

Abstract
A 57-year-old man with symptoms of fatigue, joint pains and insomnia was found to have hypercalcaemia secondary to hyperparathyroidism with a corrected calcium of 2.61 mmol/L (2.2–2.6 mmol/L) and a serum parathyroid hormone (PTH) of 86 pg/mL (10–65 pg/mL). Preoperative workup demonstrated a parathyroid adenoma in the right upper position and he proceeded to surgery. Postoperatively, however, his symptoms remained unchanged and the corrected calcium remained elevated at 2.87 mmol/L with a PTH of 59 pg/mL. He had no family history of hypercalcaemia. Further investigations revealed low 24 h urinary calcium level and a low urine calcium to creatinine ratio. Genetic testing revealed a mutation in exon 4 of the calcium sensing receptor (CaSR) confirming a diagnosis of familial hypocalciuric hyercalcaemia (FHH).
The case is an example of a rare phenomenon when a parathyroid adenoma develops in patients with FHH.
Background
This case represents one of the few cases of parathyroid adenoma development in patients with FHH and demonstrates some of the challenges in diagnosing and distinguishing between primary hyperparathyroidism and FHH.
Case presentation
A 57-year-old man was referred for surgical evaluation after he was found to have hypercalcaemia secondary to hyperparathyroidism on routine laboratory investigations with a corrected calcium of 2.61 mmol/L (2.2–2.6 mmol/L) and a serum parathyroid hormone (PTH) of 86 pg/mL (15–65 pg/mL). He had symptoms of fatigue, joint pains and insomnia at time of presentation. He had a medical history of Barrett's oesophagus and his medications included esomeprazole and ‘gaviscon’ 10 mLs nocte. He had no significant family history and in particular no family history of hypercalcaemic syndromes. Physical examination was unremarkable.
Investigations
Preoperative workup with an ultrasound neck revealed an enlarged right upper parathyroid gland and a sestamibi scan demonstrated increased tracer uptake in this parathyroid gland (figure 1). The patient underwent cervical exploration with a right upper parathyroidectomy. During surgery, all three other parathyroid glands were identified and appeared macroscopically normal. Histology confirmed the presence of a parathyroid adenoma which weighed 230 mg (normal weight 30–60 mg). He was started on calcium supplements in the immediate postoperative period.
Figure 1.
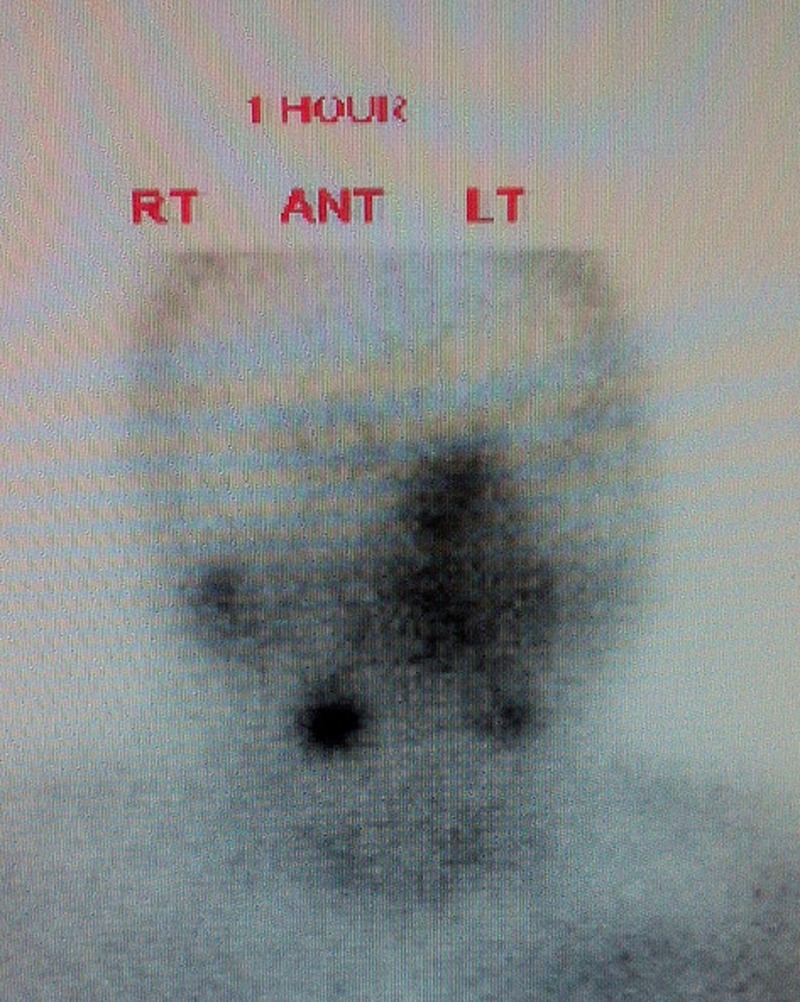
Sestamibi scan showing right parathyroid adenoma. Ant, anterior; Lt, left; Rt, right.
When he returned for follow-up his PTH level had normalised but his calcium remained elevated at 2.7 mmol/L and he was still symptomatic. The calcium supplements were discontinued but he was referred for endocrine assessment 2 months postoperatively with persistent hypercalcaemia. At this time, his corrected calcium was 2.82 mmol/L, PTH 59 pg/mL, phosphate was 0.71 mmol/L, and 25-hydroxy vitamin D was 38.7 nmol/L (table 1). He was started on vitamin D3 10 µg per day. He also had a dual energy X-ray absorptiometry (DEXA) scan performed which showed normal bone density.
Table 1.
Laboratory investigations performed in the preoperative and postoperative period
| Preoperative | 2/52 postoperative | 2/12 postoperative | 1 year postoperative (on cinacalcet) | |
|---|---|---|---|---|
| Corrected calcium (2.2–2.6 mmol/L) | 2.61 mmol/L | 2.7 mmol/L | 2.82 mmol/L | 2.53 mmol/L |
| PTH (10–65 pg/mL) | 86 pg/mL | 30 pg/mL | 59 pg/mL | |
| Vitamin D (>50 nmol/L) | 38.7 nmol/L |
PTH, parathyroid hormone.
Differential diagnosis
The differential diagnosis at this stage was parathyroid hyperplasia or familial hypocalciuric hypercalcaemia. Subsequent investigations including 24 h urine collections revealed low levels of urinary calcium of 2.3 mmol over 24 h. His calculated urine calcium to creatinine clearance ratio was less than 0.01. A second parathyroid ultrasound was carried out 6 months after surgery. This showed a 7.5 mm lesion posterior the right lobe of the thyroid and a 3 mm lesion at the superior aspect of the left lobe of the thyroid suspicious for enlarged parathyroid glands. However, follow-up sestamibi scan showed no evidence of a hyperfunctioning or an enlarged parathyroid gland.
As his biochemical results were suggestive of FHH, he underwent genetic testing. This revealed a mutation in exon 4 of the CaSR, predicting a p.CYS395Arg substitution in the patient. This pathogenic mutation confirmed the suspected diagnosis of FHH.
Treatment
He has been started on cinacalcet, a calcimimetic which binds to the CaSR and inhibits the release of PTH. Cinacalcet has been shown to cause rapid and sustained lowering of serum PTH and calcium levels as well symptomatic improvement in patients with FHH in a small number of case reports.1 2
Outcome and follow-up
His symptoms have improved and he remains on cinacalcet 30 mg once daily.
Discussion
This case is an example of primary hyperparathyroidism secondary to a solitary parathyroid adenoma in a patient with concomitant FHH. There have been a small number of similar cases reported in the literature to date.3–7 The first case of a parathyroid adenoma in a patient with FHH was reported in 2002, in a 45-year-old woman with severe symptomatic hypercalcaemia, elevated PTH levels, hypocalciuria and a family history of hypercalcaemia.3 A parathyroid adenoma was identified on parathyroid scintigraphy and genetic analysis for FHH was positive. The parathyroid adenoma was surgically resected and resulted in significant improvement in symptoms and serum calcium levels. The other reported cases were in patients who had underlying diagnoses of FHH and developed worsening hypercalcaemia on follow-up with corresponding elevations in PTH levels. They proceeded to parathyroid surgery following localisation of parathyroid adenomas with parathyroid scintigraphy and postoperatively their serum calcium levels reduced to just above the normal range.
Although surgery is not indicated in the management of FHH, these case reports of patients with a combination of FHH and parathyroid adenomas, suggest a beneficial effect from surgical resection of the adenoma with improvement in symptoms and biochemical markers following surgery.3–7 In contrast, our patient's serum calcium increased in the postoperative period and his symptoms remained unchanged. His hypercalcaemia has since been managed medically with a calcimimetic with good effect. It is unclear whether the development of parathyroid adenomas in patients with FHH is coincidental or not. Although CaSR expression is decreased in parathyroid adenomas,8 previous studies have demonstrated no mutations in the CaSR gene in parathyroid adenomas and therefore a causal link between the two conditions has not yet been established.9
It can be difficult to differentiate between primary hyperparathyroidism and FHH as there are many overlapping clinical features. Our patient was suspected to have primary hyperparathyroidism based on his age, lack of family history and his original biochemistry results. He was also taking ‘gaviscon’ which contains calcium carbonate. Larger doses of ‘gaviscon’ can cause hypercalcaemia, although this would be associated with a suppressed PTH. Although a raised serum PTH in the setting of hypercalcaemia suggests a diagnosis of primary hyperparathyroidism, PTH may also be elevated in FHH. A cross sectional study published in 2008, reported the prevalence of elevated PTH levels in patients with FHH to be 23%.10 In this patient, however, the presumed diagnosis of primary hyperparathyroidism was further corroborated when a seemingly hyperfunctioning parathyroid adenoma was observed on imaging.
Although hypercalciuria is no longer an indication for parathyroid surgery, the consensus guidelines recommend performing 24 h urinary calcium as part of the initial evaluation of patients with hypercalcaemia.11 This case highlights the importance of completing the diagnostic workup preoperatively to prevent patients with FHH from undergoing unnecessary surgery. This patient had the 24 h urine collection was done after surgery when he continued to have persistent hypercalcaemia. This showed the characteristic hypocalciuria and urinary calcium/creatinine clearance ratio <0.01 of FHH12 13 which prompted genetic testing. Although the urinary calcium/creatinine clearance ratio is the consensus biochemical test to differentiate between FHH and primary hyperparathyroidism, 15–20% of patients with FHH exhibit borderline renal clearance and have a urine calcium/creatinine clearance ratio >0.01.14 It may therefore be safer to consider genetic testing in patients with a urine calcium/creatinine ratio >0.02. Christensen et al15 have demonstrated that the diagnostic sensitivity of this cut-off is 98%.
This patient also had vitamin D insufficiency. Vitamin D insufficiency and vitamin D deficiency is more prevalent in patients with primary hyperparathyroidism and is associated with increased disease severity, in terms of clinical presentation, biochemical indices, effects on bone and weight of parathyroid adenoma.16–17 This association is incompletely understood. Increased PTH activity stimulates the conversion of vitamin D to its active metabolite 1,25-dihydroxy vitamin D which contributes to low 25 hydroxyvitamin D levels16 or it is possible that chronic vitamin D deficiency may cause autonomous parathyroid gland stimulation leading to hyperplasia and adenoma formation.18 The evidence for maintaining vitamin D sufficiency in patients with vitamin D insufficiency and primary hyperparathyroidism is limited. Grey et al19 demonstrated reduced levels of PTH and bone turnover following repletion of vitamin D stores in a small cohort of patients with primary hyperparathyroidism. Although larger randomised clinical trials are required to determine the efficacy of this treatment, most recent guidelines recommend measuring 25 hydroxyitamin D levels in all patients with suspected primary hyperparathyroidism.11
Learning points.
Calculation of the urine calcium to creatinine ratio should be performed in all patients prior to parathyroid surgery. This may prevent patients with familial hypocalciuric hyercalcaemia (FHH) from undergoing inappropriate surgery.
A diagnosis of FHH should be considered in patients who remain hypercalcaemic postremoval of a parathyroid adenoma.
Cinacalcet may have a role in the management of patients with symptomatic FHH.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Rasmussen AQ, Jørgensen NR, Schwarz P. Clinical and biochemical outcomes of cinacalcet treatment of familial hypocalciuric hypercalcemia: a case series. J Med Case Rep 2011;5:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timmers HJLM, Karperien M, Hamdy NAT, et al. Normalization of serum calcium by cinacalcet in a patient with hypercalcaemia due to a de novo inactivating mutation of the calcium-sensing receptor. J Intern Med 2006;26.2:177–82 [DOI] [PubMed] [Google Scholar]
- 3.Burski K, Torjussen B, Paulsen AQ, et al. Parathyroid adenoma is a subject familial hypocalciuric hypercalcemia: coincidence or causality? J Clin Endocrinol Metab 2002;87.3:1015–16 [DOI] [PubMed] [Google Scholar]
- 4.Brachet C, Boros E, Tenoutasse S, et al. Association of parathyroid adenoma and familial hypocalciuric hypercalcaemia in a teenager. Eur J Endocrinol 2009;161:207–10 [DOI] [PubMed] [Google Scholar]
- 5.Yabuta T, Miyauchi A, Inoue H, et al. A patient with primary hyperparathyroidism associated with familial hypocalciuric hypercalcaemia induced by a novel germline CaSR mutation. Asian J Surg 2009;32:118–22 [DOI] [PubMed] [Google Scholar]
- 6.Egan AM, Ryan J, Aziz MA, et al. Primary hyperparathyroidism in a patient with familial hypocalciuric hypercalcaemia due to a novel mutation in the calcium-sensing receptor gene. J Bone Miner Metab 2013; 31:477–80 [DOI] [PubMed] [Google Scholar]
- 7.Eldeiry LS, Ruan DT, Brown EM, et al. Primary hyperparathyroidism and FHH: relationships and Clinical implications. Endocr Pract 2012;18:412–17 [DOI] [PubMed] [Google Scholar]
- 8.Farnebo F, Höög A, Sandelin K, et al. Decreased expression of calcium-sensing receptor messenger ribonucleic acids in parathyroid adenomas. Surgery 1998;124:1094–9 [DOI] [PubMed] [Google Scholar]
- 9.Cetani F, Pinchera A, Pardi E, et al. No evidence for mutations in the calcium-sensing receptor gene in sporadic parathyroid adenomas. J Bone Miner Res 1999;14:878–82 [DOI] [PubMed] [Google Scholar]
- 10.Christensen SE, Nissen PH, Vestergaard P, et al. Plasma 25-hydroxyvitamin D, 1, 25-dihydroxyvitamin D, and parathyroid hormone in familial hypocalciuric hypercalcemia and primary hyperparathyroidism. Eur J Endocrinol, 2008;159:719–27 [DOI] [PubMed] [Google Scholar]
- 11.Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009;94:335–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath DA. Familial hypocalciuric hypercalcemia. Rev Endocr Metab Disord 2000;1:291–6 [DOI] [PubMed] [Google Scholar]
- 13.Bilezikian JP, Potts JT Jr, Fuleihan Gel-H, et al. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab 2002;87:5353–61 [DOI] [PubMed] [Google Scholar]
- 14.Shinall MC, Dahir KM, Broome JT. Differentiating familial hypocalciuric hypercalcemia from primary hyperparathyroidism. Endocr Pract 2013;19:697–702 [DOI] [PubMed] [Google Scholar]
- 15.Christensen SE, Nissen PH, Vestergaard P, et al. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin Endocrinol 2008;69:713–20 [DOI] [PubMed] [Google Scholar]
- 16.Silverberg SJ, Shane E, Dempster DW, et al. The effects of vitamin D insufficiency in patients with primary hyperparathyroidism. Am J Med 1999;107:561–7 [DOI] [PubMed] [Google Scholar]
- 17.Rao DS, Honasoge M, Divine GW, et al. Effect of vitamin D nutrition on parathyroid adenoma weight: pathogenetic and clinical implications 1. J Clin Endocrinol Metab 2000;85:1054–8 [DOI] [PubMed] [Google Scholar]
- 18.Silverberg SJ. Vitamin D deficiency and primary hyperparathyroidism. J Bone Miner Res 2007;22:V100–4 [DOI] [PubMed] [Google Scholar]
- 19.Grey A, Lucas J, Horne A, et al. Vitamin D repletion in patients with primary hyperparathyroidism and coexistent vitamin D insufficiency. J Clin Endocrinol Metab 2005;90:2122–6 [DOI] [PubMed] [Google Scholar]