffer (4x) (Life Technologies) and separated on NuPAGE® 4-12% Bis-Tris Gel (Life Technologies) and then transferred to nitrocellulose membrane (BIO-RAD). The membranes were blocked with blocking buffer (LI-COR® Biosciences, Lincoln, NE) for 1 hours and then incubated overnight at 4°C with the respective primary antibody as follows: mouse monoclonal antibody anti-PCNA (1:500, Santa Cruz Biotechnology, Dallas, TX), mouse monoclonal antibody anti-ABCC1 (MRP1) (1:500, Santa Cruz Biotechnology), mouse monoclonal antibody anti-ABCG2 (ABCG2) (1:1000, Santa Cruz Biotechnology) and goat polyclonal anti- Actin (1:5000) (Santa Cruz Biotechnology, Inc., Dallas, TX). The membrane was washed thrice for 15 minutes with TBS-T (25 mM Tris-HCl, pH 7.4, 125 mm NaCl, 0.05% Tween 20) and then incubated with Alexa Fluor® 680 goat anti-mouse IgG (H+L) (1:5000, Life Technologies) for PCNA, ABCC1 and ABCG2 and Alexa Fluor® 680 rabbit anti-goat IgG (H+L) (1:5000) (Life Technologies) for Actin for 20 minutes. Visualization and quantitation of protein expression was performed using the Odyssey imaging System (LI-COR® Biosciences). Relative expression was determined by probing the same membrane against Actin.
Cell viability and cell growth assays
For cell viability studies, cells were seeded (1 × 104/well) in collagen-coated 24-well plates. At each time point, trypan blue staining was performed and the number of viable cells expressed relative to cell counts at baseline. For cell proliferation studies, HepG2 cells were seeded (1 × 104/well) into 96-well collagen-coated plates. At indicated time points, proliferation was assessed using MTS solution (Promega, Madison, WI) and a Fluostar Omega microplate reader (BMG Labtech, Cary, NC). Background correction was performed by subtracting background fluorescence from wells without cells. For studies of chemotherapeutic stress, cells were then incubated with varying concentrations of sorafenib, camptothecin, doxorubicin or the appropriate diluent (DMSO) control.
Flow cytometry
For cell cycle analysis, cells were permeabilized with 70% ethanol, and DNA was stained with 20 μg/mL propidium iodide, 0.2 mg/mL RNase A, and 0.01mol/L PBS (pH7.4). Cellular DNA content was measured using an Accuri C6 flow cytometer (Accuri Cytometers Inc., Ann Arbor, MI), and the proportions of cells in particular phases of the cell cycle were analyzed using FCS express version 3 software (De Novo Software, Los Angeles, CA).
Transfection of plasmids
pcDNA3.1(-)-MRP1 (ABCC-1) was kindly provided by Dr. Susan Cole at Queens University Cancer Research Institute, Kingston, Ontario, Canada. pCMV6-XL5-ABCG2 was purchased from OriGene Technologies (Rockville, MD). HepG2 cells were transfected using Lipofectamine 2000 (Life Technologies) for 24 hours with 2μg ABCC1 or pcDNA3.1 control vector, or with ABCG2 or pCMV6 control vector prior to further study.
Statistical analysis
Data were expressed as the mean and standard error from at least three replicates. Comparisons between groups were performed using the two-tailed Student’s t test, and results were considered to be statistically significant when p < 0.05.
Results
Linc-VLDLR is enriched in HCC derived EVs
To identify candidate lncRNAs that could potentially function as signaling mediators through extracellular vesicle mediated mechanisms, we first sought to identify lncRNA that are enriched within extracellular vesicles. Expression profiling was performed using qRT-PCR based assays to identify lncRNA within tumor cell derived EV, and the relative change when compared with their expression within the cells of origin. Studies were performed in donor cells and EV released from these cells in two different primary liver cancer cell lines, HepG2 and MzChA1 cells (Supplementary Tables 1-3). We identified 20 lncRNAs that could be detected in EV with at least 2-fold enrichment compared with their respective donor cells. Of these, 8 lncRNAs were enriched in EV obtained from both cell lines, whereas the rest were selectively enriched in EV from one or other cell line only (Fig. 1A). Next, we examined lncRNA expression between malignant and non-malignant hepatocyte cells to identify lncRNA that are deregulated in HCC. 21 lncRNAs were identified that were aberrantly expressed by >2-log fold in malignant human HCC (HepG2) cells compared to non-malignant human hepatocytes (HH) respectively (Fig. 1B). The large intergenic non-coding RNA-VLDLR (Linc-VLDLR) was identified as amongst the most significantly up-regulated lncRNA that is also enriched within EV derived from HepG2 and MzChA1 cells. Expression of linc-VLDLR was increased in several other malignant hepatocyte cell lines by 1.9- to 2.9-fold (Fig. 1C). Thus, linc-VLDLR is selectively released in EV from tumor cells, as well as constitutively over-expressed in malignant cells.
Figure 1. LncRNA expression in liver cancer cells and extracellular vesicles.
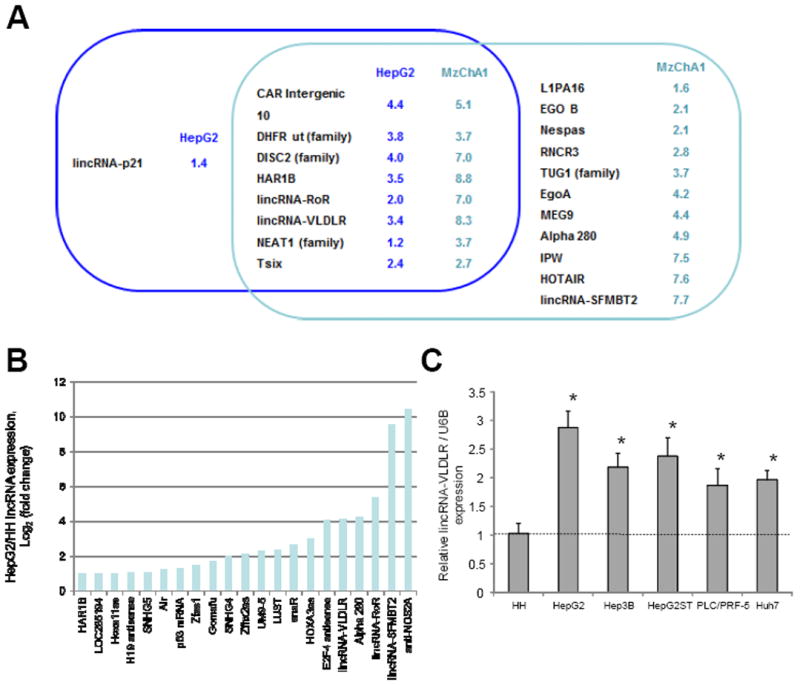
A, enrichment of lncRNA within EV was analyzed by comparing the expression of each lncRNA in either HepG2 HCC cells or Mz-ChA-1 biliary cancer cells and in EV derived from these cells. The Venn diagram illustrates lncRNA for which the EV/cell ratio was greater than 2-fold in either HepG2 cells (blue), or Mz-ChA-1 cells (green), with the overlap indicating lncRNA that were selectively enriched in EV from both tumor cell types. The numbers indicate the average log2 (fold-change) in lncRNA expression in EV relative to donor cells from three independent samples. B, lncRNA expression was performed in three independent replicates in HepG2 HCC cells and non-malignant human hepatocytes (HH). LncRNAs increased by >2-fold in HepG2 cells compared to HH cells are shown. C, RNAs were extracted and qRT-PCR for linc-VLDLR was performed in non-malignant cells (HH) and HCC cell lines. Expression of linc-VLDLR was normalized to the expression of RNU6B and is expressed relative to that in HH. Bars represent the mean ± SEM of 3 independent studies. *, p < 0.05.
Linc-VLDLR promotes cell cycle progression
To gain insight into the functional role of linc-VLDLR, we next examined the effect of linc-VLDLR knockdown using siRNA on cell proliferation and viability. Transfection with either of two different linc-VLDLR siRNA constructs reduced linc-VLDLR expression by 40 to 70% compared with non-targeting siRNA controls (Fig. 2A). Using these constructs and conditions, we assessed the effect of linc-VLDLR knockdown on cell cycle progression in HepG2 cells. siRNA to linc-VLDLR-1 significantly increased the percentage of cells in G1 phase from 50.3% to 58.2% compared with control, and decreased the percentage of cells in S and G2/M phases (data not shown). Moreover, linc-VLDLR knockdown decreased expression of PCNA, a marker of cell proliferation and S phase in HepG2 cells (Fig. 2B). Next, we investigated the effect of linc-VLDLR knockdown on cell proliferation in HepG2 and PLC/PRF-5 cells. Compared to controls, a significant reduction in cell proliferation was observed with either of two different siRNA to linc-VLDLR (Fig. 2C and D). These studies support a role of linc-VLDLR in modulating HCC cell proliferation by showing that knockdown of linc-VLDLR can result in G1/S arrest.
Figure 2. Effect of linc-VLDLR knockdown on HCC cell proliferation.
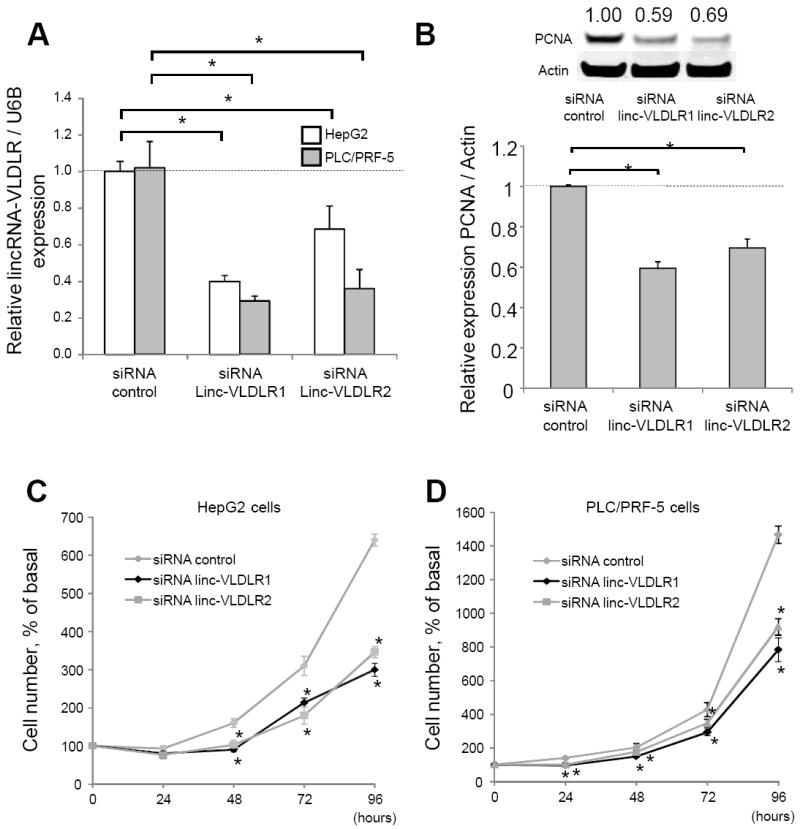
Cells were transfected with either siRNA to linc-VLDLR (siRNA linc-VLDLR 1 or siRNA linc-VLDLR 2) or non-targeting control siRNAs, and studied after 48 hours. A, linc-VLDLR was assessed in HepG2 or PLC/PRF-5 cells by qRT-PCR. B, Cells were transfected with either siRNA to linc-VLDLR (siRNA linc-VLDLR 1 or siRNA linc-VLDLR 2) or non-targeting control siRNAs. After 48 hours, western blot analysis was performed for PCNA and actin expression. A representative immunoblot is shown, along with quantitative data obtained by densitometry from three separate studies. Bars represent the mean ± SEM; *, p < 0.05. C and D, transfected HepG2 cells (C) or PLC/PRF-5 cells (D) were plated in 96 well plates, and viable cell number was assessed at the indicated times. Bars represent the mean ± SEM; *, p < 0.05.
Chemotherapeutic stress increases linc-VLDLR in cells and EV
We next sought to identify determinants of EV release of linc-VLDLR, and began by examining tumor cell responses to adverse environmental stresses such as exposure to chemotherapeutic agents. Sorafenib and doxorubicin are the most commonly used agents used in systemic or regional therapies for HCC. A significant increase in linc-VLDLR expression in HCC cells was observed during incubation with sorafenib, and doxorubicin as well as with camptothecin (Fig. 3). Furthermore, siRNA mediated knockdown of linc-VLDLR using two different siRNA (siRNA-linc-VLDLR-1 or siRNA-linc-VLDLR-2) decreased cell viability in response to sorafenib, doxorubicin or camptothecin. Thus, VLDLR represents a stress-responsive lncRNA that can be induced by exposure to chemotherapy and that can contribute to acquired chemoresistance in HCC cells. We observed that linc-VLDLR was also increased in EV released from these tumor cells exposed to these anti-cancer agents (Fig. 4A). Therefore, chemotherapeutic stress enhances cellular linc-VLDLR expression as well as release within EV.
Figure 3. Linc-VLDLR expression and therapeutic response.
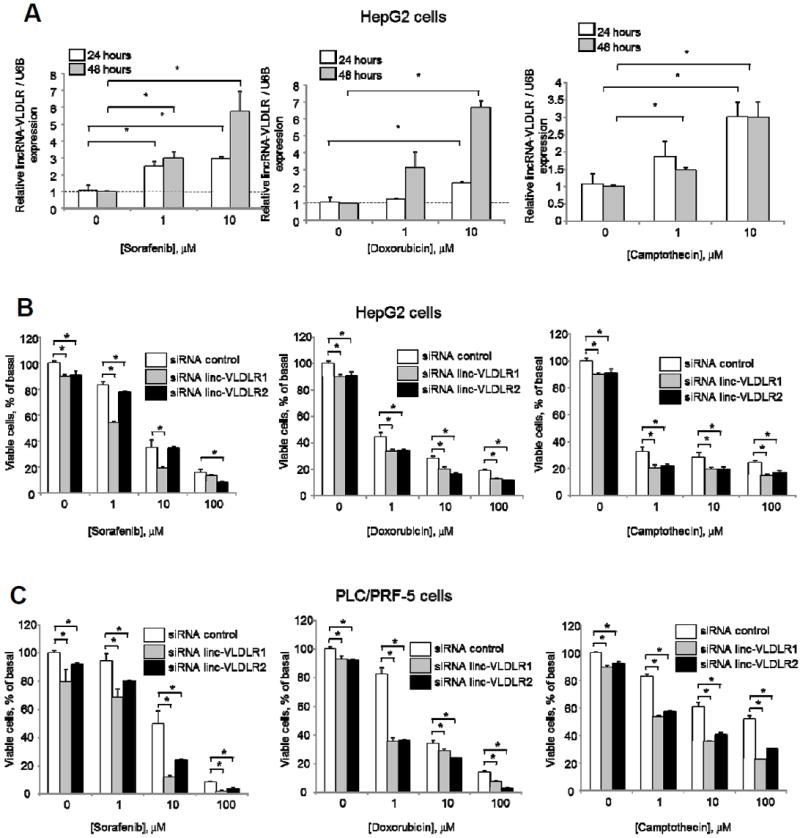
A. HepG2 cells were incubated with varying concentrations of sorafenib, doxorubicin or camptothecin for 24 or 48 hours. Linc-VLDLR expression was determined by qRT-PCR, normalized to that of RNU6B. and is represented relative to normalized expression in controls at each time point. B. HepG2 cells were transfected with siRNA to linc-VLDLR (siRNA VLDLR-1 or siRNA VLDLR-2) or non-targeting siRNA controls. Transfected cells were then incubated with varying concentrations of sorafenib, doxorubicin or camptothecin and cell viability was assessed after 48 hours. Bars represent the mean ± SEM of 3 separate studies. *, p < 0.05. C. PLC/PRF-5 cells were transfected with siRNA to linc-VLDLR (siRNA VLDLR-1 or siRNA VLDLR-2) or non-targeting siRNA controls. Transfected cells were then incubated with varying concentrations of sorafenib, doxorubicin or camptothecin and cell viability was assessed after 48 hours. Bars represent the mean ± SEM of 3 separate studies. *, p < 0.05.
Figure 4. Involvement of extracellular vesicle linc-VLDLR in tumor cell responses to chemotherapy.
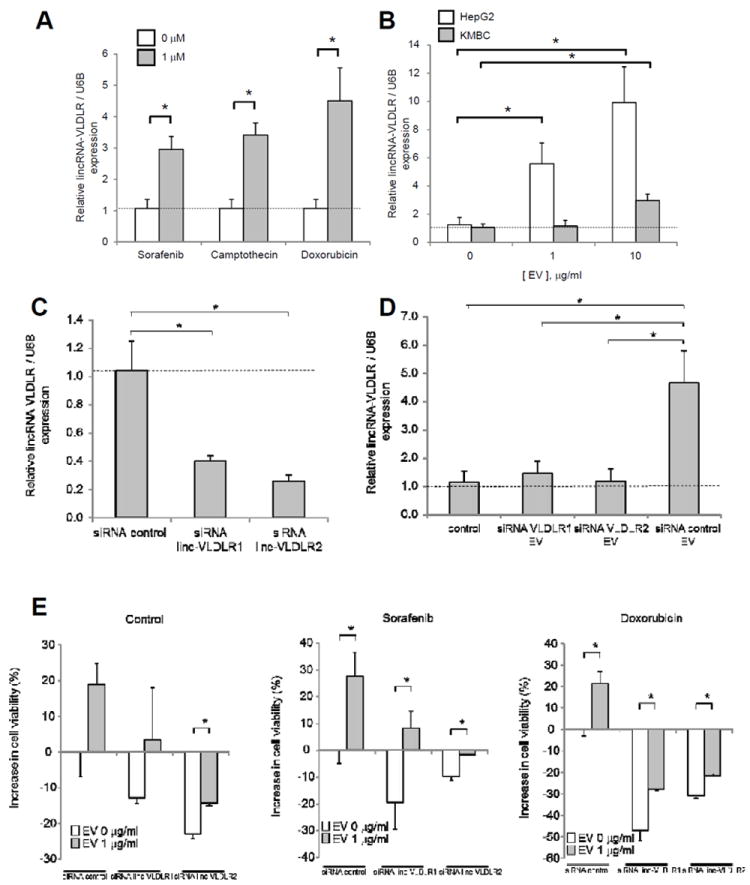
A, Cells were incubated with sorafenib, camptothecin, or doxorubicin. Extracellular vesicles (EV) were obtained after 24 hours, and qRT-PCR was performed for linc-VLDLR. B, recipient HepG2 HCC cells or KMBC biliary cancer cells were incubated with varying concentrations of EVs derived from HepG2 cells. After 24 hours, RNA was extracted and linc-VLDLR expression was assessed by qRT-PCR in recipient cells. C and D, HepG2 cells were transfected with siRNA to linc-VLDLR (siRNA VLDLR-1 or siRNA VLDLR2) or non-targeting controls. After 72 hours, EVs released from these cells were isolated. (C) RNA was isolated from EVs and qRT-PCR was performed for linc-VLDLR. (D) Recipient HepG2 cells were incubated with or without those EVs. RNA was isolated from recipient cells after 48 hours and qRT-PCR was performed for linc-VLDLR. Expression of linc-VLDLR was normalized to expression of RNU6B and expressed relative to control. Bars express the mean ± SEM of 3 separate studies. *, p < 0.05. E HepG2 cells were transfected with siRNA to linc-VLDLR (siRNA VLDLR-1 or siRNA VLDLR-2) or non-targeting controls. After 48 hours, cells were collected and plated (1 × 104/well) on 96 well-plates in vesicle-depleted medium and incubated with 0 or 1 μg/ml of HepG2 cell derived EVs. After 24 hours, cells were incubated with diluent (DMSO) control, 10 μM sorafenib, or 10 μM doxorubicin. Cell viability was assessed after 48 hours using MTS assay. The bars represent the mean ± SEM of the increase in cell viability from 3 independent studies. *, p < 0.05.
EV mediated transfer of linc-VLDLR can result in chemoresistance
We next examined if EVs could deliver linc-VLDLR to other cells similar to the inter-cellular transfer of other non-coding RNA that we and others have previously reported (14, 17). EVs were obtained from HepG2 cells and varying concentrations were incubated with either HepG2 or KMBC tumor cells. The expression of linc-VLDLR expression was assessed by qRT-PCR in these recipient cells after 24 hours. A dramatic increase in linc-VLDLR occurred in a concentration-dependent manner consistent with EV mediated transfer of this lncRNA (Fig. 4B). Linc-VLDLR expression was significantly decreased in EVs derived from HepG2 cells incubated with either one of two siRNA to linc-VLDLR compared to non-targeting siRNA control cells (Fig. 4C). An increase in linc-VLDLR was noted in recipient cells incubated with EV from control siRNA transfected cells, but not with EV from linc-VLDLR knockdown cells (Fig. 4D). Next, we examined the effect of HCC cell derived EV in recipient cell responses to either sorafenib or doxorubicin, and the contribution of linc-VLDLR to these responses. Studies were performed in recipient cells incubated with siRNA to linc-VLDLR-1 or NT controls for 48 hours prior to culture in EV depleted medium and incubation with HepG2-derived EV for 24 hours (Fig. 4E). Cell viability was decreased by linc-VLDLR siRNA-1 or linc-VLDLR siRNA-2 compared with NT siRNA controls. Cell viability was also evaluated following exposure of cells to sorafenib or doxorubicin for 48 hours. Incubation with EV increased cell viability, but this was reduced in cells transfected with linc-VLDLR. These data indicate that EV can modulate cell viability during chemotherapeutic stress, and moreover that linc-VLDLR may promote chemoresistance.
Linc-VLDLR knockdown or EVs treatment on the ATP-binding cassette (ABC) transporter superfamily
Active drug export out of the cells can maintain intracellular drug below toxic levels (18-21). Altered expression of ATP-binding cassette (ABC) transporter superfamily members involved in drug export can contribute to drug resistance through regulated transport of chemotherapeutic agents (19, 20). Incubation with tumor-cell derived EVs increased the expression of ATP-binding cassette, sub-family G member 2 (ABC-G2) (Fig. 5) suggesting a role for EV mediated mechanisms for modulation of chemoresistance through downstream effects on ABC transporters. Moreover, siRNA to Linc-VLDLR-1 resulted in a marked reduction in expression of ABCG2 mRNA and protein. Furthermore, over-expression of ABCG2 reduced the effect of linc-VLDLR1 knockdown on sorafenib-induced cell death in HepG2 cells (Fig 5). The transfer of linc-VLDLR within EV in response to chemotherapeutic stress could therefore contribute to therapeutic resistance through modulation of ABC transporter expression within adjacent tumor cells. In addition, we observed a modest but significant increase in expression of ATP-binding cassette half-transporter (ABCC1) in HepG2 cells during incubation with EV, as well as a reduction in expression of ABCC1 by siRNA mediated knockdown of VLDLR (data not shown). However, over-expression of ABCC1 did not reduce the effect of VLDLR knockdown on sorafenib-induced cell death.
Figure 5. Modulation of expression of ABCG2 by linc-VLDLR.
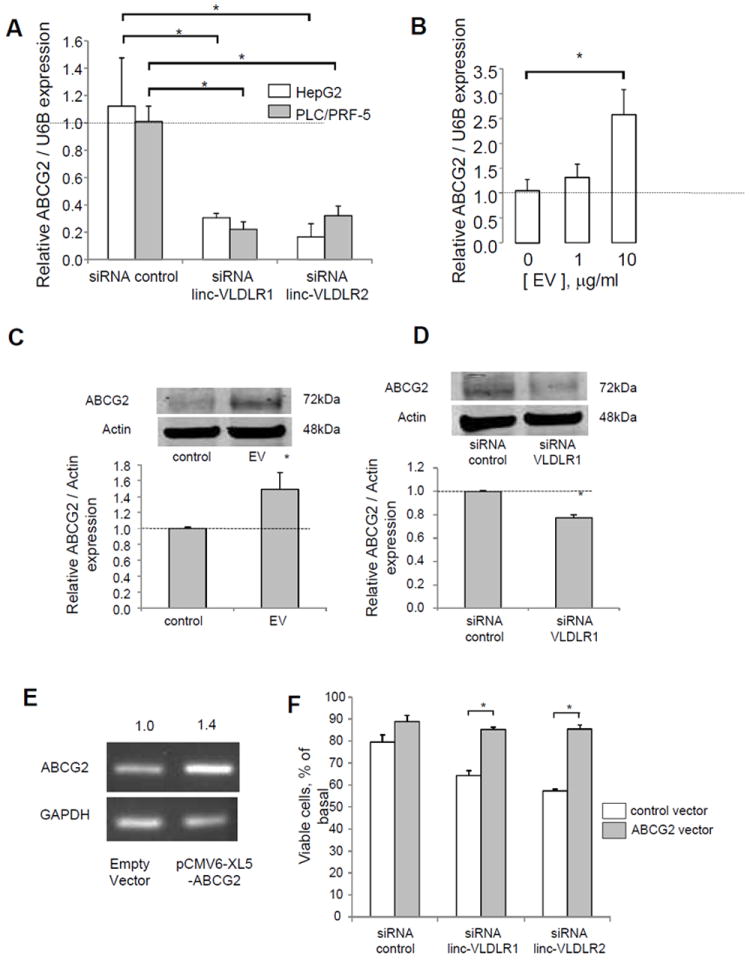
A, HepG2 or PLC/PRF-5 cells were transfected with siRNA to linc-VLDLR (siRNA VLDLR-1 or siRNA VLDLR-2) or non-targeting siRNA controls. After 48 hours, RNA was obtained for qRT-PCR analysis for ABCG2. Expression of ABCG2 gene was normalized to that of RNU6B and represented relative to control. Bars express the mean ± SEM of 3 independent determinations. *, p < 0.05. B, HepG2 cells were incubated with EVs isolated from HepG2. After 48 hours, RNA was obtained and qRT-PCR was performed for ABCG2 mRNA expression. C, HepG2 cells were incubated with 10 μg/ml of HepG2 cells derived EVs. After 48 hours, cells were lysed, and immunoblot analysis was performed using specific antibodies against ABCG2. D, HepG2 cells were transfected with siRNA to linc-VLDLR 1 or non-targeting control. After 48 hours, cells were lysed, and immunoblot analysis was performed using specific antibodies against ABCG2. A representative immunoblot and quantitative densitometric data of the mean ± SEM of ratio of ABCG2 to Actin from at least five independent experiments are shown. *, p < 0.05. E. HepG2 cells were transfected with 2μg pCMV6-XL5-ABCG2 or empty vector for 24 hours. RT-PCR was performed using primers specific for ABCG2 or GAPDH. The average normalized ratio of ABCG2/GAPDH expression from two separate determinations is shown. F. HepG2 cells were transfected with pCMV6-XL5-ABCG2 or empty vector. After 24 hours, cells were transfected with siRNA to linc-VLDLR-1 or non-targeting controls. Cells were detached 24 hours later and reseeded into 96 wells (1 × 104 cells/well). After 24 hours, cell were incubated with diluent (DMSO) control or 1 μM sorafenib. Cell viability was assessed after 48 hours. The bars represent the mean ± SEM of viable cells, % of basal from 3 independent studies. *, p < 0.05.
Taken together, these studies reveal a unique mechanism of chemotherapeutic response and resistance in HCC cells that involves both novel mediators such as the long non-coding RNA linc-VLDLR, and a novel mechanism involving inter-cellular transfer of extracellular vesicles and modulation of target gene expression resulting in acquired chemoresistance in recipient cells (Fig. 6).
Figure 6.
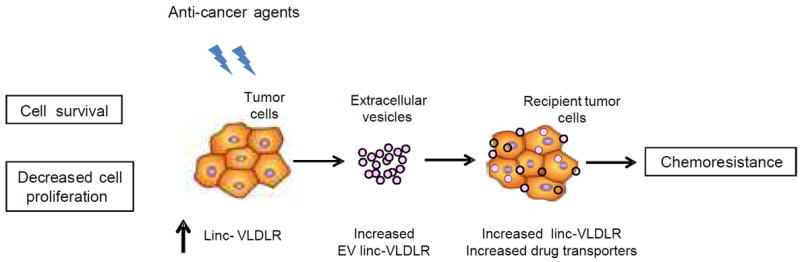
Schematic overview of the role of EV linc-VLDLR in mediating acquired chemoresistance in HCC cells.
Discussion
In this study, we identify the functional involvement on lncRNA in cell-to-cell communication through extracellular vesicles and show that Linc-VLDLR can be transferred by tumor cell derived EVs and modulate resistance to chemotherapy in recipient cancer cells. Demonstration of extracellular vesicle mediated intercellular pathways provides new insights into tumor development and resistance to environmental stresses. These studies raise the potential for linc-VLDLR and possibly other lncRNA as well to modulate biological effects in recipient cells through inter-cellular signaling roles similar to those that have been postulated for exosomal miRNAs.
The involvement of lncRNAs is being increasingly identified in human diseases and biological processes although their functional roles are still poorly understood. Although diverse mechanisms by which lncRNA can modulate gene expression have been identified, their participation as mediators involved in inter-cellular signaling in human diseases has not been characterized. In this study, we identify the large intergenic non-coding RNA linc-VLDLR as a novel signaling mediator that can contribute to chemotherapeutic stress responses in HCC through extracellular vesicle mediated inter-cellular signaling. These observations further support the potential contributions of extracellular vesicle based signaling involving biologically active RNA genes in cellular processes that are directly relevant to human cancers.(22)
Deregulated expression of several lncRNA genes has been identified in HCC but their functional contributions are still unknown. Linc-VLDLR was first identified as a large intergenic non-coding RNAs (lincRNA) with increased expression in induced pluripotent stem cells and embryonic stem cells.(23) The role of this lncRNA in cancer has not been previously established. In addition to the increased presence of linc-VLDLR in HCC derived EV, linc-VLDLR was identified as amongst the most significantly upregulated in expression in malignant human HCC cells. Thus, in addition to its effects in pluripotent cells, linc-VLDLR may have a broader functional role in human cancers that involves proliferative or chemotherapeutic stress responses. The function of linc-VLDLR is unknown.(24-26) Although the function of most lncRNA is obscure, they have been shown to regulate gene expression through very diverse mechanisms such as epigenetic regulation, chromatin remodeling, transcriptional or post-transcriptional regulation or modulation of protein function and localization.(27-31) Our studies have identified determinants of linc-VLDLR expression, release within EV and downstream contribution to acquired chemoresistance, and therefore justify further studies to define the mechanisms by which linc-VLDLR may contribute to expression of drug transporters.
Understanding the mechanisms of chemoresistance is particularly important for cancers such as HCC that respond very poorly to chemotherapeutic agents. Sorafenib, a multi-kinase inhibitor, exerts anti-angiogenic and anti-tumor effects by blocking multiple growth factor pathways.(32) This chemotherapeutic agent is the only FDA-approved treatment for patients with advanced HCC and the effectiveness of this agent is limited due to high acquired resistance.(33) Identification of mechanisms of chemotherapeutic resistance in HCC cells offers the potential to improve the results from the use of agents such as sorafenib for the treatment of HCC. The novel functional role of lncRNA in mediating cellular responses to adverse cellular stresses through extracellular vesicle mediated inter-cellular signaling could therefore provide new opportunities for approaches to enhance therapeutic efficacy.
Supplementary Material
Acknowledgments
The authors thank the members of Patel lab for their critical comments and technical assistance.
Grant Support
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers R01DK069370 and UH2TR000884. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Financial support: Supported in part by Grant R01DK069370 and UH2TR000884 from the National Institutes of Health
Abbreviations
- ABCC1
ATP-binding cassette half-transporter
- ABCG2
ATP-binding cassette, sub-family G member 2
- EV
extracellular vesicles
- HCC
hepatocellular cancer
- lncRNA
long non-coding RNA
- linc-VLDLR
long intergenic non-coding RNA VLDLR
- miRNA
microRNAs
Footnotes
Authors’ Contributions
Conception and design: K. Takahashi, T. Patel
Development of methodology: K. Takahashi, I. K. Yan, T. Patel
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): K. Takahashi, I. K. Yan, J. Wood, H. Haga
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): K. Takahashi, I. K. Yan, T. Patel,
Writing, review, and/or revision of the manuscript: K. Takahashi, I. K. Yan, H. Haga, T. Patel
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): K. Takahashi, I. K. Yan, H. Haga, T. Patel
Study supervision: T. Patel
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 3.Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–27. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22:1178–82. doi: 10.1111/j.1440-1746.2007.04997.x. [DOI] [PubMed] [Google Scholar]
- 5.Ariel I, Miao HQ, Ji XR, Schneider T, Roll D, de Groot N, et al. Imprinted H19 oncofetal RNA is a candidate tumour marker for hepatocellular carcinoma. Mol Pathol. 1998;51:21–5. doi: 10.1136/mp.51.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26:851–8. doi: 10.1038/sj.onc.1209846. [DOI] [PubMed] [Google Scholar]
- 7.Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–42. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 8.Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX, et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679–89. doi: 10.1002/hep.24563. [DOI] [PubMed] [Google Scholar]
- 9.Braconi C, Valeri N, Kogure T, Gasparini P, Huang N, Nuovo GJ, et al. Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2011;108:786–91. doi: 10.1073/pnas.1011098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S, et al. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–6. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 12.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 13.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–6. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011;54:1237–48. doi: 10.1002/hep.24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kogure T, Yan IK, Lin WL, Patel T. Extracellular vesicle-mediated transfer of a novel long noncoding RNA TUC339: A mechanism of intercellular singnaling in human hepatocellular cancer. Genes & Cancer. 2013 doi: 10.1177/1947601913499020. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marienfeld C, Tadlock L, Yamagiwa Y, Patel T. Inhibition of cholangiocarcinoma growth by tannic acid. Hepatology. 2003;37:1097–104. doi: 10.1053/jhep.2003.50192. [DOI] [PubMed] [Google Scholar]
- 17.Xin H, Li Y, Buller B, Katakowski M, Zhang Y, Wang X, et al. Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells. 2012;30:1556–64. doi: 10.1002/stem.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2:3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–17. [PubMed] [Google Scholar]
- 20.Sukowati CH, Rosso N, Pascut D, Anfuso B, Torre G, Francalanci P, et al. Gene and functional up-regulation of the BCRP/ABCG2 transporter in hepatocellular carcinoma. BMC Gastroenterol. 2012;12:160. doi: 10.1186/1471-230X-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chow EK, Fan LL, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–41. doi: 10.1002/hep.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703–19. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–7. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schonrock N, Harvey RP, Mattick JS. Long noncoding RNAs in cardiac development and pathophysiology. Circ Res. 2012;111:1349–62. doi: 10.1161/CIRCRESAHA.112.268953. [DOI] [PubMed] [Google Scholar]
- 28.Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–9. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 29.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 33.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.