

Abstract
Acute lung injury (ALI) is a major component of multiple organ dysfunction syndrome (MODS) following hemorrhagic shock (HS) resulting from major surgery and trauma. The increased susceptibility in HS patients to the development of ALI suggests not yet fully elucidated mechanisms that enhance pro-inflammatory responses and/or suppress anti-inflammatory responses in the lung. Alveolar macrophages (AMϕ) are at the center of the pathogenesis of ALI after HS. We have previously reported that HS-activated polymorphonuclear neutrophils (PMN) interact with macrophages to influence inflammation progress. In this study, we explore a novel function of PMN regulating AMϕ anti-inflammatory mechanisms involving autophagy. Using a mouse “two-hit” model of HS/resuscitation followed by intratracheal injection of muramyl dipeptide (MDP), we demonstrate that HS initiates high mobility group box 1 (HMGB1)/TLR4 signaling, which upregulates NOD2 expression in AMϕ and sensitizes them to subsequent NOD2 ligand MDP to augment lung inflammation. Additionally, upregulated NOD2 signaling induces autophagy in AMϕ, which negatively regulates lung inflammation through feedback suppression of NOD2-RIP2 signaling and inflammasome activation. Importantly, we further demonstrate that HS-activated PMN that migrate in alveoli counteract the anti-inflammatory effect of autophagy in AMϕ, possibly through NAD(P)H oxidase mediated signaling to enhance IKKγ phosphorylation, NF-κB activation, and NLRP3 inflammasome activation, and therefore augment post-HS lung inflammation. These findings explore a previously unidentified complexity in the mechanisms of ALI, which involves cell-cell interaction and receptor cross-talk.
Keywords: HMGB1, TLR4, NOD2, autophagy, inflammasome, alveolar macrophages
Introduction
Hemorrhagic shock (HS) resulting from major surgery and trauma renders patients more susceptible to the development of systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS) in response to a secondary inflammatory stimulus such as infection. This hypersusceptibility occurs through a cell priming mechanism (1). Acute lung injury (ALI) is a major component of MODS and often serves as a cause of patient death. However, the underlying mechanism by which HS primes for the development of SIRS and ALI has yet to be fully determined. Understanding these mechanisms may lead to improved prophylactic interventions for surgical and trauma patients.
Inflammation is a dynamic self-regulating process governed by interactions between pro- and counter-inflammatory factors. Alveolar macrophages (AMϕ) are at the center of the development of inflammation. Macrophages are activated via families of related pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and nucleotide-binging oligomerization domain (NOD)-like receptors (NLRs) (2-5). We have previously shown that TLR4 and TLR2 signaling plays an important role in the development of ALI following HS through activating AMϕ (6-8) and mediating high mobility group box 1 (HMGB1)-induced activation of NLRP3 inflammasome in AMϕ (9).
NOD2, the product of CARD15, is a member of a growing family of NLRs that have been implicated in the regulation of immune responses and cell death in animals and plants (10, 11). NOD2 acts as a cytosolic recognition molecule of bacterial peptidoglycan (PGN), which is found on both Gram-positive and Gram-negative bacteria, through specific detection of the conserved muramyl dipeptide (MDP) structure (12). NOD2 have been shown to associate with RIP2/RICK, via CARD-CARD interactions, which allow RIP2 to associate with TRAF6/TAK1 (13). Subsequent signaling leads to activation of NF-κB and upregulation of inflammatory mediators, such as interleukin IL-6 (13).
Recent studies have also shown that NLRs, including NOD2, regulate autophagic processes during bacterial infection, which are now recognized to influence pro- and anti-inflammatory responses in cells (14, 15). Autophagy is a basic cell biological process that occurs under physiologic circumstances in almost all human cell types. One main function of autophagy is to recycle damaged or unneeded cellular proteins to preserve cell function (16). Autophagy is upregulated by various stress conditions including those leading to inflammation (17). However, the role of autophagy in HS-induced inflammation remains unknown.
Emerging evidences have shown that reactive oxygen species (ROS) derived from NAD(P)H oxidase play an important role in mediating organ injury after HS (8, 18-22). NAD(P)H oxidase in polymorphonuclear neutrophils (PMN) is an important source of ROS mediating organ injury after HS (6, 7, 18, 21-23). NAD(P)H oxidase, a highly regulated membrane-bound enzyme complex, catalyzes the production of superoxide by the one-electron reduction of oxygen using NAD(P)H as the electron donor. Studies have suggested that ischemia-reperfusion primes circulating PMN for increased ROS production, therefore augmenting neutrophil-mediated lung injury once the PMN are sequestered in the lung (6, 24, 25). Though NAD(P)H oxidase has classically been thought of as a part of the antimicrobial armamentarium of phagocytes (26), the role of this enzyme (or its isoforms in other cell types) in signaling has been described (21, 22, 27, 28).
In this study, we demonstrate that HS upregulates NOD2 expression in AMϕ through high mobility group box 1 (HMGB1)/TLR4 signaling. Upregulated NOD2 subsequently sensitizes AMϕ to respond to NOD2 ligand MDP, which initially leads to augmented inflammation in the lung. NOD2 signaling also induces autophagy in AMϕ, which in turn exhibits a potent anti-inflammatory effect on lung inflammation at later time points, thereby negatively regulating inflammation. However, this anti-inflammatory effect was concealed by HS-activated PMN that migrated into alveoli and counteract the effects of autophagy in AMϕ. These findings explore a previously unidentified self-regulatory mechanism within AMϕ as well as a role for PMN in counteracting this anti-inflammatory effect.
Materials and Methods
Materials
Recombinant HMGB1 was purchased from R&D systems (Minneapolis, MN). Stimulating activity of recombinant HMGB1 was confirmed in mouse macrophages by assay of TNF release, with an ED50 of 3 ∼ 12 μg/ml. In some experiments, in order to exclude the effects of contaminating LPS, the recombinant HMGB1 was heated at 100 °C for 5 min or incubated with 100 μg/ml polymyxin B (PmB) at 37 °C for 2 h. Polyclonal neutralizing antibody against HMGB1 prepared as described previously (29) was provided by Dr. K. J. Tracey (Feinstein Institute for Medical Research, North Shore-LIJ Health System, Manhasset, NY). Muramyl dipeptide (MDP) was obtained from InivoGen (San Diego, CA). Nonimmune rabbit IgG (Catalog #: I5006), 3-methyladenine (3-MA) and all other chemicals were purchased from Sigma-Aldrich (St.Louis, MO), except where noted.
Mouse model of hemorrhagic shock and intratracheal injection of MDP
Male C57BL/6 wild type (WT) mice, NOD2 knockout (NOD2-/-) mice, and LC3 (Atg8E) knockout (LC3-/-) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). TLR4 knockout (TLR4-/-) mice were bred in Dr. Billiar's lab at the University of Pittsburgh; all mice used are on a C57BL/6 background. All experimental protocols involving animals were approved by Institutional Animal Care and Use Committee of VA Pittsburgh Healthcare System and University of Pittsburgh. Mice were 12-14 weeks of age at the time of experiments and were maintained on standard rodent chow and water ad libitum. The mice were not fasted before experiments. Animals were anesthetized with 50 mg/kg of ketamine and 5 mg/kg of xylazine via intraperitoneal (i.p.) administration. Femoral arteries were cannulated for monitoring of mean arterial pressure (MAP), blood withdrawal and resuscitation. HS was initiated by blood withdrawal and reduction of the MAP to 40 mmHg within 20 min. Blood was collected into a 1 ml syringe and heparinized to prevent clotting. In order to exclude the effect of heparin on immune processes, equal amounts of heparin (10 U in 0.1 ml saline) were injected into sham animals through the cannulated femoral artery during the sham operation. After a hypotensive period of 1 h, animals were resuscitated by transfusion of the shed blood and Ringer's Lactate (RL) in a volume equal to that of shed blood, over a period of 20 min. The catheters were then removed, the femoral artery was ligated, and the incisions were closed. Sham animals underwent the same surgical procedures without hemorrhage and resuscitation. At 4 h after resuscitation, MDP in a dose of 50 μg/kg by body weight was injected intratracheally (i.t.) into the mice (HS-MDP two-hit model). In some animals, autophagy inhibitor 3-MA (0.75 mg/ml in saline, 15 mg/kg B.W.) was injected i.t. 10 min prior to MDP (30, 31). The animals remained anesthetized throughout the entire experimental period. At various time points after resuscitation (0 to 18 h), either bronchoalveolar lavage (BAL) was performed and BAL fluid (BALF) was collected or lung tissue was harvested for experimental analysis.
AMϕ isolation
BAL was performed as previously described (32). The immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) was used to isolate AMϕ from BAL fluid. Magnetic nanoparticle-conjugated antibodies (anti-mouse Gr-1, anti-CD4, anti-CD8, and anti-CD45R/B220 antibodies; BD Biosciences Pharmingen, San Diego, CA) were chosen to label and remove PMN and lymphocytes. The resulting cells consisted of >98% macrophages, and cell viability was >95%.
Immunofluorescence confocal microscopy
AMϕ were cytospun onto a microscope slide and fixed with 4% paraformaldehyde for 20 min. After washing with PBS, the cells were permeabilized with 0.25% Triton X-100 in PBS for 10 min at room temperature, followed by blocking with 1% bovine serum albumin (BSA) in PBST (PBS with 0.2% Tween-20) for 2 h at room temperature to reduce nonspecific staining. The cells were then incubated with anti-LC3 antibody (1:500; Abgent, San Diego, CA) at 4°C overnight. After washing twice with PBS, the cells were incubated with Alexa Fluor 555-conjugated anti-rabbit IgG (1:500; Cell Signaling Technology, Beverly, MA) for 1 h at room temperature. Hoechst 33258 (1:200; Sigma-Aldrich, St. Louis, MO) was used to stain nuclei. The cells were then washed with PBS for 3 times, followed by confocal microscopy.
In vivo neutrophil depletion and repletion
PMN depletion was induced using RB6-8C5 monoclonal antibody (Ly-6G/Gr-1 specific) (eBioscience, San Diego, CA) (33). At approximately 16 h before performing shock or sham operation, 10 μg of the anti-mouse Ly-6G/Gr1 antibody or control antibody (rabbit anti-mouse IgG; Sigma-Aldrich) was administered i.p. to mice in 100 μl saline. Our previous studies have shown that during the period of 16 to 24 h after injection of anti-mouse Ly-6G/Gr1 antibody the circulating PMN count in the antibody-treated group was decreased to 0.08 ± 0.02% of total white blood cells vs. 22.2 ± 1.9% in the control group (28). There were no statistically significant differences in the number of peripheral lymphocytes, atypical lymphocytes, monocytes, or eosinophils between the antibody-treated and control groups (28). In order to determine the role of PMN NAD(P)H oxidase in interacting AMϕ, PMN repletion in neutropenic mice was performed by tail vein injection of PMN (∼2 × 106 cells) isolated from the blood of WT or gp91phox-/- mice that were subjected to either HS or sham operation. An immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) (34) was used to isolate PMN. Viability of the isolated PMN was > 95%, and PMN purity was >95% as assessed by trypan blue exclusion and Wright-Giemsa staining, respectively.
Coimmunoprecipitation and immunoblotting analysis
Mouse AMϕ were lysed (∼1 × 106 cells/ml) in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 20 mM PMSF). Protein levels were quantified, and 600 μg of total protein for each sample was then immunoprecipitated with anti-ASC antibody (Santa Cruz Biotechnologies, CA), anti-NOD2 antibody (Santa Cruz Biotechnologies, CA), or anti-p62/SQSTM1 antibody (Sigma-Aldrich, St. Louis, MO). The immunoprecipitated proteins were separated on a 10% SDS-PAGE gel, and were then electroblotted onto PVDF membrane and blocked for 1 h at room temperature with Tris-buffered saline containing 3% non-fat dried milk. NLRP3 and RIP2 protein was detected by probing the membranes with anti-NLRP3 and anti RIP2 antibodies (Santa Cruz Biotechnologies, CA) at 1:500 dilution, respectively, and detected with Clean-Blot IP Detection Reagent (Thermo Scientific, Rockford, IL) following the manufacturer's instructions. Blots were then stripped and reprobed with anti-ASC antibody or anti-NOD2 antibody, and detected with Clean-Blot IP Detection Reagent. Caspase-1 cleavage in the AMϕ was measured by detecting its p10 fragment in Western blot using rabbit polyclonal anti-mouse caspase-1 p10 (Santa Cruz Biotechnologies, CA).
Western Blot Analysis
Aliquots of AMϕ lysates were separated on a 10% SDS-PAGE under non-reducing condition. Equivalent loading of the gel was determined by quantitation of protein as well as by reprobing membranes for actin detection. Separated proteins were electroblotted onto PVDF membrane and blocked for 1 h at room temperature with Tris-buffered saline containing 1% BSA. The membranes were then probed with primary antibody (polyclonal anti-NOD2, -NOD1, -IKKγ (phospho Ser31), -MIP-2, -MIF or −LC3 antibody purchased from Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature for 1 h. After washing, primary antibodies associated with the membranes were detected on autoradiographic film by horseradish peroxidase-conjugated secondary antibodies and the ECL plus chemiluminescent system (Amersham, Arlington Heights, IL) according to the manufacturer's instructions. Blots were quantitated using Scion Image software (Scion Corp., Frederick, MD) and normalized to actin. Caspase-1 cleavage in the AMϕ was measured by detecting its p10 fragment in Western blot using rabbit polyclonal anti-mouse caspase-1 p10 (Santa Cruz Biotechnologies, CA).
Measurement of cytokines
IL-1β, TNFα, and IL-6 in AMϕ, BALF, cell supernatant, and cell culture media were measured using ELISA Ready-Set-Go kit for mouse IL-1β, TNFα, and IL-6 (eBioscience, San Diego, CA), respectively, following the manufacturer's instructions.
RNA extraction and quantitative real-time PCR
Total RNA was isolated from AMϕ by TRI-REAGENT (Molecular Research Center, Cincinnati, OH) following manufacture's instruction. Real time RT-PCR was done using a PrimerPCR™ SYBR® Green Assay kit (BIO-RAD, HerculesCA) in a Bio-Rad iQ5 real time PCR machine (Bio-Rad Laboratories, Hercules, CA) using SYBR Green detection protocol. The specific primers for mouse NOD2, TNFα, IL-6 and IL-1β real time RT-PCR were also purchased from BIO-RAD, and the assays were performed follow the manufacturer's instruction. After amplification protocol was over, PCR product was subjected to melt curve analysis using Bio-Rad iQ5 software. Fold change was calculated using the ΔΔCT method (35) and the value for the GAPDH gene, which was normalized to untreated mouse AMϕ.
NF-κB p65 assay
NF-κB activation in AMϕ was measured by detecting p65 in cell nuclear extracts using TransAM™ NFκBp65 assay kit obtained from Active Motif (Carlsbad, CA), following manufactory instruction. Nuclear protein extracts were prepared from AMϕ by the method of Deryckere and Gannon (36).
Data presentation and statistical analysis
The data are presented as mean ± SEM of the indicated number of experiments. Statistical significance among group means was assessed by ANOVA. Student Neuman- Keuls post-hoc test was performed. Differences were considered significant at p<0.05.
Results
HS upregulates NOD2 expression in AMϕ through HMGB1/TLR4 signaling
We observed a TLR4-dependent upregulation of NOD2 expression in AMϕ after HS. C57BL/6 (wild type, WT) mice and TLR4-/- mice were subjected to HS, and at 1 - 8 h after resuscitation BAL fluid was collected and AMϕ were isolated using an immunomagnetic separation system (7). NOD2 mRNA and protein levels in the AMϕ were measured using real-time RT-PCR and Western blotting, respectively. As shown in Figure 1A and 1B, NOD2 expression was increased in AMϕ isolated from WT/HS mice by 4 h, but not in AMϕ from TLR4-/-/HS mice.
Figure 1. HS upregulates NOD2 expression in AMϕ.
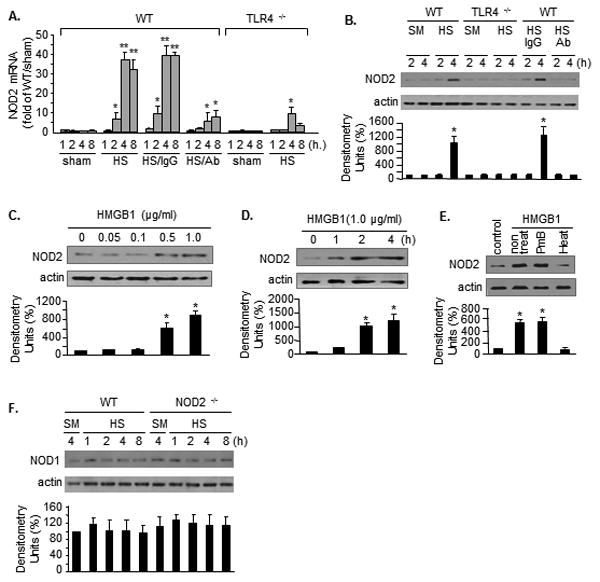
NOD2 mRNA (A) or protein (B) in AMϕ in WT and TLR4-/- mice at 1 - 8 h after hemorrhagic shock (HS) or sham surgery. In some experiments, neutralizing antibody to HMGB1 (Ab) or non-specific IgG (IgG) was injected into mice 10 min before HS. n=4/gp, **(or *) P<0.01 vs. the indicated groups. NOD2 expression in WT AMϕ stimulated with increasing concentrations of HMGB1 (C) or 1.0μg/mL HMGB1 for up to 4h (D). WT AMϕ were also treated with heated HMGB1 (100 °C, 5 min) (Heat) or PmB treated HMGB1 (PmB) for 4h (E). NOD1 expression (F) in WT and NOD2-/- AMϕ after sham (SM) surgery of HS up to 8h. Images are representative of three independent experiments. The graphs depict the mean ± SEM of the % changes in NOD2 or NOD1 expression from 3 – 4 experiments. *P < 0.01 compared with the groups with no asterisk.
Our previous studies have shown that HMGB1 is an important mediator of AMϕ activation during HS (37, 38). To determine if extracellular HMGB1 is responsible for the HS-induced NOD2 expression, neutralizing antibody to HMGB1 was administered to mice (600 μg/mouse) 10 min before HS. Treatment with anti-HMGB1 antibody markedly prevented the HS-induced NOD2 expression in AMϕ (Figure 1A and 1B). Furthermore, AMϕ isolated from normal WT mice were treated with recombinant HMGB1 in vitro for up to 4 h, and NOD2 expression in the AMϕ was then assessed. HMGB1 treatment resulted in a dose- and time-dependent induction of NOD2 in AMϕ (Figure 1C and 1D). In order to exclude the effects of contaminating LPS in recombinant HMGB1, we pre-treated the recombinant HMGB1 with either heating at 100 °C for 5 min or incubation with 100 μg/ml polymyxin B (PmB) at 37 °C for 2 h. As shown in Figure 1E, pre-heating disabled the effect of HMGB1 on up-regulating NOD2, whereas, PmB did not alter the effect of HMGB1. Since NOD 1 and NOD2 share many similar functions, NOD1 expression in AMϕ in response to HS was also measured. As shown in Figure 1C, by contrast, HS did not alter NOD1 expression in AMϕ (Figure 1F).
Upregulated NOD2 signaling mediates HS-primed AMϕ activation
To determine the pathophysiological significance of NOD2 upregulation in AMϕ activation, we applied the HS-MDP two-hit mouse model. MDP is a NOD2 ligand and constituent of peptidoglycan from both Gram positive and Gram negative bacteria. Mice were first subjected to HS for 2 h, and at 4 h after resuscitation, MDP (50 μg/kg B.W.) was injected i.t. to stimulate the upregulated NOD2 in AMϕ. AMϕ were recovered from BAL fluid at 4 h after MDP stimulation for measurement of cytokine expression. The results demonstrate that in WT AMϕ, but not in TLR4-/- and NOD2-/- AMϕ, antecedent HS significantly enhanced the expression of the mRNA and protein of TNFα (Fig. 2A) and IL-6 (Fig. 2B), as well as MIP2 and MIF (Fig. 2C) in response to MDP, suggesting that NOD2 upregulation may contribute to hyperinflammation after HS.
Figure 2. Upregulated NOD2 signaling mediates HS-primed inflammatory responses in AMϕ.
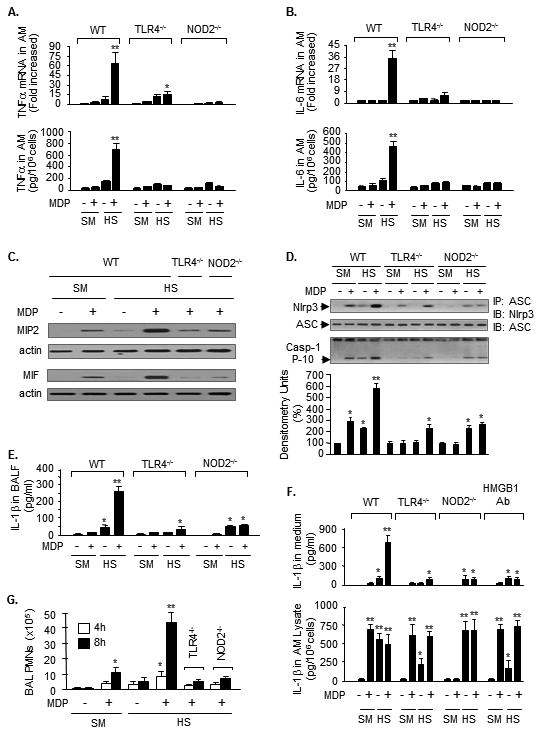
WT, TLR4-/- and NOD2-/- mice were subjected to HS or sham surgery (SM) followed by i.t. MDP or saline. In some mice, neutralizing antibody to HMGB1 (HMGB1 Ab) was injected 15 min prior to HS. AMϕ were recovered 4 h after MDP or saline, and TNFα mRNA and protein in AMϕ (A), IL-6 mRNA and protein in AMϕ (B), and MIP2 and MIF protein in AMϕ (C), were measured by real time RT-PCR and Western blot, respectively. ** (*): P<0.01 vs. the indicated groups; n=3/gp. The images are representative of three independent studies. D. AMϕ collected at 4 h after MDP or saline were subjected to immunoprecipitation (IP) by anti-ASC antibody followed by immunobloting for Nlrp3 and ASC, and caspase-1 p10 fragments were detected by Western blot. The images are representative of four independent studies. The grap depicts the mean SEM of the % changes in caspase-1 p10 fragments from four experiments. *: P< 0.01 vs. the groups with no asterisk; **: P<0.01 vs. all other groups. E. IL-1β in BALF was measured by ELISA 4 h after the MDP or saline. ** (*): P<0.01 vs. indicated groups; n=3/gp. F. Mice were subjected to HS, and at 4 h after resuscitation, AMϕ were then isolated from BALF and incubated with MDP (1μg/ml) in vitro for 4 h. IL-1β in cell culture media and in AMϕ lysates was measured by ELISA. Mean ± SEM, n=3/gp, ** P< 0.01 vs. all other groups; * P< 0.01 vs. the groups labeled with no asterisk. G. Mice were subjected to HS-MDP two-hit and PMN in the BAL fluid were counted at 4 or 8 h after MDP. Mean ± SEM, n=3/gp, ** P< 0.01 vs. all other groups; * P< 0.01 vs. the groups labeled with no asterisk.
IL-1β importantly contributes to the development of post-HS SIRS (21, 39-41), and the production of active IL-1β is tightly controlled by the formation and activation of the inflammasome (42-44). Thus, the effect of upregulated NOD2 signaling on Nlrp3 inflammasome activation in AMϕ was also evaluated. Using the HS-MDP two-hit mouse model, we found that at 4 h after MDP, AMϕ from WT/HS animals exhibited a noticeable increase in the association between Nlrp3 and ASC, cleavage of caspase-1, and IL-1β level in BAL fluid as compared with that in the AMϕ from WT/sham animals (Fig. 2D and 2E). In contrast, genetic deficiency of TLR4 and NOD2 in AMϕ significantly decreased inflammasome activation and IL-1β release into BAL fluid compared to WT in response to HS-MDP (Figure 2D and 2E).
The alterations in IL-1β expression and secretion from AMϕ were further recapitulated in ex vivo experiments. Mice were first subjected to HS model, and at 4 h after resuscitation, AMϕ were then isolated from BAL fluid collected from these mice and treated with MDP (1μg/ml) in vitro for 4 h. IL-1β in cell culture media and in AMϕ lysates was measured by ELISA. As shown in Figure 2F, sequential treatments of HS-MDP induced a significant IL-1β secretion into the medium from WT/HS AMϕ, but not from TLR4-/- and NOD2-/- AMϕ, although HS-MDP increased intracellular IL-1β expression in all of the WT, TLR4-/-, and NOD2-/- AMϕ. In vivo administration of neutralizing antibody against HMGB1 in WT mice prior to HS attenuated IL-1β secretion from the AMϕ in response to in vitro MDP stimulation (Figure 2F).
Moreover, PMN infiltration in the lung was evaluated by counting PMN in BAL fluid at 4 h and 8 h after HS-MDP. Figure 2G shows that antecedent HS primed for an accelerated and enhanced PMN infiltration in the lung in response to MDP, as compared to sham animals. However, genetic deficiency of TLR4 or NOD2 prevented this HS-MDP-induced PMN pulmonary infiltration. Taken together, these results indicate an important role for HMGB1/TLR4-mediated upregulation of NOD2 in mediating HS-primed lung inflammation.
NOD2 signaling induces autophagy in AMϕ
Since NOD2 signaling regulates autophagic processes (14, 15, 45), we therefore further determined the effect of upregulated NOD2 on autophagy formation in AMϕ. We observed in the HS-MDP two-hit model that upregulated NOD2 signaling induced autophagy in AMϕ. AMϕ were recovered from BAL fluid at 1-8 h after i.t. MDP or saline. Microtubule-associated protein1 light chain 3 (LC3) puncta, a marker of autophagy induction, and LC3-II, an activated form of LC3, were detected in the AMϕ by confocal microscopy and Western blot, respectively. As shown in Figure 3A, HS induced LC3 puncta formation in up to 15% of AMϕ by 8 h after i.t. saline. By contrast, HS primed for increased LC3 puncta formation in 55% of WT AMϕ in response to MDP, and this increase was significantly attenuated by giving i.t. autophagy inhibitor 3-MA (15 mg/Kg B.W.) 10 min prior to MDP. NOD2 deficiency prevented HS-MDP-induced AMϕ increases in autophagy (Figure 3A). Figure 3B shows that at 8 h after MDP, LC3-II, an indication of LC3 activation, was detected in the AMϕ from WT/HS mice.
Figure 3. HS induces AMϕ autophagy via NOD2 signaling.
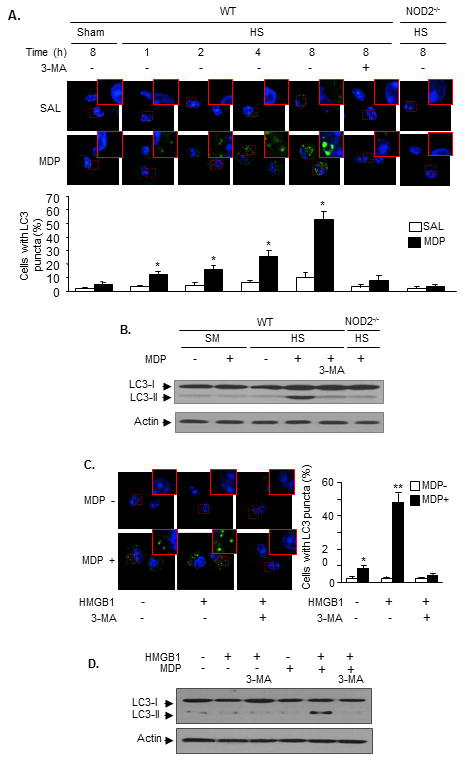
Mice were subjected to the HS-MDP two-hit model. In some animals 3-MA was injected (15 mg/kg B.W., i.t.) 10 min prior to MDP. AMϕ were then isolated from BALF at 1 - 8 h after MDP. LC3 puncta in AMϕ was detected using immunofluorescence confocal microscopy (A), and LC3-I and LC3-II in AMϕ were measured by Western blot (B). n=3/gp, mean ± SD, * P< 0.01 vs. the groups with no MDP treatment. AMϕ isolated from non-surgically treated WT mice were sequentially treated with HMGB1 for 4 h then with MDP for 8 h. In some groups, 3-MA (5 mmol/L) was added to the cells prior to MDP. LC3 puncta in the AMϕ was detected using immunofluorescence confocal microscopy (C), and LC3-I and LC3-II in AMϕ were measured by Western blot (D). Images are representative of three independent studies. n=3/gp, mean ± SD, ** (*): P<0.01 vs. indicated groups.
To confirm that the MDP/NOD2-mediated autophagy is secondary to HMGB1 signaling, AMϕ isolated from non-surgically treated WT mice were sequentially treated with HMGB1 for 4 h then with MDP for 8 h. As shown in Figure 3C, sequential treatment of HMGB1-MDP induced LC3 puncta formation in ∼50% of the AMϕ, which was decreased to 10% of the AMϕ by adding 3-MA (5 mmol/L)10 min prior to MDP. Figure 3D shows that sequential treatment of HMGB1 and MDP induced formation of LC3-II, which diminished by 3-MA. Collectively, the results indicate a NOD2-dependent activation of autophagy in AMϕ at a late phase of HS-MDP-induced onset of inflammation.
Autophagy inhibits inflammatory processes in AMϕ and these effects are counteracted by PMN
Next, we addressed the significance of autophagy induction in the post-HS inflammation. We observed an important anti-inflammatory role of autophagy in regulating MDP-induced inflammation in AMϕ in vitro. As shown in Figure 4A to 4C, at 12 h after MDP treatment, the expression of TNFα and IL-6 mRNA and protein, and IL-1β mRNA in HMGB1 pretreated AMϕ were markedly decreased compared to levels at 4 h after MDP. These decreases in cytokine expression at 12 h were prevented in LC3-/- AMϕ. These data suggest a novel function of autophagy in downregulating cytokine expression at a transcriptional level. Autophagy has also been shown previously to reduce inflammasome activation (46, 47). Similarly, in our model Nlrp3 inflammasome assembly and caspase-1 cleavage also declined at 12 h after MDP as shown by immunoprecipitation with inflammasome component ASC (Figure 4D).
Figure 4. Autophagy inhibits inflammatory processes in AMϕ and these are counteracted by PMN.
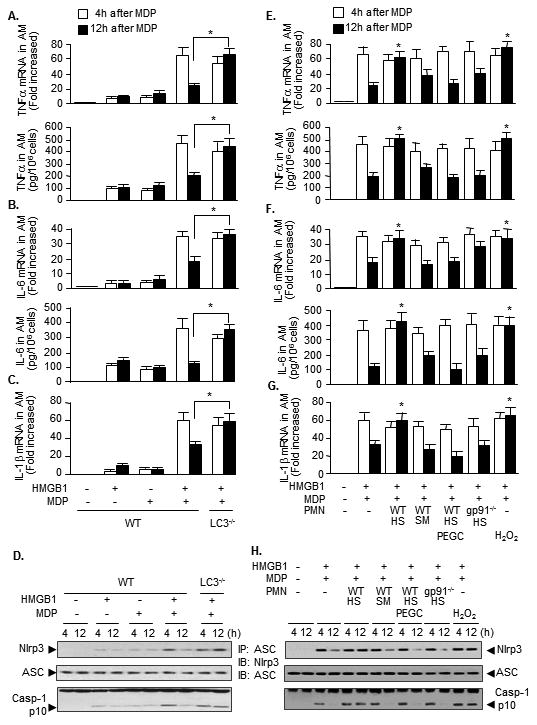
AMϕ isolated from WT or LC3-/- mice were sequentially treated with HMGB1 for 4 h then with MDP for 4 h or 12 h. The expression of mRNA and protein of TNFα (A) and IL-6 (B), and mRNA of IL-1β (C) in AMϕ were then measured by real time RT-PCR and Western blot, respectively (A to C). The association of Nlrp3 and ASC was detected by immunoprecipitation with anti-ASC antibody followed by immunobloting (IB) for Nlrp3 and ASC; caspase-1 cleavage product p10 fragments were detected by Western blot (D). AMϕ from WT mice were pretreated with HMGB1 for 4 h and then cocultured with PMN isolated from HS or sham (SM) mice 2 h after resuscitation in the presence or absence of MDP (1 μg/ml) for 4 h and 12 h. In some groups, PEG catalase (PEGC, 1,000 U/ml) was added to the cocultures. In some other groups, H2O2 (250 μM), in stead of PMN, was added to AMϕ. The expression of mRNA and protein of TNFα (E) and IL-6 (F), and mRNA of IL-1β (G) in AMϕ were then measured by real time RT-PCR and Western blot. The association of Nlrp3 and ASC was detected by IP with anti-ASC antibody followed by IB for Nlrp3 and ASC; caspase-1 cleavage product p10 fragments were detected by Western blot (H). * P< 0.01 vs. other groups at 12 h time point; n=3. Images are representative of three independent studies.
We have previously shown that HS-activated PMN sensitize AMϕ and lung endothelial cell (EC) to bacterial products by upregulating TLR2 signaling in AMϕ and EC as well as NAD(P)H oxidase activation in EC, and thereby, promote lung inflammation following HS(7, 8, 20, 21, 28). In this study we further addressed whether HS-activated PMN affect NOD2-induced inflammatory responses in AMϕ. AMϕ from WT mice were pretreated with HMGB1 for 4 h and then cocultured with PMN isolated from HS mice 2 h after resuscitation, in the presence or absence of MDP for 4 h and 12 h. HS-activated PMN maintained levels of TNFα and IL-6 mRNA and protein (Figure 4E and 4F) and IL-1β mRNA (Figure 4G) and so seem to counteract the anti-inflammatory effect of autophagy on cytokine expression at 12 h. ROS scavenger PEG catalase blocked the influence of PMN on sustained cytokine expression. Furthermore, PMN from WT/sham or gp91-/-/HS mice failed to counteract the effect of autophagy at 12 h (Figure 4E-4G). Additionally, H2O2 (250 μM) also enhances the cytokines expression (Figure 4E to 4G). These data suggest an important role for PMN NAD(P)H oxidase-derived ROS in influencing AMϕ inflammatory response. The effect of HS-activated PMN on Nlrp3 inflammasome assembly and caspase-1 cleavage is consistent with the alterations in the cytokines expression (Figure 4H).
The in vitro observations led us to further investigate the interaction of PMN and AMϕ in vivo. As shown in Figure 5A and 5B, in vivo treatment of WT mice with HS-MDP increased TNFα and IL-6 expression in AMϕ, which reached a peak by 12 h after MDP. However, depletion of circulating PMN in WT mice prior to HS-MDP resulted in significantly lowered TNFα and IL-6 expression in AMϕ, reaching a peak at 4 h after MDP, followed by continuous decline (Figure 5A and 5B). These results suggested an in vivo role for PMN in counteracting autophagy-derived anti-inflammatory effects in AMϕ.
Figure 5. PMN counteract autophagy-derived anti-inflammatory effects in vivo.
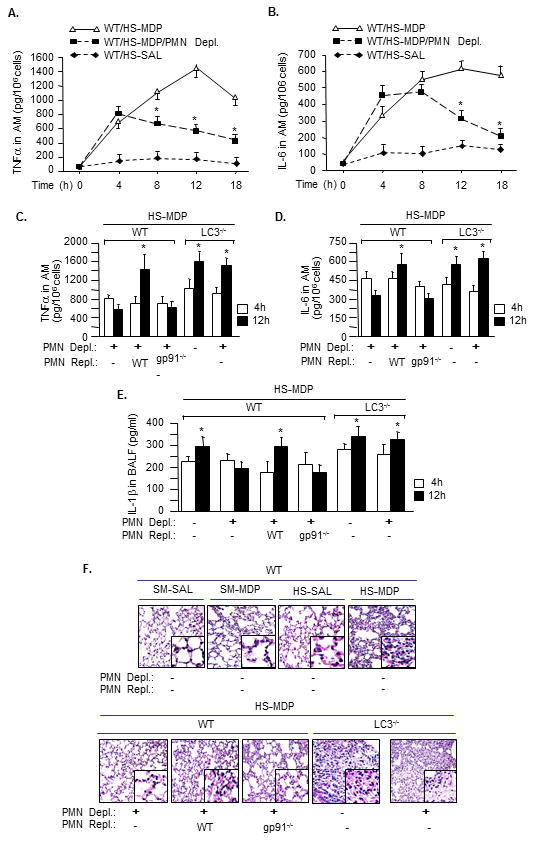
PMN depletion and/or repletion were performed in WT and LC3-/- mice, which were then subjected to HS-MDP two-hit model. The PMN for repletion were isolated from WT or gp91phox-/- mice. AMϕ were isolated from BALF at indicated times after MDP, and TNFα, IL-6 and IL-1β were measured by ELISA (A to E). * P< 0.01 vs. other groups at the same time point; n=3/gp. F. Lung tissue was harvested at 18 h after HS-MDP, and lung histology was assessed with H&E staining. Images are representative of three independent studies.
PMN NAD(P)H oxidase regulates inflammatory response in AMϕ
Previous studies suggested that PMN NAD(P)H oxidase is critical in mediating PMN interaction with other cells(7, 8, 20, 21, 28). To elucidate the role of PMN NAD(P)H oxidase in counteracting the effect of autophagy in AMϕ, we depleted circulating PMN prior to subjecting mice to HS-MDP, and in some cases we replenished mice made neutropenic with PMN isolated either from WT or gp91phox-/- mice. As shown in Figure 5C and 5D, depletion of PMN resulted in a decrease in expression of TNFα and IL-6 in AMϕ at 12 h in WT mice, but not in LC3-/- mice, indicating an autophagy-derived anti-inflammatory role. Repletion of WT PMN in WT neutropenic mice restored expression of TNFα and IL-6 in AMϕ at 12 h, whereas replenishing neutropenic mice with gp91phox -/- PMN failed to do so (Figure 5C and 5D). The maintained increases in IL-1β in BAL fluid in LC3-/- mice, as shown in Figure 5E, demonstrate a role of autophagy in suppressing IL-1β release at 12 h after MDP. Similarly a role for PMN NAD(P)H oxidase counteracting the effects of autophagy is suggested, as replenishing the neutropenic mice with WT PMN, but not gp91phox deficient PMN, increased IL-1β release in response to HS-MDP at 12 h. Figure 5F demonstrates the lung histological changes at 18 h after HS-MDP. HS-MDP induced PMN infiltration in alveoli and interstitial edema in WT mice. LC3 deficiency worsened the HS-MDP-induced lung inflammation. PMN depletion improved the pathological changes; however, replenishing neutropenic mice with WT PMN caused more severe alveolar PMN infiltration and interstitial edema; whereas, replenishing with gp91phox -/- PMN did not worsen the lung inflammation.
In aggregate, these results indicate an important role for PMN NAD(P)H oxidase in mediating PMN counteraction with AMϕ autophagy-derived anti-inflammatory effects.
Autophagy and PMN target NF-κB signaling and inflammasome via different pathways
We next investigated the mechanism by which autophagy and PMN regulate AMϕ inflammatory responses to HS-MDP. We found that PMN depletion and repletion did not alter AMϕ autophagy formation in response to HS-MDP (Figure 6A). This finding excludes a direct role for PMN in suppressing autophagy formation.
Figure 6. Autophagy and PMN target NF-κB signaling and inflammasome via different pathways.
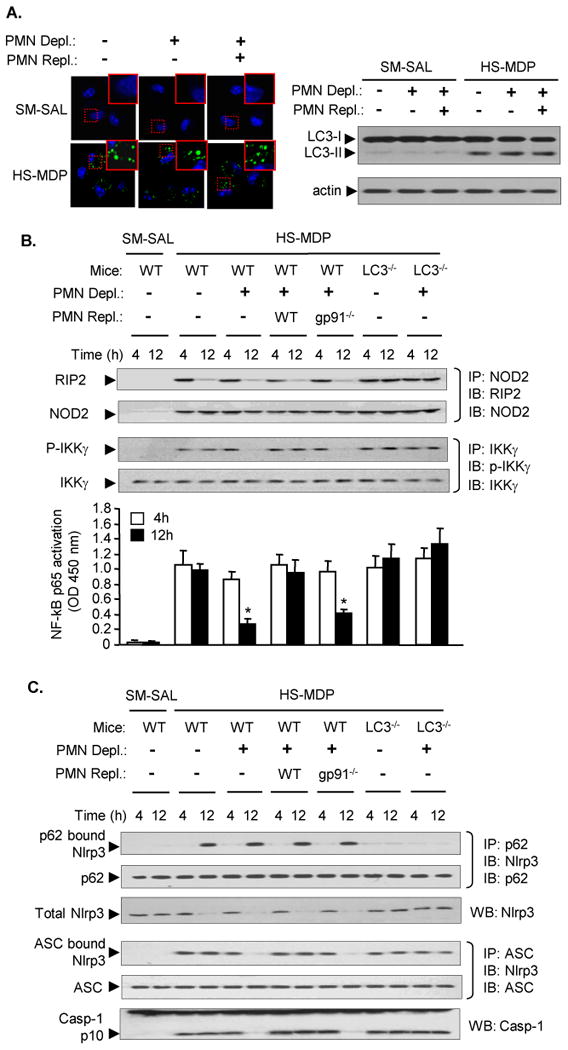
A. PMN depletion and repletion were performed in WT mice, which were then subjected to HS-MDP two-hit model. The PMN for repletion were isolated from WT mice. AMϕ were isolated from BALF at 8h after MDP. LC3 puncta in AMϕ was detected using immunofluorescence confocal microscopy, and LC3-I and LC3-II in AMϕ were measured by Western blot. B and C. PMN depletion and repletion was performed in WT and LC3-/- mice, which were then subjected to HS-MDP two-hit model. At 4h and 12 h after MDP, AMϕ were isolated from BALF. B. The association of RIP2 and NOD2 was detected by immunoprecipitation (IP) with anti-NOD2 antibody followed by immunoblotting (IB) for RIP2 and NOD; the phosphorylated IKKγ was detected by IP with anti- IKKγ antibody followed by IB for phosphorylated IKKγ and IKKγ. NF-κB activation in AMϕ was measured by detecting p65 in cell nuclear extracts using TransAM™ NF Bp65 assay kit. * P< 0.01 vs. other groups at the same time point; n=3/gp. C. The association of Nlrp3 and p62 was detected by IP with anti-p62 antibody followed by IB for Nlrp3 and p62. Association of Nlrp3 and ASC was detected by IP with anti-ASC antibody followed by IB for Nlrp3 and ASC. Total Nlrp3, as well as caspase-1 cleavage product p10 fragments were detected by Western blotting. Images are representative of three independent studies.
NOD2 through RIP2-I-κB kinase (IKK)-NF-κB signaling induces inflammatory cytokines expression (48-52). We measured the association of NOD2 and RIP2 as well as IKKγ phosphorylation in AMϕ following in vivo HS-MDP treatments by immunoprecipitation. We found that in WT and LC3-/- animals the association of RIP2 and NOD2 could be detected at 4 h after MDP, and decreased at 12 h after MDP in WT mice, but remained increase in LC3-/- mice (Figure 6B). PMN depletion and repletion did not affect the HS-MDP-induced association of RIP2 and NOD2. PMN depletion in WT mice decreased IKKγ phosphorylation and NF-κB p65 activity, whereas, repletion of WT PMN, but not gp91phox-/- PMN, restored IKKγ phosphorylation and NF-κB p65 activity. These results suggest that AMϕ autophagy down-regulates inflammation through suppression of association of RIP2 and NDO2, whereas PMN NAD(P)H oxidase acts through different pathway induce IKKγ phosphorylation and NF-κB activation.
In order to address whether autophagy-suppressed association of Nlrp3 and ASC is caused by p62-induced degradation of Nlrp3, we measured the physical binding between Nlrp3 and p62. We found that at 12 h after MDP treatment in WT AMϕ, but not in LC3-/- AMϕ, there was a binding between Nlrp3 and p62, and this binding associated with a decrease in total Nlrp3 protein (Figure 6C). These alterations were not affected by PMN depletion and repletion (Figure 6C), suggesting that PMN are not involved in autophagy regulation of Nlrp3 amount. Nonetheless, the decrease in total Nlrp3 protein at 12 h after MDP did not consequently reduce the association of Nlrp3 and ASC as well as caspase-1 cleavage, the markers of inflammasome activation, unless in the animals that underwent PMN depletion or PMN depletion followed by repletion of gp91phox-/- PMN Figure 6C). Taken together, these results suggest a p62-mediated decrease in Nlrp3 availability and suppression of inflammasome activation following autophagy development; while, PMN promote inflammasome assembly and activation through a mechanism other than affecting Nlrp3 abundance.
Discussion
Following survival from resuscitation, HS patients often face a risk of the development of SIRS and ALI. We have previously reported that augmented activation of innate immunity is an important mechanism underlying HS-primed ALI (7, 32, 53). In this study we explored a novel role for PMN in augmenting post-HS lung injury through counteracting AMϕ anti-inflammatory mechanisms. The current study supports a model, as illustrated in Figure 7, that HS acting through HMGB1/TLR4 signaling upregulates NOD2 expression in AMϕ and subsequent sensitization of AMϕ to NOD2 ligand MDP, which leads to augmented inflammation in the lung. Additionally, upregulated NOD2 signaling induces autophagy in AMϕ, which in turn negatively regulates lung inflammation via a mechanism that involves suppression of NOD2-RIP2 signaling and inflammasome activation. PMN counteract this anti-inflammatory effect of autophagy, possibly via a NAD(P)H oxidase-derived ROS mechanism, and therefore PMN enhance post-HS lung inflammation.
Figure 7. Model of PMN counteraction of autophagic anti-inflammatory mechanisms to augment ALI following HS.
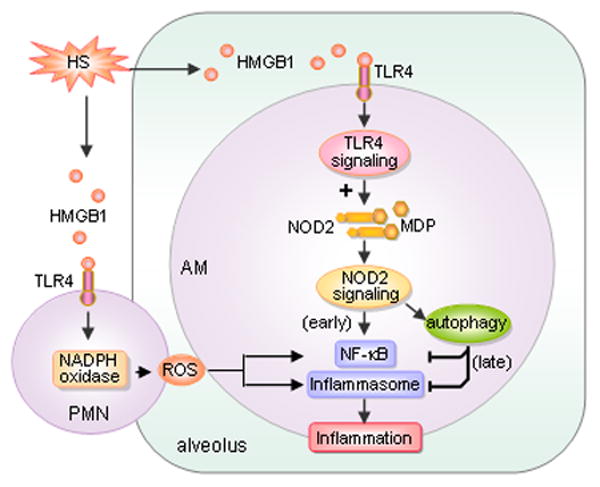
HS increases HMGB1/TLR4 signaling upregulates NOD2 expression in AMϕ, with a subsequent sensitization of AMϕ to NOD2 ligand MDP, which leads to augmented inflammation in the lung. Additionally, upregulated NOD2 signaling induces autophagy in AMϕ, which in turn negatively regulates lung inflammation by suppressing NOD2-RIP2 signaling and inflammasome activation. PMN counteract the anti-inflammatory effect of autophagy, possibly via NAD(P)H oxidase-derived ROS, and therefore enhance post-HS lung inflammation.
NOD2 ligand muramyl dipeptide (MDP) is a molecule derived from peptidoglycan (PGN), which exists in both Gram positive and Gram negative bacteria; thus, NOD2 plays an important role in sensing a wide range of bacteria and regulating host response to infection. The HS/MDP model that was used in the study mimics a clinical scenario in which bacterial ligands of NOD2 may be derived from a wound or the gut after hemorrhage (54-56) or exist in the surrounding environment.
Previous reports have shown that HMGB1 contributes to augmented lung injury by increasing the release of chemokines and proinflammatory cytokines, as well as subsequent PMN sequestration following HS and resuscitation (7, 8, 37, 57). In the present study we further observed that HMGB1/TLR4 signaling upregulated NOD2 expression in AMϕ. This cross-talk between TLR4 and NOD2 in AMϕ resulted in amplification of expression of cytokines and chemokines, as well as Nlrp3 inflammasome activation in response to the bacterial product PGN-derived MDP. This enhanced activation of AMϕ subsequently led to enhanced PMN sequestration in the lung. The role of HMGB1/TLR4 signaling in regulating NOD2 expression was clearly delineated in our studies using neutralizing antibody against HMGB1 and TLR4 knockout mice as shown in Figure 1. Furthermore, direct treatment of AMϕ with HMGB1 induced NOD2 expression in a dose- and time-dependent manner. Upregulation of NOD2 seems necessary for AMϕ to augment expression and release of pro-inflammatory cytokines, since MDP only able to induce enhanced AMϕ activation in HS-stimulated AMϕ in which NOD2 has been upregulated. Our findings therefore suggest an important physiological mechanism of activation of a positive regulatory signal, NOD2, leading to amplified AMϕ activation and lung inflammation in response to microbial products.
However, there is a second critical effect on lung inflammation via NOD2 induction of autophagy in AMϕ, which then negatively regulates lung inflammation. We observed that MDP did not induce autophagy in AMϕ from sham animals, in which NOD2 was not upregulated. Similarly, NOD2 deficiency prevented HS-MDP-induced autophagy in AMϕ. These observations support the hypothesis that upregulation of NOD2 is required for autophagy induction in AMϕ in response to MDP. The mechanism underlying NOD2 activation of autophagy is not fully elucidated here, but others have suggested that NOD2 detects bacterial MDP in the cytoplasm and then directs the autophagic machinery by recruiting ATG16‐like 1 (ATG16L1) to the plasma membrane at the site of bacterial entry(45, 58). NOD2‐assisted localization of ATG16L1 at the plasma membrane is consistent with the role of ATG16L1 in the formation of a portion of autophagic precursors from the plasma membrane (59). Nonetheless, a recent report showed that ATG16L1 functions at an upstream step of the NOD signaling pathway by negatively regulating the activation of RIP2 and the incorporation of RIP2 into large Nodosome complexes; and this NOD-regulatory capacity of ATG16L1 was independent of its role in canonical autophagy(60). Thus, the mechanism underlying NOD2 activation of autophagy remains to be addressed.
Autophagy has been suggested to suppress inflammatory responses to pathogens (61-63). In this study, we demonstrate an anti-inflammatory role for autophagy in regulating HS-primed responses to MDP. The role of autophagy in our studies was defined using LC3-/- mice. We showed that following autophagy formation in WT AMϕ, pro-inflammatory cytokine expression and release were markedly reduced at 12 h after MDP, which we term ‘late phase’. However, these reductions in cytokine expression were prevented in LC3-/- AMϕ. The anti-inflammatory role for autophagy was further supported by our studies showing that LC3 deficiency resulted in the most severe lung histological changes in response to HS-MDP. Importantly, the anti-inflammatory role for autophagy was hidden by PMN-AMϕ interactions, which occur following PMN migration into alveoli. A significant increase in BAL fluid PMN counts was seen at 8 h after HS-MDP, a time when AMϕ autophagy occurred in ∼55% of AMϕ in BAL fluid. In vitro PMN-AMϕ coculture study demonstrated an important role for PMN in counteracting the autophagy-derived anti-inflammatory effects as shown in Figure 4. Noteworthy, our results suggest that the function of PMN is mediated by NAD(P)H oxidase-derived ROS, as PMN isolated from HS-stimulated gp91phox-/- mice were not able to counteract the anti-inflammatory effect of autophagy. An ROS scavenger could also cancel out the effect of WT PMN. In vivo studies using PMN depletion-repletion approaches further confirmed the role of PMN and PMN NAD(P)H oxidase in counteracting autophagy-derived anti-inflammatory mechanism.
We also investigated the mechanisms by which autophagy negatively regulates AMϕ inflammatory responses. NOD2 through interaction with RIP2 activates IKKγ and subsequently NF-κB, which transcriptionally promotes inflammatory cytokines expression (48-52). The data from this study demonstrated that in the late phase (12 h after MDP), induction of autophagy in AMϕ decreased the association of NOD2 and RIP2, and this was prevented by LC3 deficiency. Interestingly, these changes were not affected by PMN depletion and repletion, which suggests separate mechanisms of regulation. However, with the decrease in the association of NOD2 and RIP2, the corresponding decrease in IKKγ phosphorylation and NF-κB activation were only observed when PMNs were depleted, or PMNs depleted and then repleted with gp91phox-/- PMN. These results support a notion that PMN promote IKKγ activation and this counteracts the role of autophagy in suppression of NOD2-RIP2 interaction.
The p62 protein, also called sequestosome 1 (SQSTM1), is an ubiquitin-binding scaffold protein that is able to polymerize via an N-terminal PB1 domain, and interact with ubiquitinated proteins via the C-terminal UBA domain (58). Also, p62 binds directly to LC3 via a specific sequence motif and undergoes degradation by autophagy and so serves to link ubiquitinated proteins to the autophagic machinery to enable their degradation in the lysosome (58). NLRP3 protein deubiquitination has been suggested as an important mechanism for NLRP3 inflammasome priming and activation (61, 64). Our current study suggests that autophagy suppresses inflammasome activation by promoting degradation of ubiquitinated Nlrp3 via p62-mediated pathway. However, we have previously shown that in HS PMN promote inflammasome activation in lung EC through PMN NAD(P)H oxidase-derived ROS, a mechanism other than affecting Nlrp3 abundance (21). We also showed that ROS promote the association of thioredoxin-interacting protein (TXNIP) with Nlrp3 to subsequently induce inflammasome activation and IL-1β secretion from the EC. Further study will be needed to confirm if this mechanism is also valid in AMϕ.
In summary, the present study identifies a previously unrecognized HMGB1/TLR4-NOD2-autophagy axis that serves as a macrophage self-regulatory mechanism governing post-HS inflammatory responses to bacterial products. The study also explored a novel function of PMN NAD(P)H oxidase-derived oxidant signaling in enhancing HS-primed lung injury. PMN NAD(P)H oxidase activates transcellular oxidant signaling through its ability to counteract the autophagy-induced anti-inflammatory mechanisms, and therefore enhancing post-HS lung inflammation and injury. In the broadest sense, these findings may also be valid in other human diseases in which macrophages play a role, including diseases associated with acute and chronic inflammation.
Acknowledgments
1 This work was supported by the National Institutes of Health Grant R01-HL-079669 (J.F. and M.A.W.), National Institutes of Health Center Grant P50-GM-53789 (T.R.B. and J.F.), a VA Merit Award (J.F.), the Key Project of 12th Five-Year Plan of the PLA.BWS 12J027 (X.S.), the Natural Science Foundation of China Grant No.81372013 (X.S.), the Project of Shanghai Leading Talent (X.S.), the Natural Science Foundation of China Grant No. 81400052 (Z.W.), and Shanghai Pujiang Program No. 14PJD030 (Z.W.).
Footnotes
These authors contributed equally.
The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations used in this paper: 3-MA, methyladenine; ALI, Acute lung injury; AMϕ, alveolar macrophages; ASC, apoptosis-associated speck-like protein containing a CARD domain; HMGB1, high mobility group box 1; HS, hemorrhagic shock; MODS, multiple organ dysfunction syndrome; LC3, light chain 3; NLRs, NOD-like receptors; NLRP, nucleotide-binding oligomerization domain-like receptor protein; PRRs, pattern recognition receptors; ROS, reactive oxygen species; SIRS, Systemic inflammatory response syndrome.
References
- 1.Rotstein OD. Modeling the two-hit hypothesis for evaluating strategies to prevent organ injury after shock/resuscitation. J Trauma. 2003;54:S203–206. doi: 10.1097/01.TA.0000064512.62949.92. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 3.Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–454. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity. 2008;29:178–181. doi: 10.1016/j.immuni.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Hansen JD, Vojtech LN, Laing KJ. Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol. 2011;35:886–897. doi: 10.1016/j.dci.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, Yang H, Tracey KJ, Billiar TR, Wilson MA. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178:6573–6580. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 7.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage-primed lung inflammation. American journal of physiology. Lung cellular and molecular physiology. 2006;290:L738–L746. doi: 10.1152/ajplung.00280.2005. [DOI] [PubMed] [Google Scholar]
- 8.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Neutrophil NAD(P)H oxidase is required for hemorrhagic shock-enhanced TLR2 up-regulation in alveolar macrophages in response to LPS. Shock. 2007;28:213–218. doi: 10.1097/shk.0b013e318033ec9d. [DOI] [PubMed] [Google Scholar]
- 9.Xu P, Wen Z, Shi X, Li Y, Fan L, Xiang M, Li A, Scott MJ, Xiao G, Li S, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock augments Nlrp3 inflammasome activation in the lung through impaired pyrin induction. J Immunol. 2013;190:5247–5255. doi: 10.4049/jimmunol.1203182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girardin SE, Sansonetti PJ, Philpott DJ. Intracellular vs extracellular recognition of pathogens--common concepts in mammals and flies. Trends Microbiol. 2002;10:193–199. doi: 10.1016/s0966-842x(02)02334-x. [DOI] [PubMed] [Google Scholar]
- 11.Scott MJ, Chen C, Sun Q, Billiar TR. Hepatocytes express functional NOD1 and NOD2 receptors: a role for NOD1 in hepatocyte CC and CXC chemokine production. Journal of hepatology. 2010;53:693–701. doi: 10.1016/j.jhep.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, Inohara N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. Embo J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 15.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature medicine. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 16.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunological reviews. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdelrahman M, Mazzon E, Bauer M, Bauer I, Delbosc S, Cristol JP, Patel NS, Cuzzocrea S, Thiemermann C. Inhibitors of NADPH oxidase reduce the organ injury in hemorrhagic shock. Shock. 2005;23:107–114. doi: 10.1097/01.shk.0000151028.15377.f7. [DOI] [PubMed] [Google Scholar]
- 19.Sun Q, Gao W, Loughran P, Shapiro R, Fan J, Billiar TR, Scott MJ. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J Biol Chem. 2013;288:15947–15958. doi: 10.1074/jbc.M112.426791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan J, Frey RS, Rahman A, Malik AB. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-alpha -induced NF-kappa B activation and intercellular adhesion molecule-1 expression in endothelial cells. J Biol Chem. 2002;277:3404–3411. doi: 10.1074/jbc.M110054200. [DOI] [PubMed] [Google Scholar]
- 21.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J. Hemorrhagic Shock Activation of NLRP3 Inflammasome in Lung Endothelial Cells. J Immunol. 2011;187:4809–4817. doi: 10.4049/jimmunol.1102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang M, Yin L, Li Y, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock activates lung endothelial reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase via neutrophil NADPH oxidase. Am J Respir Cell Mol Biol. 2011;44:333–340. doi: 10.1165/rcmb.2009-0408OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehnert M, Arteel GE, Smutney OM, Conzelmann LO, Zhong Z, Thurman RG, Lemasters JJ. Dependence of liver injury after hemorrhage/resuscitation in mice on NADPH oxidase-derived superoxide. Shock. 2003;19:345–351. doi: 10.1097/00024382-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation: an early vulnerable window. Surgery. 1995;118:358–364. doi: 10.1016/s0039-6060(05)80345-9. discussion 364-355. [DOI] [PubMed] [Google Scholar]
- 25.Hogg JC. Neutrophil kinetics and lung injury. Physiol Rev. 1987;67:1249–1295. doi: 10.1152/physrev.1987.67.4.1249. [DOI] [PubMed] [Google Scholar]
- 26.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovascular research. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 28.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. The Journal of clinical investigation. 2003;112:1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, Zhao Y, Wang YG, Zhang GG, Yang P, Guo F, Sun Y, Jiang CY. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell death & disease. 2011;2:e159. doi: 10.1038/cddis.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan J, Marshall JC, Jimenez M, Shek PN, Zagorski J, Rotstein OD. Hemorrhagic shock primes for increased expression of cytokine-induced neutrophil chemoattractant in the lung: role in pulmonary inflammation following lipopolysaccharide. J Immunol. 1998;161:440–447. [PubMed] [Google Scholar]
- 33.Assier E, Jullien V, Lefort J, Moreau JL, Di Santo JP, Vargaftig BB, Lapa e Silva JR, Theze J. NK cells and polymorphonuclear neutrophils are both critical for IL-2-induced pulmonary vascular leak syndrome. J Immunol. 2004;172:7661–7668. doi: 10.4049/jimmunol.172.12.7661. [DOI] [PubMed] [Google Scholar]
- 34.Cotter MJ, Norman KE, Hellewell PG, Ridger VC. A novel method for isolation of neutrophils from murine blood using negative immunomagnetic separation. The American journal of pathology. 2001;159:473–481. doi: 10.1016/S0002-9440(10)61719-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- 37.Liu Y, Yuan Y, Li Y, Zhang J, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Interacting neuroendocrine and innate and acquired immune pathways regulate neutrophil mobilization from bone marrow following hemorrhagic shock. J Immunol. 2009;182:572–580. doi: 10.4049/jimmunol.182.1.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang M, Yuan Y, Fan L, Li Y, Li A, Yin L, Scott MJ, Xiao G, Billiar TR, Wilson MA, Fan J. Role of macrophages in mobilization of hematopoietic progenitor cells from bone marrow after hemorrhagic shock. Shock. 2012;37:518–523. doi: 10.1097/SHK.0b013e318249b81d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roumen RM, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, Hendriks T, Goris RJ. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Critical care medicine. 1995;23:474–480. doi: 10.1097/00003246-199503000-00010. [DOI] [PubMed] [Google Scholar]
- 40.Shenkar R, Abraham E. Effects of hemorrhage on cytokine gene transcription. Lymphokine Cytokine Res. 1993;12:237–247. [PubMed] [Google Scholar]
- 41.Abraham E, Richmond NJ, Chang YH. Effects of hemorrhage on interleukin-1 production. Circ Shock. 1988;25:33–40. [PubMed] [Google Scholar]
- 42.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 43.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 44.Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA, Lien E. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Travassos LH, Carneiro LA, Girardin S, Philpott DJ. Nod proteins link bacterial sensing and autophagy. Autophagy. 2010;6:409–411. doi: 10.4161/auto.6.3.11305. [DOI] [PubMed] [Google Scholar]
- 46.Mizumura K, Cloonan SM, Haspel JA, Choi AM. The emerging importance of autophagy in pulmonary diseases. Chest. 2012;142:1289–1299. doi: 10.1378/chest.12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, Keilty JJ, Gosselin ML, Robison KE, Wong GH, Glucksmann MA, DiStefano PS. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 49.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 50.Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 51.Lecine P, Esmiol S, Metais JY, Nicoletti C, Nourry C, McDonald C, Nunez G, Hugot JP, Borg JP, Ollendorff V. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem. 2007;282:15197–15207. doi: 10.1074/jbc.M606242200. [DOI] [PubMed] [Google Scholar]
- 52.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 53.Li Y, Xiang M, Yuan Y, Xiao G, Zhang J, Jiang Y, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic Shock Augments Lung Endothelial Cell Activation: Role of Temporal Alterations of TLR4 and TLR2. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1670–R1680. doi: 10.1152/ajpregu.00445.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sori AJ, Rush BF, Jr, Lysz TW, Smith S, Machiedo GW. The gut as source of sepsis after hemorrhagic shock. American journal of surgery. 1988;155:187–192. doi: 10.1016/s0002-9610(88)80691-3. [DOI] [PubMed] [Google Scholar]
- 55.Rush BF, Jr, Sori AJ, Murphy TF, Smith S, Flanagan JJ, Jr, Machiedo GW. Endotoxemia and bacteremia during hemorrhagic shock. The link between trauma and sepsis? Annals of surgery. 1988;207:549–554. doi: 10.1097/00000658-198805000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koziol JM, Rush BF, Jr, Smith SM, Machiedo GW. Occurrence of bacteremia during and after hemorrhagic shock. J Trauma. 1988;28:10–16. [PubMed] [Google Scholar]
- 57.Kim JY, Park JS, Strassheim D, Douglas I, Diaz Del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E. HMGB1 contributes to the development of acute lung injury after hemorrhage. American journal of physiology. Lung cellular and molecular physiology. 2005 doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 58.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–317. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sorbara MT, Ellison LK, Ramjeet M, Travassos LH, Jones NL, Girardin SE, Philpott DJ. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity. 2013;39:858–873. doi: 10.1016/j.immuni.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 61.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Molecular cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 63.Birmingham CL, Higgins DE, Brumell JH. Avoiding death by autophagy: interactions of Listeria monocytogenes with the macrophage autophagy system. Autophagy. 2008;4:368–371. doi: 10.4161/auto.5594. [DOI] [PubMed] [Google Scholar]
- 64.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]