

Abstract
Calcific aortic stenosis is a common disease, and some of its early causes are the activation and differentiation of resident fibroblasts to myofibroblasts in response to transforming growth factor β1 (TGF-β1). The aim of this study was to understand how TGF-β1 and its downstream effector, OB-cadherin [cadherin 11 (CDH11)], regulate porcine myofibroblast phenotypes. Based on whole-genome microarrays, 95 and 107 genes are up- and down-regulated at both the early (8 h) and the late (24 h) time points of TGF-β1 treatment. Gene functions related to cell adhesion, skeletal system development, and extracellular matrix are up-regulated by TGF-β1, whereas oxidation-reduction and steroid metabolic process are down-regulated. Notably, one of the cell adhesion molecules, CDH11, is up-regulated by ∼2-fold through both the Smad2/3 and the ERK pathways elicited by TGF-β1. CDH11 mediates cell-cell contacts in both valvular fibroblasts and myofibroblasts. Knockdown of CDH11 by small interfering RNA increases the myofibroblast phenotype, including an ∼2-fold increase in α-smooth muscle actin (α-SMA) expression and stress fiber formation. In contrast, increased binding of CDH11 through antibody treatment inhibits α-SMA expression. This study presents gene functional changes in response to TGF-β1 at the systems level and supports an inhibitory role of CDH11 in myofibroblast differentiation.—Wang, H., Leinwand, L. A. and Anseth, K. S. Roles of transforming growth factor-β1 and OB-cadherin in porcine cardiac valve myofibroblast differentiation.
Keywords: aortic valve fibrosis, genome-wide transcription, α-smooth muscle actin
Calcific aortic stenosis (CAS) is a predominant cardiac valve disease in Western populations, affecting 2–3% of people older than 65 yr (1). It is characterized by gradual stiffening and calcific nodule formation in aortic valves. A number of risk factors, including old age, male sex, smoking, hypertension, and high serum lipid levels, have been identified (2). However, recent studies suggest that this disease is not purely a result of tissue degeneration, but an active process mediated by the resident cells, named valvular interstitial cells (VICs; ref. 2).
VICs are the main cell population in valve leaflets and play important roles in both tissue homeostasis and disease progression. They are largely composed of fibroblasts that can differentiate into pathogenic myofibroblasts or osteoblast-like cells (3–5). VICs produce the major extracellular matrix (ECM) components of the valves, including collagen, fibronectin, elastin, and proteoglycans, and secrete ECM-remodeling enzymes, such as matrix metalloproteases (MMPs) and tissue inhibitors of MMPs (6, 7). During valve development, VICs actively participate in tissue formation by secreting ECM-related proteins (8, 9). On tissue damage, these cells are activated to a myofibroblast phenotype, which is characterized by increased deposition of collagen and increased contractility, mediated by α-smooth muscle actin (α-SMA)-positive stress fibers (10). Myofibroblasts are important regulators of tissue repair in multiple mesenchymal tissues, such as skin, lung, and liver (11–13). However, if the myofibroblast phenotype persists, excessive collagen is deposited, leading to tissue fibrosis and stiffening (14, 15). Therefore, understanding regulation of the myofibroblast phenotypes may shed light on the mechanism of disease progression in various valvular diseases, including CAS.
Many extracellular cues regulate activation of VICs to myofibroblasts, including chemical factors, mechanical stress, and cell-cell interactions. Despite the fact that a number of chemical cues can activate myofibroblasts, transforming growth factor β1 (TGF-β1) is one of the most common and potent chemokines to induce this phenotype in different tissues (16). It initiates the phosphorylation and translocation of Smad2/3 into the nucleus, where they promote transcription of important myofibroblast genes, including α-SMA, collagen 1A1 (Col1A1), and connective tissue growth factor (CTGF; refs. 17–21). TGF-β1 can also activate the noncanonical pathways, e.g., p38 mitogen-activated protein kinase (MAPK; ref. 22) or phosphatidylinositol 3-kinase (23), to promote myofibroblast differentiation. In addition to chemical factors, mechanical cues, such as dynamic stretching (24, 25) or elasticity (26–31), can modulate myofibroblast activation. Although much progress has been made in understanding how chemical and mechanical cues regulate VIC activation, little is known about the role of cell-cell contact in myofibroblast differentiation.
Cadherins mediate calcium-dependent cell-cell interaction, and they play critical roles in tissue formation and cell sorting during development (32). In addition to these major functions, cadherins also regulate other cellular functions, including migration, proliferation, and differentiation. During myofibroblast differentiation in skin, fibroblast cells had increased expression of OB-cadherin [cadherin 11 (CDH11)], but decreased expression of N-cadherin [cadherin 2 (CDH2); ref. 33]. Anti-CDH11 peptides, but not anti-CDH2 peptides, inhibited skin myofibroblast contraction (33). In addition, a recent study showed that CDH11-null mice had reduced pulmonary fibrosis induced by bleomycin (34). Collectively, these results support the possibility that CDH11 may regulate the pathogenic myofibroblast phenotype.
Because VICs in their native matrix constantly receive many different types of signals, it is important to understand systematically how cells process these signals to give the appropriate phenotypic output. Further, because TGF-β1 is a pleiotropic factor and has cell type-specific and time-dependent effects, we decided to examine the gene expression program elicited by TGF-β1 over time in porcine aortic VICs. Through microarrays, we found that TGF-β1 induced distinct gene programs in VICs after 8 and 24 h of treatment. Intriguingly, genes involved in cell adhesion were up-regulated by TGF-β1 at 24 h of treatment, with CDH2 and CDH11 increased by ∼2-fold. Further, when CDH11 was knocked down by small interfering RNA (siRNA), myofibroblast differentiation was increased, as assessed by α-SMA-positive stress fibers; in contrast, increased CDH11 engagement induced by CDH11 antibody treatment inhibited α-SMA expression. However, the effects of CDH11 were overridden by high doses of TGF-β1 treatment. This study suggests a unique mechanism as to how CDH11, a downstream target of TGF-β1, regulates myofibroblast differentiation. Whereas the overall effect of TGF-β1 is to promote myofibroblast differentiation, there may be downstream targets (e.g., CDH11) of TGF-β1 that act to inhibit or modulate the differentiation process and the signaling pathways.
MATERIALS AND METHODS
VIC cell culture
Fresh porcine hearts were obtained from Hormel Foods Corp. (Austin, MN, USA) within 24 h of euthanasia, and aortic valve leaflets were excised. Primary VICs were harvested from porcine aortic valve leaflets based on sequential collagenase digestion as described previously (35). The isolated cells were cultured in growth medium [medium 199, 15% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.5 μg/ml Fungizone] and expanded up to passage 3 (P3). P3 VICs were seeded directly or after transfection on plastic plates at different densities in low-serum medium (medium 199, 1% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.5 μg/ml Fungizone]. Samples were collected after culture in the low-serum medium for 2 d. For the small molecule inhibitor treatment in Fig. 3C, cells were seeded at 400 cells/mm2 and treated with SB431542 (S4317; Sigma-Aldrich, St. Louis, MO, USA) at 10 μM or CI1040 (S1020; Selleck Chemicals, Houston, TX, USA) at 2 μM with or without porcine TGF-β1 (5 ng/ml; R&D Systems; Minneapolis, MN, USA) for 24 h. The dimethyl sulfoxide (DMSO) vehicle was treated at 0.1% as a control.
Figure 3.

TGF-β1 treatment up-regulates expression of CDH11 in VICs through both the Smad2 and the ERK signaling pathways. A) Normalized expression value of different cadherins in VICs detected by microarrays. CDH2, CDH5, CDH11, and CDH13 were the abundantly expressed cadherins in VICs. However, only CDH2 and CDH11 were significantly up-regulated after 24 h of TGF-β1 treatment. T8, TGF-β1 treatment for 8 h; T24, TGF-β1 treatment for 24 h. *P < 0.05 vs. control. B) Confirmed by qRT-PCR, TGF-β1 treatment for 24 h increased mRNA level of CDH11 and also up-regulated expression of the fibrogenic genes, including Col1A1, CTGF, FN1, and MMP1. This is representative of ≥3 independent qRT-PCR experiments. Error bars = sd. C) At the protein level, TGF-β1 treatment (24 h), which activated pSmad2, also up-regulated CDH11 protein expression. The effect of TGF-β1 on CDH11 expression was blocked by inhibiting either Smad2/3 signaling via SB431542 or ERK signaling via CI1040. Con, control (untreated), *P < 0.05 vs. Con + DMSO; #P < 0.05 vs. TGF-β1 + DMSO.
Porcine genome microarray
P3 VICs were seeded at 400 cells/mm2 in low-serum medium supplemented with 1% FBS on plastic plates. All samples were collected at 32 h after cell seeding, and 3 experimental conditions were performed: cells without TGF-β1 treatment (control); cells treated with TGF-β1 (5 ng/ml) for the last 8 h of culture (T8); and cells treated with TGF-β1 (5 ng/ml) for the last 24 h of culture (T24). Total RNA was extracted using an RNeasy Mini Kit (74104; Qiagen, Valencia, CA, USA). All the RNA samples had a 260/280 ratio ≥2.0 and RNA integrity number ≥9.2 verified by an RNA bioanalyzer. Total RNA was amplified and labeled using a Gene Chip 3′ IVT Express Kit (901228; Affymetrix, Santa Clara, CA, USA) and hybridized to porcine genome microarrays (900624; Affymetrix). Three biological replicates were performed for each condition. Microarray data were analyzed using Spotfire (Tibco, Palo Alto, CA, USA), GeneSpring (Agilent Technologies, Santa Clara, CA, USA), the Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation bioinformatics analysis (U.S. National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA; http://david.abcc.ncifcrf.gov/), and Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Inc., Redwood, CA, USA; http://www.ingenuity.com). The microarray data have been deposited in the U.S. National Center for Biotechnology Information (Bethesda, MD, USA) Gene Expression Omnibus (GEO; http://www.ncbi/nlm/nih.gov) archives with accession number GSE48839.
Real-time quantitative reverse transcriptase PCR (qRT-PCR)
For each condition, total RNA was isolated based on the RNeasy Mini Kit (74104; Qiagen) or according to the manufacturer's protocol for TRI Reagent (T9424; Sigma-Aldrich). The RNA concentration and integrity were measured and verified by a NanoDrop 1000 system (Thermo Scientific, Waltham, MA, USA). cDNA was synthesized from total RNA with SuperScript III reverse transcriptase (18080-051; Life Technologies, Grand Island, NY, USA) and random hexamer primers. Gene expression was determined by SYBR Green-based qRT-PCR using gene-specific primer sets (shown in Table 1) and a 7500 real-time PCR machine (Applied Biosystems, Foster City, CA, USA).
Table 1.
Gene primer sequences for qRT-PCR
| Gene | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| 18S | GCCGCTAGAGGTGAAATTCTT | CTTTCGCTCTGGTCCGTCTT |
| Human α-SMA | GCAAACAGGAATACGATGAAGCC | AACACATAGGTAACGAGTCAGAGC |
| Human Col1A1 | GGGCAAGACAGTGATTGAATACA | GGATGGAGGGAGTTTACAGGAA |
| CTGF | CTGGTCCAGACCACAGAGTGG | GCAGAAAGCGTTGTCATTGG |
| FN1 | GGCATTGATGAAGAACCCTTG | GCCTCCACTATGATGTTGTAGGTG |
| MMP1 | GGCATCCAGGCCATCTATG | CACTTGTGGGGTTTGTGGG |
| OB-CDH | GGGTCCCTGAGCTCCTTAGA | CGAGGTCCCCAGTTCTGTAG |
| TUFT1 | GGAGAAGATCCACCACTTGGA | GTGTCCTTTGACTGGATCACAG |
| RASAL2 | ACACGAGCTTTCGGCTTCC | GGCTCAGCAAGGATTCATGTG |
| CHSY1 | CGCCCAGAAATACCTGCAGA | CGACGTGTCTGAACCCTCACTAG |
| ITGA5 | CCAAAGGGAACCTCACCTACG | ACCTGTTCCCCTGAGAAGTTGTAG |
| PDK4 | CCACATTGGCAGCATTGAC | ACAGAGCATCTTGGAACACTCAA |
| MYLIP | CTGTGCTGTGAGGGCGAGA | CACTCGCGACCTGCAAACG |
| ADAMTS1 | GTGATCCCAGTAGAAGCTGCTC | CATTGCTCGGCATCATCATG |
| ACSS3 | CTGACTTAGGCTGGGTTGTCG | CGGAAATAAGCACCAGCATCC |
| ID2 | CGCTGACCACCCTAAATACG | GAGCGCTTTGCTGTCACTTG |
| Smad7 | CCTGTGTGCTGAGCTCTGC | GAATCCTTCTTGGTGGGAAG |
Immunocytochemistry
VICs were fixed with 4% paraformaldehyde (15 min at 25°C), permeabilized in 0.1% Triton X-100, and blocked with 3% bovine serum albumin (BSA). Mouse anti-α-SMA antibody (ab7817; Abcam, Cambridge, MA, USA) was diluted at 1:100 in phosphate-buffered saline (PBS) with 1% BSA and 5% normal goat serum and incubated with the samples overnight at 4°C. After washes in PBS with 0.05% Tween 20 (PBST), samples were labeled with goat anti-mouse Alexa Fluor 488 secondary antibody (A-11001; Life Technologies). For double-staining of CDH11 with either α-SMA or F-actin, samples were not permeabilized and were stained with CDH11 (clone 15F7, a generous gift from Dr. Michael Brenner, Brigham and Women's Hospital, Boston, MA, USA) first. Samples were then fixed with 4% paraformaldehyde (10 min at 25°C), permeabilized, and blocked for staining with α-SMA or phalloidin-tetramethylrhodamine B isothiocyanate (P1951; Sigma-Aldrich). Samples were mounted and subsequently imaged on an LSM 710 laser scanning microscope (Carl Zeiss GmbH, Jena, Germany) with a ×20 objective. Images of different fluorescence channels were compiled with Zen (Carl Zeiss) or ImageJ (U.S. National Institutes of Health, Bethesda, MA, USA) software.
Western blot
VICs were scraped into the radioimmunoprecipitation assay buffer (20-188; Upstate, Temecula, CA, USA) with protease inhibitor cocktail (539134; EMD Millipore, Billerica, MA, USA) and phosphatase inhibitors (04906845001; Roche, Indianapolis, IN, USA). Cell lysates were subsequently rotated at 4°C for 30 min and centrifuged at 12,000 rpm for 15 min. The supernatant was saved for SDS-PAGE. The protein concentrations of the samples were measured by a microBCA kit (23235; Thermo Scientific). The same amount of total protein for each sample was loaded into 10% TGX mini gels (456–1033; Bio-Rad Laboratories, Hercules, CA, USA) and separated by electrophoresis for 1 h at 200 V. The proteins were subsequently transferred onto a nitrocellulose membrane using tank transfer at 100 V for 1 h. The nitrocellulose membrane was blocked with PBST with 5% nonfat milk for 1 h at room temperature and then was incubated with primary antibodies overnight at 4°C. After washes with PBST, secondary antibodies conjugated with horseradish peroxidase (HRP) were applied for 1 h at room temperature. Protein bands were visualized by applying a chemiluminescent substrate of HRP (34075; Thermo Scientific) and exposing the membrane to X-ray films. Protein abundance was subsequently quantified by ImageJ. The following antibodies were used in this study: glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 2118; Cell Signaling Technology, Danvers, MA, USA), α-SMA (ab7817; Abcam), phospho-Smad2 (Ser465/Ser467, 3108; Cell Signaling Technology), phospho-extracellular signal-regulated kinase 1/2 (ERK1/2; Thr202/Tyr204, 9106; Cell Signaling Technology), ERK1/2 (9102; Cell Signaling Technology), pan-cadherin (4068; Cell Signaling Technology), and CDH11 (clone 3H10, a generous gift from Dr. Michael Brenner, Brigham and Women's Hospital). For each condition, ≥3 biological replicates were examined by Western blotting.
siRNA transfection
P3 VICs were transfected with siRNAs with the U23 protocol in Amaxa Nucleofector (Lonza, Basel, Switzerland). In brief, cells were prepared in a single cell suspension at 2–3 × 106 cells/100 μl of the transfection buffer provided by the kit (VPI-1002; Lonza). siRNA against CDH2 (sequence CUGUGUCUGUCACAGUUAUUU) and siRNA against CDH11 (sequence CAGACUUGGACUAUGACUAUU), both of which were customer-designed based on porcine mRNA sequences (Dharmacon RNA Technologies, Lafayette, CO, USA) or nontargeting siRNA (D-001210-02-20; Dharmacon), were used at 2 μM in the transfection buffer. After transfection, cells were recovered in growth medium with 15% FBS for 15 min in the incubator and seeded at 350–400 cells/mm2 (accounting for 50% survival of cells after the transfection) on plastic plates in low serum medium (1% FBS) for 2 d. In Fig. 5, transfected cells were treated with or without porcine TGF-β1 (5 ng/ml) for 48 h before sample collection.
Figure 5.

Knocking down CDH11 (OB-CDH) promotes α-SMA protein expression and stress fiber formation in VICs, which is overridden by TGF-β1. VICs transfected with an OB-siRNA or an NT-siRNA were treated with or without TGF-β1 (5 ng/ml) and were examined at d 3 for myofibroblast activation. A) CDH11 mRNA was significantly knocked down by siRNA at d 3, based on qRT-PCR. *P < 0.05. B) Protein expression of CDH11 was also significantly reduced by OB-siRNA independent of TGF-β1 treatment. Meanwhile, α-SMA protein expression was increased ∼2-fold with OB-siRNA treatment, compared with control. However, α-SMA was increased to similar levels by the TGF-β1 treatment. C) Densitometry analysis of 3 independent Western blots. D) Myofibroblasts characterized by α-SMA stress fibers were consistently increased in OB-siRNA-transfected cells compared with the NT-siRNA condition (quantified in bar graph, right panel). However, TGF-β1 overrode the effect of OB-siRNA and induced similar percentages of myofibroblasts in both conditions. Green, α-SMA; red, CDH11. blue, nuclei. Scale bars= 100 μm.
Antibody treatment
Untreated plastic plates were incubated with the antibody solution containing human IgG1 Fc (110-HG; R&D Systems) or human CDH11 Fc (1790-CA; R&D Systems) antibodies at 10 μg/ml overnight at 4°C. Before cell seeding, the antibody solution was aspirated off, and the plates were washed once with PBS. Cells were seeded at 300–400 cells/mm2 and collected 48 h after treatment.
Statistics
A Student's t test was used to compare data sets with 2 conditions and a 1-way analysis of variance (ANOVA) with a Newman-Keuls post hoc test was used to compare data sets with >2 conditions. A value of P < 0.05 was considered statistically significant.
RESULTS
TGF-β1-induced common and distinct gene expression changes globally in VICs after 8 and 24 h
Gene expression in VICs treated with TGF-β1 over time was measured by porcine genome arrays. Data were filtered (coefficient of variation, <50%), normalized based on GeneChip robust multiarray average (GCRMA), and analyzed based on hierarchical clustering, as shown in Fig. 1A. The different experimental groups spontaneously clustered based on their specific treatment. Two major types of regulation were observed: genes up-regulated or down-regulated by TGF-β1 over time. For the differentially regulated genes after 24 h of TGF-β1 treatment (fold change ≥2, P<0.05), DAVID functional annotation analysis showed that gene functions related to cell adhesion, skeletal system development, and ECM were significantly up-regulated, whereas those involved in oxidation-reduction and steroid metabolic process were significantly down-regulated (Fig. 1B). The lists of genes in each functional category in Fig. 1B are summarized in Supplemental Tables S1−S5. Different numbers of genes were differentially regulated at the 2 time points of TGF-β1 treatment (Fig. 1C). Specifically, 127 and 178 genes were significantly up-regulated by TGF-β1 at 8 and 24 h, respectively, with 95 overlapping genes. In contrast, 154 and 225 genes were significantly down-regulated by TGF-β1 at 8 and 24 h, respectively, with 107 genes in common. Microarray data were consistently validated by qRT-PCR based on 5 genes up-regulated by TGF-β1 [tuftelin 1 (TUFT1) RASAL2, fibronectin 1 (FN1), CHSY1, and ITGA5] and 5 genes down-regulated by TGF-β1 (PDK4, MYLIP, ADAMTS1, ACSS3, and ID2; Supplemental Fig. S1).
Figure 1.

Global gene expression of porcine VICs in response to TGF-β1 over time. VICs cultured on plastic plates were treated with TGF-β1 at 5 ng/ml for 8 h (T8) and 24 h (T24). Total RNA from 3 biological replicates of each condition was collected for microarray analysis. A) Hierarchical clustering of GCRMA-normalized and ANOVA-analyzed (P<0.05) microarray data showed that the control (Con) condition and 2 different durations of TGF-β1-treated conditions segregated into 3 clusters. B) DAVID functional annotation analysis of genes differentially regulated by TGF-β1 at 24 h (with fold change ≥2 and P<0.05) showed that genes associated with cell adhesion, skeletal system development, and ECM were up-regulated by TGF-β1, whereas oxidation-reduction and steroid metabolic process were down-regulated. C) Venn diagrams showing the number of gene probes that were differentially regulated by TGF-β1 treatment at different time points with fold change of ≥2 and P < 0.05. At both time points, 95 gene probes were up-regulated, and 107 gene probes were down-regulated.
TGF-β1 signaling was significantly up-regulated based on IPA
Some known TGF-β1-responsive genes, including JUNB, TGFB1, KLF10, and SMURF2, were increased in a time-dependent manner based on the microarray data (Fig. 2A). In addition, IPA in Fig. 2B showed that TGF-β1 signaling was activated with more downstream targets being up-regulated over time, validating the microarray results.
Figure 2.

TGF-β1 signaling is up-regulated in the microarrays. A) Known TGF-β1-responsive genes, JUNB, TGFB1, KLF10, and SMURF2, were up-regulated by TGF-β1 treatment over time in the microarrays. Con, control (untreated); T8, TGF-β1 treatment for 8 h; T24, TGF-β1 treatment for 24 h. *P < 0.05 vs. control. B) IPA based on the differentially regulated genes from microarrays revealed that a number of genes associated with TGF-β1 signaling were significantly up-regulated. Red, up-regulated genes; green, down-regulated genes.
TGF-β1 treatment up-regulated the expression of CDH11 in VICs through both the Smad2/3 and the ERK signaling pathways
From the microarrays, 4 types of cadherins (CDH2, CDH5, CDH11, and CDH13) were abundantly expressed in VICs (Fig. 3A). Among these, CDH2 and CDH11 were up-regulated ∼2-fold after 24 h of TGF-β1 treatment based on the microarrays (Fig. 3A). Up-regulation of CDH11 was then validated by both qRT-PCR and Western blot. With TGF-β1 treatment for 24 h, Col1A1, CTGF, FN1, MMP1, and CDH11 were all up-regulated 3- to 7-fold (Fig. 3B). At the protein level, TGF-β1 increased expression of CDH11 relative to the internal control, GAPDH (Fig. 3C). However, the effect of TGF-β1 in inducing CDH11 expression was blocked by inhibiting either the Smad2/3 pathway via SB431542 or the ERK pathway via CI1040 (Fig. 3C). Furthermore, the existing microarray data also showed that CDH11 mRNA was up-regulated with the myofibroblast phenotype in a number of disease models, such as pulmonary fibrosis and liver fibrosis (Supplemental Fig. S2).
CDH11 expression was increased with cell density and localized at cell-cell contacts in VICs
To confirm the role of CDH11 in mediating VIC-VIC adhesion, we showed that CDH11 expression was increased with cell density in VICs (Fig. 4A). Only when VICs were plated at high densities (>200 cells/mm2), which enable cell-cell contacts, did they have significant CDH11 expression. Based on immunostaining images, CDH11 was increased with cell density and localized to both cell-cell contacts (arrowheads) and leading edges of the cells (Fig. 4B, arrows).
Figure 4.

CDH11 (OB-CDH) expression increases with cell density and localizes to cell-cell contacts in VICs. A) Brightfield images of VICs seeded at increasing densities from 50 to 400 cells/mm2. As the cells formed more cell-cell contacts with increasing densities, CDH11 expression was also increased. *P < 0.05 vs. 50 cells/mm2. B) Staining of CDH11 (green) showed increased expression of this protein at 400 vs. 50 cells/mm2. Double-staining of CDH11 (green) and F-actin (red) showed that CDH11 localized to cell-cell adhesion (arrowheads) and sometimes leading edges of the cells (arrows). Scale bars = 100 μm.
Knocking down CDH11 promoted α-SMA protein expression and stress fiber formation in VICs, which was overridden by TGF-β1
VICs were transfected with siRNAs designed against porcine OB-cadherin [OB-cadherin siRNA (OB-siRNA)] or nontargeting siRNA (NT-siRNA). OB-siRNA transfection caused a significant reduction in CDH11 at both the mRNA level (Fig. 5A) and the protein level (Fig. 5B), compared with NT-siRNA transfection. Meanwhile, knocking down CDH11 in VICs increased expression of α-SMA protein, which was overridden by TGF-β1 treatment at 5 ng/ml (Fig. 5B). Further, cells treated with OB-siRNA had reduced CDH11 staining, but an increased percentage of myofibroblasts (marked by α-SMA stress fibers), compared with those treated with NT-siRNA (Fig. 5C). However, TGF-β1 treatment induced similar levels of myofibroblast activation in NT-siRNA and OB-siRNA conditions (Fig. 5C). In addition, CDH11 knockdown did not affect the transcriptional response to TGF-β1 of some genes (including CTGF, Col1A1, and Smad7; Supplemental Fig. S4). To rule out the possibility that CDH2 may compensate for the function of CDH11, we showed that when CDH11 was knocked down, CDH2 expression was not induced in VICs (Supplemental Fig. S5A). In addition, when we knocked down both CDH2 and CDH11, VICs had increased α-SMA protein expression and increased numbers of myofibroblasts with α-SMA stress fibers as well (Supplemental Fig. S5).
Increased CDH11 engagement induced by antibody treatment inhibited α-SMA expression
The effect of increasing CDH11 engagement on valvular myofibroblast differentiation was examined by coating plates with CDH11-mimicking antibodies, which has been shown previously to increase CDH11 binding (36). When CDH11 antibodies were presented to VICs for 48 h, α-SMA mRNA and protein expression was inhibited, compared with the control (Fig. 6).
Figure 6.
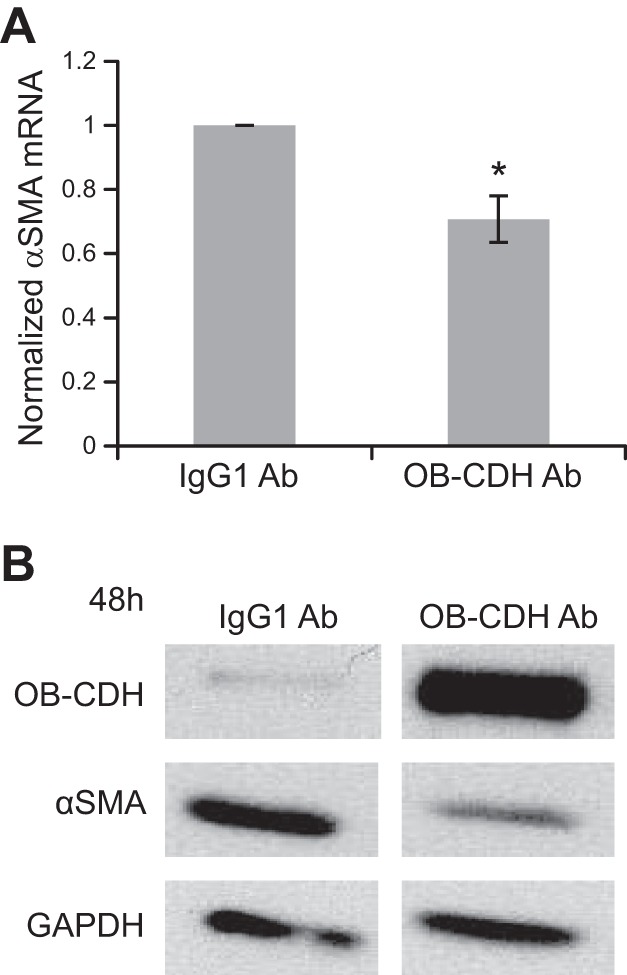
CDH11 antibody (OB-CDH Ab) engagement leads to down-regulation of α-SMA protein expression. A) VICs were cultured on plates coated with either IgG1 control antibody or CDH11 Fc antibody for 2 d. At both mRNA and protein levels, CDH11 antibody treatment decreased α-SMA expression. B) Because the CDH11 antibody has a molecular weight similar to that of endogenous CDH11, we confirmed that the antibody coating was still present on the plate when we collected the protein samples by probing CDH11 on Western blots.
DISCUSSION
Myofibroblasts are critical during wound healing, serving to deposit more ECM and to contract the wound opening. However, if these cells are not effectively turned over at the end of wound healing, their persistence can lead to fibrosis in various organs (11, 13). Understanding the regulation of the myofibroblast phenotype by chemokines can help reveal a common molecular pathology of various fibrotic diseases. In this study, gene expression changes initiated by TGF-β1, one of the most prevalent cytokines in fibrotic loci (37), were examined longitudinally by whole-genome microarrays. TGF-β1 was found to elicit some common and some distinct genes after 8 and 24 h of treatment. These genes were involved in a range of cellular functions, and, relevant to the focus of this study, cell adhesion was up-regulated by TGF-β1 at the later time point. Specifically, CDH11 was up-regulated by TGF-β1. Knockdown or increased binding of CDH11 demonstrated its direct inhibitory role in the expression of α-SMA, a hallmark of myofibroblast differentiation. However, the effect of CDH11 was overridden by high doses of TGF-β1 treatment. This study suggests that TGF-β1 can initiate different early and late gene responses in VICs. One of the late genes induced by TGF-β1, CDH11, could inhibit myofibroblast differentiation.
Microarrays based on sound experimental design and validation are powerful tools for detecting system changes in gene expression and revealing novel gene targets and signaling pathways for future exploration. In this study, we used porcine genome microarrays to examine mRNA changes in VICs treated with TGF-β1 for 8 and 24 h. Based on our experience working with VICs and the previous literature (30), it takes ∼48 h of culture in low-serum medium for the majority of the cells to differentiate into myofibroblasts. We chose 8 and 24 h for the microarray experiments so that both early and late gene responses could be detected during myofibroblast differentiation. TGF-β1 activates various signaling pathways with different kinetics. It can induce Smad2/3-mediated transcription within 10 min (38) but may induce activation of MAPK signaling with much slower kinetics (39). By examining gene expression changes at both 8 and 24 h, we aimed to capture a bigger spectrum of gene regulation by TGF-β1 during the differentiation process.
The microarray data have been validated in multiple ways. Ten of 10 genes were validated positively by qRT-PCR (Supplemental Fig. S1), and known TGF-β1-responsive genes were up-regulated (Fig. 2A). Hierarchical clustering also confirmed that the different treatment groups cluster together (Fig. 1A). Further, we found that shared sets of 95 and 107 gene probes were up- or down-regulated, respectively, by TGF-β1 at both time points. These genes probably play very important roles in the process of TGF-β1-induced myofibroblast differentiation and are valuable targets to examine in future studies. For example, TUFT1 (Supplemental Fig. S1) may be a novel gene target of TGF-β1 in fibroblasts, where its functions are unknown. In addition, TGF-β1 induced more genes with significant expression changes at 24 h than at 8 h (Fig. 1C). Based on gene functional annotation, we found that cell adhesion, skeletal system development, and ECM were significantly up-regulated by TGF-β1 (see the gene lists in Supplemental Tables S1−S3). Many of the known TGF-β1 targets, such as Col1A1, FN1, and tenascin C, were present in the gene lists of cell adhesion and ECM. For genes in the down-regulated functional categories, 2 were enzymes that directly catalyze fatty acid oxidation (acyl-coenzyme A dehydrogenase and acetyl-coenzyme A acyltransferase 2), indicating that TGF-β1 may negatively regulate this process in VICs (Supplemental Tables S4 and S5). Using IPA, we showed that TGF-β1 signaling was activated, and more downstream targets of TGF-β1 were activated or inhibited with time (Fig. 2B). Notably, signaling changes associated with tumor necrosis factor α (TNF-α) and p53 were also detected by IPA (data not shown), indicating that these pathways may be involved in the phenotypic changes of myofibroblasts induced by TGF-β1.
In the cell adhesion functional category, many genes were involved in cell-matrix adhesion (e.g., integrin α5 and FN1), and CDH11 was the only cell-cell adhesion molecule (Supplemental Table S1). Previous studies have suggested important regulation of CDH11 in fibrosis and activation of fibroblasts. It was found that CDH11-neutralizing antibodies had therapeutic benefits toward inflammatory arthritis (40) and pulmonary fibrosis in vivo (34). In vitro, blocking CDH11 reduced skin myofibroblast contraction (33, 41) or calcific nodule formation of VICs (42). Because CDH11 was the most abundantly expressed cadherin in VICs based on microarrays (Fig. 3A), and CDH11 expression was detected in normal and diseased aortic valves (42, 43), we chose to follow up with CDH11 and to examine its role during myofibroblast activation of VICs. At both mRNA and protein levels, CDH11 was up-regulated by TGF-β1. This result is consistent with previous findings by Hutcheson et al. (42) and Schneider et al. (34) that CDH11 mRNA increased with TGF-β1 treatment in VICs and lung epithelial cells, respectively. Because TGF-β1 primarily activates its canonical pathway (Smad2/3), we asked whether this pathway was involved in TGF-β1-induced CDH11 up-regulation. Using SB431542 (an inhibitor of the TGF-β1 type I receptors ALK4, ALK5, and ALK7), we found that inhibition of phosphorylation of Smad2, but not of ERK1/2, at 24 h of treatment (Fig. 3C). We also observed that SB431542 blocked the up-regulation of CDH11 by TGF-β1, suggesting that Smad2/3 may be required for the TGF-β1 effect (Fig. 3C). In addition, Hutcheson et al. (42) found that induction of CDH11 by TGF-β1 was inhibited by U0126, a mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor, in VICs. Because of this observation, we also investigated whether the ERK1/2 pathway was involved in the TGF-β1-induced CDH11 expression. Notably, although CDH11 was significantly decreased when VICs were treated with both TGF-β1 and CI1040 (an inhibitor of MEK1/2) compared with VICs treated with TGF-β1 alone, CDH11 was still induced by TGF-β1 in a comparison of VICs treated with CI1040 and those treated with CI1040 and TGF-β1. This result suggests that ERK activation is required for maintaining the basal expression of CDH11 and is only partially responsible for TGF-β1-induced CDH11 expression. The discrepancies between our observation and that of Hutcheson et al. (42) may be due to the differences in serum concentrations in medium and different dosages of TGF-β1; however, our data provided an understanding as to the regulation of CDH11 by TGF-β1 that was different and unique from that in the previous article. Furthermore, p38 MAPK is also important for myofibroblast activation and can be activated by TGF-β1 (22, 44); thus, an interesting future direction will be to test whether this pathway is involved.
Surprisingly, when we knocked down CDH11 by siRNAs, myofibroblast differentiation was enhanced in VICs as characterized by an increased α-SMA protein level and increased stress fiber organization (Fig. 5B, C). In contrast, when CDH11 engagement was induced by antibody treatment, the α-SMA protein expression was inhibited (Fig. 6B). These data suggest that CDH11 plays an inhibitory role in α-SMA protein expression during differentiation of valvular myofibroblasts. Consistently, Li et al. (45) observed that CDH11 overexpression inhibited actin filament formation in carcinoma cells and Masur et al. (46) found that high density, which should correlate with higher expression of cadherins than low density, inhibited myofibroblast differentiation (46). We also observed that knocking down both CDH11 and CDH2 promoted the myofibroblast phenotype in VICs (Supplemental Fig. S5), supporting the inhibitory role of these cadherins during myofibroblast differentiation. It seems contradictory that the role of CDH11 is contrary to that of TGF-β1 toward myofibroblast differentiation. However, it is possible that TGF-β1 activates negative regulators of myofibroblast differentiation. In addition, Varelas et al. (47) showed that high seeding density prevented Smad2/3 nuclear localization and activation by TGF-β1 in mouse mammary epithelial cells, and this was reversed by Ca2+ depletion or knockdown of α-catenin, indicating that cell-cell adhesion is inhibitory toward TGF-β1 signaling (47). It is possible that CDH11-mediated cell-cell adhesion plays a similar inhibitory role toward signaling responses to autocrine TGF-β1 in VICs. Notably, the effects of CDH11 knockdown were overridden by TGF-β1 at 5 ng/ml (Fig. 5B, C), but not at a low dosage of 1 ng/ml (data not shown). This observation indicates that in a diseased environment where TGF-β1 is highly expressed, it can outweigh the inhibitory effect of CDH11 on myofibroblast differentiation, which may otherwise predominate in a normal tissue environment with low levels of autocrine TGF-β1.
The influence of cell-cell contact on myofibroblast differentiation is an underinvestigated but relevant question for the progression of fibrosis. Fibroblasts are usually in closer contact with the surrounding matrix than with other cells, so there has been much focus on how the cell-matrix interaction regulates fibrotic properties of the cells (48). However, as fibrotic loci develop, resident fibroblasts and myofibroblasts proliferate and migrate toward the loci, increasing cell density and formation of adherent junctions through cadherins (49). Therefore, it is consistent that CDH11 expression is increased with fibrosis in various tissues, including skin (33), lung (34), and joint (50). However, the role of cadherins during myofibroblast differentiation and the different roles that different cadherins may play is still under investigation. Studies have shown that CDH11 bonds can resist ∼2-fold higher forces than CDH2 bonds (51), mediate mechanical coupling between myofibroblasts (52), and promote cellular tension-dependent nodule formation (42). So, it seems reasonable to hypothesize that a reduction in CDH11 connections in myofibroblasts will decrease their tension and activation. In fact, blocking peptides against CDH11 caused reduced contraction and expression of α-SMA in diseased dermal myofibroblasts (41). However, a similar effect was not observed in normal dermal fibroblasts (41), suggesting that CDH11 may play different roles in these 2 different cell phenotypes. In this study, we examined the role of CDH11 as the cells differentiated from fibroblasts into myofibroblasts, mimicking cellular changes at the early stage of fibrosis. However, a lot of the previous studies examined the role of CDH11 in mature myofibroblasts or in animal models with fibrosis, in which multiple cell types and factors contributed to the disease. It is possible that CDH11 plays roles in early myofibroblast differentiation that are different from those in mature myofibroblasts. In addition, CDH11 engagement can stimulate the production of TGF-β1 in alveolar macrophages (36) and of interleukin-6 in synovial fibroblasts (36). Thus, CDH11 could also modulate cellular functions in different cell types to regulate fibrogenic development in vivo, and its roles in fibrosis warrant further investigation. Nevertheless, our results suggest that myofibroblast activation is a complex process in which multiple signaling pathways and molecules interact. The overall effects of TGF-β1 and other extracellular signals, such as cell-cell contact and cell-matrix interaction, on VIC myofibroblast differentiation depend on the signaling intensities of different pathways, their mode of interplay (inhibitory, synergistic, or parallel), and the temporal regulation of the pathways.
CONCLUSIONS
TGF-β1 is a potent chemical factor that mediates wound healing by activating local fibroblasts to myofibroblasts, but it can also induce fibrotic lesions if the signaling pathways go awry. To understand the specific role of TGF-β1 in inducing myofibroblast differentiation of VICs, we examined whole-genome gene expression changes in response to TGF-β1 at an early (8 h) and a late (24 h) time point. Based on the microarrays, distinct early and late gene programs were activated by TGF-β1. Signaling pathway analysis showed that TGF-β1 signaling was activated, as well as pathways related to TNF-α and p53. Consistent with previous findings, CDH11 expression was increased with TGF-β1 treatment, and it was dependent on both the ERK1/2 and the Smad2/3 pathways. Using gene knockdown and induced binding of CDH11, we found that CDH11 inhibited valvular myofibroblast differentiation as characterized by α-SMA expression. However, the effects of CDH11 knockdown were overridden by high doses of TGF-β1 treatment. These results suggest that TGF-β1 can up-regulate both positive and negative regulators to modulate myofibroblast differentiation and functions. We present new information on how TGF-β1 regulates myofibroblast differentiation through up-regulation of CDH11. TGF-β1 elicits both the canonical Smad2/3 pathway and the noncanonical cascades (e.g., ERK1/2 and p38 MAPK) and induces expression of genes with different cellular functions (e.g., cell adhesion, extracellular matrix, and oxidation reduction) to coordinately regulate myofibroblast cell fate and to determine the switch between normal tissue repair and fibrosis.
Supplementary Material
Acknowledgments
The authors appreciate scientific discussions with Dr. Nicholas Farina and Dr. Bradley Olwin regarding microarray analysis and thank Dr. Bradley Olwin for generously sharing software resources. The authors also thank Dr. Michael Brenner (Brigham and Women's Hospital, Boston, MA, USA) for generously sharing CDH11 antibodies.
This work was supported by the U.S. National Institutes of Health (grants R01 HL089260 and R01 GM29090) and the Howard Hughes Medical Institute.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- α-SMA
- α-smooth muscle actin
- ANOVA
- analysis of variance
- BSA
- bovine serum albumin
- CAS
- calcific aortic stenosis
- CDH2
- cadherin 2 (N-cadherin)
- CDH11
- cadherin 11 (OB-cadherin)
- Col1A1
- collagen 1A1
- CTGF
- connective tissue growth factor
- DAVID
- Database for Annotation, Visualization and Integrated Discovery
- DMSO
- dimethyl sulfoxide
- ECM
- extracellular matrix
- ERK
- extracellular signal-regulated kinase
- FBS
- fetal bovine serum
- FN1
- fibronectin 1
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GCRMA
- GeneChip robust multiarray average
- HRP
- horseradish peroxidase
- IPA
- Ingenuity Pathway Analysis
- MAPK
- mitogen-activated protein kinase
- MEK1/2
- mitogen-activated protein kinase kinase 1/2
- MMP
- matrix metalloprotease
- NT-siRNA
- nontargeting small interfering RNA
- OB-siRNA
- OB-cadherin small interfering RNA
- qRT-PCR
- quantitative reverse transcriptase PCR
- PBS
- phosphate-buffered saline
- siRNA
- small interfering RNA
- TGF-β1
- transforming growth factor β1
- TNF-α
- tumor necrosis factor α
- TUFT1
- tuftelin 1
- VIC
- valvular interstitial cell
REFERENCES
- 1. Roger V. L., Go A. S., Lloyd-Jones D. M., Adams R. J., Berry J. D., Brown T. M., Carnethon M. R., Dai S., de Simone G., Ford E. S., Fox C. S., Fullerton H. J., Gillespie C., Greenlund K. J., Hailpern S. M., Heit J. A., Ho P. M., Howard V. J., Kissela B. M., Kittner S. J., Lackland D. T., Lichtman J. H., Lisabeth L. D., Makuc D. M., Marcus G. M., Marelli A., Matchar D. B., McDermott M. M., Meigs J. B., Moy C. S., Mozaffarian D., Mussolino M. E., Nichol G., Paynter N. P., Rosamond W. D., Sorlie P. D., Stafford R. S., Turan T. N., Turner M. B., Wong N. D., Wylie-Rosett J., and American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2011) Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 123, e18–e209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajamannan N. M., Evans F. J., Aikawa E., Grande-Allen K. J., Demer L. L., Heistad D. D., Simmons C. A., Masters K. S., Mathieu P., O'Brien K. D., Schoen F. J., Towler D. A., Yoganathan A. P., Otto C.M. (2011) Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Circulation 124, 1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mohler E. R., Gannon F., Reynolds C., Zimmerman R., Keane M. G., Kaplan F. S. (2011) Bone formation and inflammation in cardiac valves. Circulation 103, 1522–1528 [DOI] [PubMed] [Google Scholar]
- 4. Chen J.-H., Yip C. Y., Sone E. D., Simmons C. A. (2009) Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am. J. Pathol. 174, 1109–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walker G. A., Masters K. S., Shah D. N., Anseth K. S., Leinwand L. A. (2004) Valvular myofibroblast activation by transforming growth factor-β1: implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 95, 253–260 [DOI] [PubMed] [Google Scholar]
- 6. Fondard O., Detaint D., Iung B., Choqueux C., Adle-Biassette H., Jarraya M., Hvass U., Couetil J. P., Henin D., Michel J. B., Vahanian A., Jacob M. P. (2005) Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur. Heart J. 26, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 7. Liu A. C., Joag V. R., Gotlieb A. I. (2007) The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 171, 1407–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hinton R. B., Jr., Lincoln J., Deutsch G. H., Osinska H., Manning P. B., Benson D. W., Yutzey K. E. (2006) Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ. Res. 98, 1431–1438 [DOI] [PubMed] [Google Scholar]
- 9. Aikawa E., Whittaker P., Farber M., Mendelson K., Padera R. F., Aikawa M., Schoen F. J. (2006) Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation 113, 1344–1352 [DOI] [PubMed] [Google Scholar]
- 10. Rabkin E., Aikawa M., Stone J. R., Fukumoto Y., Libby P., Schoen F. J. (2001) Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104, 2525–2532 [DOI] [PubMed] [Google Scholar]
- 11. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 12. Duffield J. S., Lupher M., Thannickal V. J., Wynn T. A. (2013) Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barnes J. L., Gorin Y. (2011) Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 79, 944–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wynn T. A. (2008) Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biernacka A., Dobaczewski M., Frangogiannis N. G. (2011) TGF-β signaling in fibrosis. Growth Factors 29, 196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Igarashi A., Okochi H., Bradham D. M., Grotendorst G. R. (1993) Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 4, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu L., Zhu Y. J., Yang X., Guo Z. J., Xu W. B., Tian X. L. (2007) Effect of TGF-β/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol. Sin. 28, 382–391 [DOI] [PubMed] [Google Scholar]
- 19. Meyer-Ter-Vehn T., Gebhardt S., Sebald W., Buttmann M., Grehn F., Schlunck G., Knaus P. (2006) p38 inhibitors prevent TGF-β-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest. Ophthalmol. Vis. Sci. 47, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 20. Garrett Q., Khaw P. T., Blalock T. D., Schultz G. S., Grotendorst G. R., Daniels J. T. (2004) Involvement of CTGF in TGF-β1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Invest. Ophthalmol. Vis. Sci. 45, 1109–1116 [DOI] [PubMed] [Google Scholar]
- 21. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Furukawa F., Matsuzaki K., Mori S., Tahashi Y., Yoshida K., Sugano Y., Yamagata H., Matsushita M., Seki T., Inagaki Y., Nishizawa M., Fujisawa J., Inoue K. (2003) p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 38, 879–889 [DOI] [PubMed] [Google Scholar]
- 23. Conte E., Fruciano M., Fagone E., Gili E., Caraci F., Iemmolo M., Crimi N., Vancheri C. (2011) Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS One 6, e24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J., Chen H., Seth A., McCulloch C. A. (2003) Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 285, H1871–H1881 [DOI] [PubMed] [Google Scholar]
- 25. Blaauboer M. E., Smit T. H., Hanemaaijer R., Stoop R., Everts V. (2011) Cyclic mechanical stretch reduces myofibroblast differentiation of primary lung fibroblasts. Biochem. Biophys. Res. Commun. 404, 23–27 [DOI] [PubMed] [Google Scholar]
- 26. Balestrini J. L., Chaudhry S., Sarrazy V., Koehler A., Hinz B. (2012) The mechanical memory of lung myofibroblasts. Integr. Biol. 4, 410–421 [DOI] [PubMed] [Google Scholar]
- 27. Chen J.-H., Chen W. L., Sider K. L., Yip C. Y., Simmons C. A. (2011) β-Catenin mediates mechanically regulated, transforming growth factor-β1 induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler. Thromb. Vasc. Biol. 31, 590–597 [DOI] [PubMed] [Google Scholar]
- 28. Liu F., Mih J. D., Shea B. S., Kho A. T., Sharif A. S., Tager A. M., Tschumperlin D. J. (2010) Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 190, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Olsen A. L., Olsen A. L., Bloomer S. A., Chan E. P., Gaça M. D., Georges P. C., Sackey B., Uemura M., Janmey P. A., Wells R. G. (2011) Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G110–G118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kloxin A. M., Benton J. A., Anseth K. S. (2010) In situ elasticity modulation with dynamic substrates to direct cell phenotype. Biomaterials 31, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang H., Haeger S. M., Kloxin A. M., Leinwand L. A., Anseth K. S. (2012) Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS One 7, e39969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niessen C. M., Leckband D., Yap A. S. (2011) Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 91, 691–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hinz B., Pittet P., Smith-Clerc J., Chaponnier C., Meister J. J. (2004) Myofibroblast development is characterized by specific cell-cell adherens junctions. Mol. Biol. Cell 15, 4310–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schneider D. J., Wu M., Le T. T., Cho S. H., Brenner M. B., Blackburn M. R., Agarwal S. K. (2012) Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-β production and epithelial to mesenchymal transition. FASEB J. 26, 503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson C. M., Hanson M. N., Helgeson S. C. (1987) Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J. Mol. Cell. Cardiol. 19, 1185–1193 [DOI] [PubMed] [Google Scholar]
- 36. Chang S. K., Noss E. H., Chen M., Gu Z., Townsend K., Grenha R., Leon L., Lee S. Y., Lee D. M., Brenner M. B. (2011) Cadherin-11 regulates fibroblast inflammation. Proc. Natl. Acad. Sci. U.S.A. 108, 8402–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leask A., Abraham D. J. (2044) TGF-β signaling and the fibrotic response. FASEB J. 18, 816–827 [DOI] [PubMed] [Google Scholar]
- 38. Vindevoghel L., Kon A., Lechleider R. J., Uitto J., Roberts A. B., Mauviel A. (1988) Smad-dependent transcriptional activation of human type VII collagen gene (COL7A1) promoter by transforming growth factor-β. J. Biol. Chem. 273, 13053–13057, 1998 [DOI] [PubMed] [Google Scholar]
- 39. Javelaud D., Mauviel A. (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: implications for carcinogenesis. Oncogene 24, 5742–5750 [DOI] [PubMed] [Google Scholar]
- 40. Chang S. K., Gu Z., Brenner M. B. (2010) Fibroblast-like synoviocytes in inflammatory arthritis pathology: the emerging role of cadherin-11. Immunol. Rev. 233, 256–266 [DOI] [PubMed] [Google Scholar]
- 41. Verhoekx J. S. N., Verjee L. S., Izadi D., Chan J. K., Nicolaidou V., Davidson D., Midwood K. S., Nanchahal J. (2013) Isometric contraction of dupuytren's myofibroblasts is inhibited by blocking intercellular junctions. J. Invest. Dermatol. 133, 2664–2671 [DOI] [PubMed] [Google Scholar]
- 42. Hutcheson J. D., Chen J., Sewell-Loftin M. K., Ryzhova L. M., Fisher C. I., Su Y. R., Merryman W. D. (2013) Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler. Thromb. Vasc. Biol. 33, 114–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang H., Sridhar B., Leinwand L. A., Anseth K. S. (2013) Characterization of cell subpopulations expressing progenitor cell markers in porcine cardiac valves. PLoS One 8, e69667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hutcheson J. D., Ryzhova L. M., Setola V., Merryman W. D. (2012) 5-HT2B antagonism arrests non-canonical TGF-β1-induced valvular myofibroblast differentiation. J. Mol. Cell. Cardiol. 53, 707–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li L., Ying J., Li H., Zhang Y., Shu X., Fan Y., Tan J., Cao Y., Tsao S. W., Srivastava G., Chan A. T., Tao Q. (2012) The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/β-catenin signaling and silenced in common carcinomas. Oncogene 31, 3901–3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Masur S. K., Dewal H. S., Dinh T. T., Erenburg I., Petridou S. (1996) Myofibroblasts differentiate from fibroblasts when plated at low density. Proc. Natl. Acad. Sci. U.S.A. 93, 4219–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B. G., Rossant J., Wrana J. L. (2010) The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell. 19, 831–844 [DOI] [PubMed] [Google Scholar]
- 48. Margadant C., Sonnenberg A. (2010) Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hinz B., Gabbiani G. (2003) Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb. Haemost. 90, 993–1002 [DOI] [PubMed] [Google Scholar]
- 50. Lee D. M., Kiener H. P., Agarwal S. K., Noss E. H., Watts G. F., Chisaka O., Takeichi M., Brenner M. B. (2007) Cadherin-11 in synovial lining formation and pathology in arthritis. Science 315, 1006–1010 [DOI] [PubMed] [Google Scholar]
- 51. Pittet P., Lee K., Kulik A. J., Meister J. J., Hinz B. (2008) Fibrogenic fibroblasts increase intercellular adhesion strength by reinforcing individual OB-cadherin bonds. J. Cell Sci. 121:877–886, 2008 [DOI] [PubMed] [Google Scholar]
- 52. Follonier L., Schaub S., Meister J. J., Hinz B. (2008) Myofibroblast communication is controlled by intercellular mechanical coupling. J. Cell Sci. 121, 3305–3316 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.