

Abstract
The tumor necrosis factor (TNF) receptor family member CD40 plays an essential role in the activation of antigen-presenting cells, B cell maturation, and immunoglobulin (Ig) class switching critical for adaptive immunity. Although the bioactive sphingolipid metabolite sphingosine-1-phosphate (S1P) and the kinase that produces it, sphingosine kinase 1 (SphK1), have long been implicated in the actions of TNF mediated by engagement of TNFR1, nothing is yet known of their role in CD40-mediated events. We have now found that ligation of CD40 activates and translocates SphK1 to the plasma membrane, leading to generation of S1P. SphK1 inhibition in human tonsil B cells, as well as inhibition or deletion of SphK1 in mouse splenic B cells, significantly reduced CD40-mediated Ig class switching and plasma cell differentiation ex vivo. Optimal activation of downstream CD40 signaling pathways, including NF-κB, p38, and JNK, also required SphK1. In mice treated with a SphK1 inhibitor or in SphK1−/− mice, isotype switching to antigen-specific IgE was decreased in vivo by 70 and 55%, respectively. Our results indicate that SphK1 is important for CD40-mediated B cell activation and regulation of humoral responses and suggest that targeting SphK1 might be a useful therapeutic approach to control antigen-specific IgE production.—Kim, E. Y., Sturgill, J. L., Hait, N. C., Avni, D., Valencia, E. C., Maceyka, M., Lima, S., Allegood, J., Huang, W.-C., Zhang, S., Milstien, S., Conrad, D., Spiegel, S. Role of sphingosine kinase 1 and sphingosine-1-phosphate in CD40 signaling and IgE class switching.
Keywords: B cells, NF-κB, sphingolipids, inflammation
Activation of CD40, a tumor necrosis factor (TNF) receptor family member, which is expressed constitutively on B cells, macrophages, and dendritic cells, is important for efficient activation of humoral and cell-mediated immune responses (1–3). Engagement of CD40 on B cells by surface CD154, known as CD40 ligand (CD40L), expressed on CD4+ T cells leads to B-cell proliferation and maturation, and ultimately to immunoglobulin (Ig) class switching and development of memory and plasma cells (1–3). The key role of the CD40-CD40L dyad in isotype switching has been highlighted in mice and humans. Mice deficient in CD40 or CD40L fail to undergo isotype switching in response to thymus-dependent antigens (4). Moreover, agonistic anti-CD40 antibody bypasses the requirement for T cells in IL-4–driven IgE isotype switching in vitro (5). Genetic defects affecting CD40 cause a rare form of hyper-IgM syndrome, a disorder characterized by defects in isotype switching associated with recurrent infections (6). As such, dysregulations in the CD40-CD40L pathway play an important role in many inflammatory disorders ranging from various autoimmune diseases to airway inflammation and allergic responses.
CD40 engagement leads to the activation of the stress-activated protein kinases, JNK and p38, and the transcription factor NF-κB, which up-regulate the expression of cytokines and other factors that promote immune responses. These signaling pathways are required for germinal center (GC) and memory B-cell formation. Despite abundant knowledge of key requirements for the maintenance of GC cell viability, much less is known about the environmental cues involved in regulation of GC size. Interestingly, a recent study demonstrated that the bioactive sphingolipid metabolite sphingosine-1-phosphate (S1P) regulates the survival and positioning of GC B cells, thus promoting GC homeostasis (7). S1P, a ligand for 5 G-protein–coupled receptors, referred to as S1PR1–5, has long been implicated in inflammatory and immune responses (8, 9). S1P has various functions as a central mediator of lymphocyte trafficking, including egress of mature single positive T cells out of thymus (10), T-cell migration into and out of secondary lymph nodes (11), and B-cell entry into marginal zones (12). In addition, ligation of TNFR1 by the proinflammatory cytokine TNF activates sphingosine kinase 1 (SphK1), one of the isoenzymes that produce S1P (13–15). S1P, in turn, can be exported out of cells and activate numerous signaling pathways, including NF-κB and mitogen-activated protein kinase cascades, through its cell surface receptors. It was also suggested that intracellular S1P can modulate the E3 ligase activity of tumor-necrosis factor receptor-associated factor 2 (TRAF2), a key component in NF-κB signaling triggered by TNF (16). Similarly, S1P enhances the E3 ligase activity of the cellular inhibitor of apoptosis 2 (cIAP2), resulting in K63-linked polyubiquitination of the transcription factor IRF1 that is essential for IL-1-induced production of chemokines CXCL10 and CCL5 (17). The E3 ligase activities of TRAF2 and cIAP2 are also required for activation of the canonical and alternative NF-κB pathways by CD40 (18, 19). Surprisingly, however, nothing is known about the role of SphK1 and S1P in CD40-mediated events. In this study, we found that, like activation of TNFR1, in B cells, CD40 activates and translocates SphK1 to the plasma membrane where its substrate sphingosine resides, leading to increased S1P. Using pharmacological and genetic approaches, we demonstrated that SphK1 and S1P are important for optimal CD40-mediated B-cell activation and differentiation and Ig isotype switching in vitro and for regulation of T-cell-dependent humoral responses and antigen-specific IgE production in vivo.
MATERIALS AND METHODS
Cell culture and reagents
HEK293 cells stably transfected with murine CD40 (HEK-CD40), kindly provided by Dr. Michael Karin (University of California, San Diego, CA, USA), were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). SphK1 was down-regulated by transfection with 100 nM On-TargetPlus SmartPool siRNA (Dharmacon Fisher Scientific, Pittsburgh, PA, USA) using Oligofectamine or Lipofectamine 2000 (Invitrogen, Gaithersburg, MD, USA). V5-SphK1 was overexpressed by transfection with Lipofectamine Plus (Invitrogen). Human tonsilar B cells and mouse splenic B cells were cultured in RPMI supplemented with 10% heat-inactivated (56°C, 30 min) FBS (Atlanta Biologicals, Flowery Branch, GA, USA), 2 mM l-glutamine, 50 μg/ml penicillin, 50 μg/ml streptomycin, 1 mM sodium pyruvate, 50 μg/ml amphotericin B, 50 μM 2-mercaptoethanol, and 20 mM HEPES (Invitrogen). The SphK1-specific inhibitor PF543 was synthesized by Dr. Shijun Zhang. SK1-I (BML-EI411) was obtained from Enzo Life Sciences (Farmingdale, NY, USA).
Mice and ovalbumin (OVA) immunization
C57BL/6 wild-type and SphK1−/− mice were originally from Dr. Richard L. Proia (National Institute of Diabetes and Digestive and Kidney Diseases, U.S. National Institutes of Health, Bethesda, MD, USA). Mice 8 to 15 wk of age with littermate controls to ensure the same genetic and environmental background were used for all experiments. All mouse protocols were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee. Mice were given acidified, antibiotic-treated drinking water for ≥2 wk prior to immunization by subcutaneous injection of 10 mg OVA in 4 mg alum adjuvant, followed by intraperitoneal injection after 2 wk. Mice were euthanized 7–8 d following the second immunization. Where indicated, mice were treated with SK1-I (10 mg/kg, i.p.) every other day, beginning on the same day as immunization.
B-cell purification and culture
Human B cells were purified from tonsils as described previously (20). Tonsils obtained from the Virginia Commonwealth University Tissue Data Acquisition and Analysis Core were disrupted in medium supplemented with antibiotics with a homogenizer (Seward Stomacher 80 Biomaster Lab Blender; Brinkmann, Westbury, NY, USA). After underlaying with Ficoll-Hypaque (GE Healthcare, Pittsburgh, PA, USA) and centrifugation (20 min at 400 g), cells at the interphase were removed and washed with PBS. Cells were incubated with FITC-mouse anti-human IgD (BD Pharmingen, San Jose, CA, USA) for 30 min on ice, and naive B cells were isolated with anti-FITC magnetic microbeads according to the manufacturer's protocol (Miltenyi, San Diego, CA, USA). B cell preparations were >95% pure IgD+ by FACS analysis.
Mouse splenic B-cell suspensions were prepared by grinding spleens between 2 frosted glass slides. Cells were washed and filtered through a 40 μm filter to remove extracellular matrix and debris. CD43-naive B cells were isolated by negative selection after removal of CD43+ cells with magnetic beads (MACS; Miltenyi).
B cells were cultured in RPMI 1640 containing 10% heat-inactivated FBS, 2 mM l-glutamine, 50 μg/ml penicillin, 50 μg/ml streptomycin, 1 mM sodium pyruvate, 50 μg/ml amphotericin B, 50 μM 2-mercaptoethanol, and 20 mM HEPES.
Proliferation assay
To measure proliferation, human B cells were cultured with anti-CD40 (1 μg/ml, clone G28.5), IL-4 (10 ng/ml; R&D Systems, Minneapolis, MN, USA), and IL-21 (200 ng/ml; R&D Systems) for 96 h and pulsed during the last 16 h with 1 μCi [3H]thymidine (GE Healthcare) as described previously (20). Mouse B cells were cultured with anti-CD40 (1 μg/ml, clone 3/23; BD Pharmingen) and IL-4 (10 ng/ml) for 72 h and pulsed during the last 16 h with [3H]thymidine for proliferation assays. Cells were then harvested onto a Unifilter 96 plate (Packard Instrument Co., Meridian, CT, USA) using a Filtermate 96 plate harvester (Packard), and [3H]thymidine incorporation into DNA was measured with a TopCount model B9902 (Packard).
ELISA
For measurement of antibody production by ELISA, mouse and human B cells were cultured for 8 and 14 d, respectively, conditions for optimal antibody production in vitro (20, 21). Murine B cells class switch from IgG1 to IgE. Ig production was measured by standard sandwich ELISA as we described previously (22). Briefly mouse IgE ELISA utilized paired rat anti-mouse IgE mab B1E3 and biotinylated R1E4 along with alkaline phosphatase (AP)-conjugated streptavidin (Southern Biotech, Birmingham, AL, USA), and mouse IgM and IgG1 ELISA utilized paired goat anti-mouse IgM and IgG1 and AP-conjugated goat anti-mouse IgM and IgG1 (Southern Biotech). Color was generated by addition of AP substrate (Sigma, St. Louis, MO, USA). Serum OVA-specific Ab ELISA was performed by coating the plate with OVA in carbonate buffer. Samples were detected with either AP-conjugated IgG1 (Southern Biotech) or horseradish peroxidase (HRP)-conjugated goat-anti-mouse IgE (Abcam, Cambridge, MA, USA). Plates were read on a SpectraMax 250 ELISA plate reader at either 405 nm for AP or 450 nm for HRP detection, and data were analyzed using SOFTmax Pro (Molecular Devices, Downingtown, PA, USA).
Human B cells class switch from IgG4 to IgE. Although some humans will make IgG4, which is the direct comparison of mouse IgG1, not all humans make IgG4 in vitro, and thus total IgG was determined. Human Ig was also measured by ELISA as we described previously (20). Briefly, capture antibodies (goat anti-human IgM and IgG; Southern Biotech) and mouse anti-human IgE (4.15)) were coated, and samples and standard IgM and IgG (Sigma) and IgE (purified from JW8 hybridoma) were detected with HRP-conjugated goat anti-human IgM, goat anti-human IgG, and rabbit anti-human IgE (Southern Biotech). TMB substrate (BD Pharmingen) was used to develop the reaction, which was stopped with 0.18M H2SO4.
Plasma cell determination by flow cytometry
Human tonsil B cells and mouse splenic B cells were cultured for 7 or 6 d, respectively, for optimal plasma cell differentiation, which was assessed by flow cytometry as described previously (21). Briefly, cells were pelleted and washed once and resuspended in cold FACS buffer (PBS with 0.5% FBS and 0.03% sodium azide). Fc receptors were blocked using commercial human FcR block (Miltenyi Biotec), and cells were then stained with mouse anti-human CD38 PE or anti-mouse CD138 PE (Biolegend, San Diego, CA, USA), respectively. Cells were incubated for 30 min on ice, washed in cold FACS buffer, and then resuspended in 1 ml of FACS buffer. Prior to analysis, propidium iodide (PI) stain was added to determine cell viability. Data were collected on a BD FACSCanto II and analyzed using BD FACS-Diva software ver. 6.1.3 (BD Biosciences). Unstained cells and PI-stained cells were analyzed to ensure only live plasma cells were used for determination of plasma cell differentiation.
In some experiments, purified human B cells were incubated with 1.5 mM carboxyfluorescein diacetate succinimidyl ester (Sigma) for 8 min before quenching with FBS for an additional 5 min and washed before culturing for 7 d. Human B cells were than labeled with anti-human CD138 (eBioscience, San Diego, CA, USA). Differentiation of human B cells to plasma cells was assessed by monitoring the frequency of CD138+, proliferated (CFSElow) B cells (23).
Immunofluorescence
Immunofluorescent localization was performed essentially as described previously (24). Briefly, 105 HEK-CD40 cells were seeded onto poly-d-lysine-coated glass coverslips in 6-well dishes and transfected the next day with V5-SphK1 using Lipofectamine Plus. Transfection efficiency was routinely between 60 and 70%. The next day, cells were treated as described and then fixed with 3% paraformaldehyde for 10 min, washed extensively with 10 mM glycine in PBS, and permeabilized with 0.5% Triton X-100 for 3 min. After washing, coverslips were incubated with mouse anti-V5 antibody (1:200 dilution; Life Technologies, Gaithersburg, MD, USA) in 10% IgG-free BSA in PBS for 20 min, washed 3 times, and then incubated with Alexa 488 labeled anti-mouse antibody (1:500 dilution; Life Technologies) and Hoechst 33342 (0.8 mg/ml; fluoropure grade from Life Technologies) in 10% IgG-free BSA in PBS for 20 min. After washing, coverslips were mounted on slides with 10 mM n-propylgallate in 100% glycerol and visualized on a Zeiss LSM 510 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Western blotting
Cells were washed with ice-cold PBS and lysed in 20 mM Tris (pH 7.4), 1 mM EDTA, 1 mM sodium orthovanadate, 0.5 mM 4-deoxypyridoxine, 15 mM NaF, 40 mM b-glycerophosphate, 150 mM NaCl, 10% glycerol, 1:500 protease inhibitor cocktail (Sigma), and 0.5% Nonidet P-40. Equal amounts of proteins were separated by SDS-PAGE, transblotted to PVDF, incubated with the indicated primary antibodies overnight, washed, and incubated with appropriate HRP-conjugated secondary antibodies (1:5000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Immunocomplexes were visualized by enhanced chemiluminescence (Pierce Thermo Scientific, Rockford, IL, USA). Antibodies used include SphK1 (Abcam), p-SphK1 (ECM Biosciences, Versailles, KY, USA), and JNK (Santa Cruz Biotechnology, Santa Cruz, CA, USA). p-IKK, IKK, pIκBα, IκBα, pJNK, p-p65, tubulin, and p-p38 were from Cell Signaling (Danvers, MA, USA). Optical densities of bands were quantified with ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov) and normalized to the optical densities of their respective endogenous control.
SphK1 activity and analysis of sphingolipids by mass spectrometry
SphK1 activity was determined with 50 μM sphingosine and ATP (1 mM) containing MgCl2 (10 mM) in the presence of 0.25% Triton X-100, which inhibits SphK2, as described previously (25). SphK2 activity was determined similarly when sphingosine was added as a complex with 4 mg/ml BSA in the presence of 1 M KCl, which inhibits SphK1 activity (25). S1P formation was measured by electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS; ABI 4000 QTRAP; ABSciex, Farmingham, MA USA). Some experiments were carried out in the presence of [32P]ATP as described previously (25) and [32P]S1P was extracted and then separated by TLC on silica gel G60 with chloroform/acetone/methanol/acetic acid/H2O (10:4:3:2:1, v/v) as solvent. Radioactive bands corresponding to S1P were quantified with a FX Molecular Imager (Bio-Rad, Hercules, CA, USA). SphK-specific activity is expressed as picomoles S1P formed per minute per milligram protein. It should be noted that SphK activity determined in the presence of 1 M KCl or in 0.25% Triton X-100 is only an operational differentiation method developed with overexpressed SphK1 and SphK2 (25), and a small amount of cross-contamination cannot be ruled out when these protocols are used to determine the endogenous isoenzymes.
For mass measurements of sphingolipid metabolites, cells were washed, lipids were extracted, internal standards were added (0.5 nmol each; Avanti Polar Lipids, Alabaster, AL, USA), and sphingolipids were quantified by LC-ESI-MS/MS as described previously (26).
Statistical analysis
Statistical analyses were performed using an unpaired 2-tailed Student's t test for comparison of 2 groups and analysis of variance (ANOVA) for analyzing experiments consisting of ≥3 groups (GraphPad Prism; GrahPad, San Diego, CA, USA). Significant ANOVA results were followed by post hoc tests for multiple comparisons. A value of P < 0.05 was considered significant. When primary human B cells were used, data are from a representative donor, and similar results were obtained from ≥3 additional donors.
RESULTS
CD40 stimulation leads to activation of SphK1 and production of S1P
Activation of SphK1, which catalyzes S1P synthesis, has long been implicated in the actions of the proinflammatory cytokine TNF, mediated by engagement of TNFR1 (13, 14, 16, 27–30). Thus, we examined whether ligation of CD40, another TNFR family member, also activates SphK1 and increases S1P. To this end, we first used HEK-CD40 cells stably expressing CD40 (18, 19). Similar to previous studies with TNF (13, 14), stimulation of CD40 with an agonistic anti-CD40 antibody induced translocation of SphK1 from the cytosol to the plasma membrane within 5 min, as determined by confocal immunofluorescence microscopy (Fig. 1A). Both SphK1 and SphK2 are expressed in HEK293 cells and can be qualitatively distinguished by differences in their enzymatic activities (25). CD40 stimulated the enzymatic activity of SphK1, reaching a maximum within 10 min and decreasing thereafter (Fig. 1B). In contrast, CD40 ligation did not affect SphK2 activity (Fig. 1C). Moreover, CD40 also increased phosphorylation of SphK1 on serine 225 (Fig. 1D), which is known to enhance its enzymatic activity (14). Because TNF induces phophorylation and activation of SphK1 in an ERK-dependent manner (14), and CD40 has been shown to activate ERK1/2 (31, 32), we sought to examine whether activation of SphK1 and phosphorylation at serine 225 following CD40 ligation is mediated via ERK1/2. U0126, a highly selective inhibitor of MEK1/2, as expected markedly reduced ERK1/2 phosphorylation induced by CD40. This also inhibited CD40-mediated activation of SphK1 and its phosphorylation (Fig. 1B, D).
Figure 1.

CD40 ligation translocates and activates SphK1. A) HEK-CD40 cells were transfected with V5-SphK1 and stimulated with agonistic anti-CD40 antibody for the indicated times. Cells were stained with Hoechst (blue) and V5 antibody (green) and visualized by confocal microscopy. Scale bars = 10 μm. B, C) Serum-starved HEK-CD40 cells were stimulated with anti-CD40 antibody for the indicated times in the presence of vehicle or U0126 (1 μM). SphK1 activity (B) and SphK2 activity (C) in cell lysates were measured by formation of S1P. Data are expressed as means ± sd from triplicate determinations. Similar results were obtained in 2 additional experiments. D) In duplicate cultures, phospho-Ser225-SphK1, total SphK1, and pERK1/2 were evaluated by Western blot analysis with the indicated antibodies. One set of representative data is shown from 3 independent experiments. Quantified data by densitometry are expressed as mean ± sd fold vs. time 0; n = 3.
We next examined whether CD40 engagement enhanced SphK1 activity and S1P formation in B cells. Immunoblotting of human tonsilar B cells showed that CD40 ligation also rapidly increased phosphorylation of SphK1 (Fig. 2A). Similarly, in purified splenic B cells from SphK1+/+ mice, stimulation of CD40 robustly increased SphK1 activity within 10 min and maximum stimulation was observed at 30 min (Fig. 2B). It should be noted that other factors in addition to phophorylation at serine 225 can also affect SphK1 activity (33). Notably, CD40 ligation did not increase SphK activity in splenic B cells null for SphK1, and the observed residual activity is probably due to SphK2 (Fig. 2B). In agreement, S1P levels in SphK1-null B cells were only 30% lower than in wild-type B cells (Fig. 2C). Concomitantly with the increased SphK1 activity, CD40 stimulation also markedly increased S1P levels in SphK1+/+ B cells (Fig. 2C), but not in the SphK1−/− B cells. However, although in vitro SphK activity remained elevated for 60 min, S1P levels in cells returned to basal levels at this time, likely due to degradation by phosphatases and S1P lyase (34). CD40 induced a slight decrease in the levels of the substrate sphingosine only in the SphK1+/+ B cells (Fig. 2D). Taken together, this is the first demonstration that CD40 stimulation leads to activation of SphK1 and elevation of S1P.
Figure 2.

CD40 stimulation activates SphK1 and increases S1P levels in B cells. A) Human tonsil B cells were stimulated with anti-CD40 antibody, and levels of phospho-Ser225-SphK1 and total SphK1 were evaluated by Western blot analysis. One set of representative data is shown from 2 independent experiments. Quantified data by densitometry are expressed as mean ± sd fold vs. time 0; n = 2. *P < 0.05 vs. time 0. B) Splenic B cells purified from SphK1+/+ and SphK1−/− mice were stimulated with agonistic CD40 antibody for the indicated times. B) SphK1 activity was measured by formation of [32P]S1P, and data are expressed as means ± sd from triplicate determinations. Where not shown, the size of the symbols indicates the sd. C, D) Mass levels of S1P (C) and sphingosine (D) were measured by LC-ESI-MS/MS. Data are expressed as means ± sd from triplicate determinations. Similar results were obtained in 2 additional independent experiments. *P < 0.05 vs. time 0.
SphK1 is involved in CD40-mediated B-cell activation, differentiation, and isotype switching in vitro
Having established that CD40 stimulation leads to activation of SphK1 and formation of S1P, we next investigated the involvement of SphK1 in CD40-mediated B-cell functions. As a first approach, specific SphK1 inhibitors were utilized. Primary human tonsilar B cells cultured in vitro with agonistic anti-CD40 in the presence of IL-4 and IL-21, cytokines that augment plasma cell development, produced high levels of IgM, IgG, and IgE (Fig. 3A). Treatment with the specific SphK1 inhibitor SK1-I (35) had only a small effect on IgM production but reduced IgG levels in a concentration-dependent manner with significant reduction at a concentration as low as 1 μM, while IgE production was completely blocked at 0.5 μM SK1-I (Fig. 3A). These results suggest that inhibition of SphK1 impairs isotype switching in human B cells, as demonstrated by the major reduction in IgE production. Moreover, inhibition of SphK1 diminished the differentiation of activated B cells into plasma cells, as demonstrated by decreased numbers of cells with high expression of CD138 (Fig. 3B) or CD38 (also known as syndecan-1; Fig. 3C), commonly used markers for identification of plasma cells (36, 37).
Figure 3.

Effect of SphK1 inhibition in human B cells on CD40-mediated isotype switching and plasma cell differentiation. Human B cells (A, B) were isolated from tonsils by positive selection using IgD-specific beads by MACS sorting. B cells were cultured with anti-CD40 antibody, IL-4, and IL-21 for 14 d (A) or 7 d (B, C) in the absence or presence of the indicated concentration of SK1-I. A) Antibody levels were measured by antibody-specific ELISAs. Data are expressed as means ± sd from triplicate determinations. Similar results were obtained in 2 additional independent experiments. *P < 0.05. B, C) Plasma cell differentiation was analyzed by FACS. B) Representative flow cytometry plots. Circled numbers indicate frequency of plasma cells (CD138+ and CFSElow). C) Percentage plasma cells from another donor. Data are expressed as means ± sd from triplicate determinations.
Murine splenic B cells were also sensitive to inhibition of SphK1, and a significant decrease in IgG1 production was observed at a concentration of 0.25 μM SK1-I (Fig. 4A). Similarly, PF543, a structurally different SphK1 inhibitor (38), also significantly decreased IgG1 production induced by CD40 (Fig. 4B). There was a dramatic reduction of IgE production by treatment of the splenic B cells with either SK1-I or PF543 (Fig. 4A, B). Similar to the results with human B cells, SphK1 inhibition did not significantly alter CD40-mediated IgM production (Fig. 4A, B). Because CD40 ligation induces B-cell proliferation (39), and isotype switching might be linked to proliferation, we examined whether the inhibition of IgE production by SK1-I or PF543 is accompanied by inhibition of proliferation. Both SK1-I and PF543 at concentrations ≥ 0.5 μM significantly reduced proliferation of murine B cells (Fig. 4C, D).
Figure 4.

SphK1 inhibitors suppress mouse B-cell isotype switching ex vivo. Splenic B cells from C57BL/6 mice were isolated by negative selection using anti-CD43 beads by MACS sorting. B cells were cultured with agonistic anti-CD40 antibody and IL-4 for 8 d (A, B) or 3 d (C, D) in the presence of the indicated concentrations of SK1-I (A, C) or PF543 (B, D). A, B) Antibody levels were measured by ELISA. C, D) DNA synthesis was measured by [3H]thymidine uptake. Data are expressed as means ± sd from triplicate determinations. Similar results were obtained in 2 additional experiments. *P < 0.05.
To substantiate the effects of these pharmacological inhibitors, we also examined CD40-mediated antibody switching in B cells devoid of SphK1 expression compared to wild-type littermate mice. Production of IgM and IgG1 was similar to splenic B cells isolated from SphK1−/− mice compared to B cells from wild-type littermates. However, deletion of SphK1 decreased IgE production by 48% (Fig. 5A). Although proliferation of SphK1-null B cells was not significantly lower than wild-type B cells after stimulation with anti-CD40 and IL-4 for 3 d (Fig. 5B), accumulation of plasma cells after 6 d (optimal time for differentiation) was significantly impaired (Fig. 5C).
Figure 5.
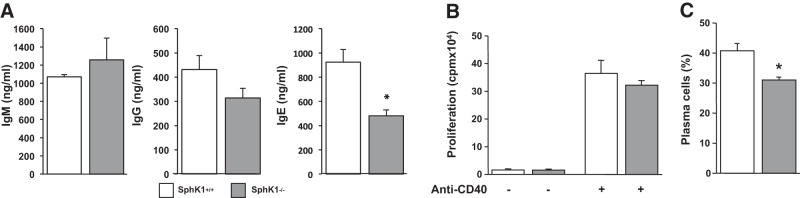
SphK1 deletion in B cells impairs differentiation to plasma cells and isotype switching. Splenic B cells from SphK1−/− mice and wild-type littermates were isolated by negative selection using anti-CD43 beads by MACS sorting and cultured with agonistic anti-CD40 antibody and IL-4. A) After 8 d, antibody levels were measured by ELISA. B) After 3 d, DNA synthesis was measured by [3H]thymidine uptake. C) After 7 d, differentiation of B cells to plasma cells was determined by FACS. Data are expressed as means ± sd from triplicate determinations. Similar results were obtained in 2 additional experiments. *P < 0.05.
SphK1 is important for optimal activation of downstream CD40 signaling pathways
CD40 stimulation leads to activation of both canonical and noncanonical NF-κB signaling, as well as JNK and p38 MAPK pathways (2). In agreement with previous studies (19), CD40 ligation of HEK-CD40 cells rapidly activated IKK, which led to phosphorylation of IκBα and JNK, as determined with phospho-specific antibodies (Fig. 6A, B). Inhibition of SphK1 with SK1-I or PF543 greatly reduced CD40-mediated phosphorylation of IKK and IκBα (Fig. 6A, B). Phosphorylation of JNK (Fig. 6A) and p65 (Fig. 6B) were also decreased by these SphK1 inhibitors. Similarly, in splenic B cells devoid of SphK1, phosphorylation of IκBα, p65, and p38 was diminished compared to wild-type littermates (Fig. 6C). However, IκBα degradation was similar in SphK1+/+ and SphK1−/− B cells (Fig. 6C).
Figure 6.

SphK1 is important for optimal CD40 signaling. A, B) Serum-starved HEK-CD40 cells were pretreated with vehicle, SK1-I (2.5 μM), or PF543 (5 μM) for 30 min and then stimulated with anti-CD40 antibody for various times as indicated. C) Splenic B cells purified from SphK1+/+ and SphK1−/− mice were stimulated with anti-CD40 antibody for various times. A–C) Equal amounts of cell lysate proteins were analyzed by Western blotting with the indicated antibodies. One set of representative data is shown from 3 independent experiments. Quantified data by densitometry are expressed as mean ± sd fold vs. time 0; n = 3. *P < 0.05 vs. time 0.
Role of SphK1 in isotype switching to antigen-specific IgE
It was then of interest to investigate the role of SphK1 in humoral responses. To directly assess antigen-specific antibody production, mice were immunized with thymus-dependent antigen OVA together with the adjuvant alum, with a boost 2 wk later. During this time, mice were treated with saline or SK1-I (10 mg/kg, i.p.) every other day (Fig. 7A). In agreement with our previous results (40, 41), blood levels of S1P were significantly decreased in SK1-I treated mice, whereas sphingosine levels were increased (Fig. 7B). Administration of SK1-I reduced serum levels of IgE (Fig. 7C). Moreover, OVA-specific IgE levels were significantly decreased by inhibition of SphK1 with SK1-I (Fig. 7C). To substantiate the results of pharmacological inhibition of SphK1, we next utilized SphK1−/− mice. In agreement with other reports (42–44), although serum levels of S1P were significantly lower in SphK1−/− mice, there were no obvious differences in blood and splenic B cell profiles between SphK1−/− and wild-type mice (Fig. 7D, E). Examination of T-cell-dependent isotype switching in SphK1−/− mice and littermate controls revealed that serum levels of IgE were not markedly altered, whereas OVA-specific IgE production was significantly decreased by 57% compared to wild-type mice (Fig. 7F). These data further support the notion that the switch to antigen-specific IgE is regulated by SphK1 in vivo.
Figure 7.

SphK1 is involved in antigen-specific isotype switching in vivo. A, B) C57BL/6 mice were immunized with OVA/alum (open arrows) and then treated with saline (vehicle) or SK1-I (10 mg/kg, i.p.) every other day (solid arrows), given an OVA boost on d 15, and euthanized on d 21. B) Blood levels of S1P and sphingosine were determined by LC-ESI-MS/MS. Data are means ± sd; n = 3 mice/group. C) Serum IgE and OVA-specific IgE levels (1:10 dilutions) were determined by ELISA. Data are means ± se; n = 5 mice/group. B, C) Similar results were obtained in 3 independent experiments. *P < 0.05 vs. vehicle treatment. D–F) SphK1+/+ and SphK1−/− mice were immunized with OVA/alum, given an OVA boost on d 15, and euthanized on d 21. D) Blood levels of S1P and sphingosine of SphK1+/+ and SphK1−/− mice were determined by LC-ESI-MS/MS. E) FACS analysis of B220+ B cells in blood and spleen of SphK1+/+ and SphK1−/− mice. F) Serum IgE and OVA-specific IgE levels (1:10 dilutions) were determined by ELISA. Data are representative of 3 independent experiments and are means ± se; n = 5 mice/group. *P < 0.05 vs. WT mice.
DISCUSSION
Here we report that CD40, like TNFR1, activates and translocates SphK1 to the plasma membrane, leading to increased formation of S1P. Our results demonstrate that SphK1 activation in B cells is important for CD40-mediated Ig class switching to IgE. Inhibition of SphK1 with 2 specific inhibitors or its deletion reduced activation of CD40-mediated signaling pathways, including NF-κB, p38, and JNK, and differentiation of B cells into plasma cells. Treatment of CD40-activated B cells with the SphK1 inhibitors did not affect IgM production but caused a concentration-dependent reduction in IgG1 and severe shutdown of IgE production. Similarly, genetic ablation of SphK1, although not significantly affecting IgM, drastically reduced production of IgE by B cells. Intriguingly, in immunized mice treated with a SphK1 inhibitor or in SphK1-null mice, antigen-specific IgE production was also strongly decreased. This has important clinical implications, as antigen-specific IgE plays an essential role in various allergic diseases, including allergic asthma, allergic rhinitis, food allergy, and atopic dermatitis. IgE production is also critical in anaphylactic reactions. Early studies showed that inhalation of nonisozyme-specific SphK inhibitors attenuates airway inflammation, bronchial hyperresponsiveness to inhaled methacholine, and goblet cell hyperplasia in an asthmatic mouse model (45). However, a different nonspecific SphK inhibitor ameliorated antigen-induced bronchial smooth muscle hyperresponsiveness, but not airway inflammation (46). We have recently shown that the specific SphK1 inhibitor SK1-I attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma, as well as activation of NF-κB in the lungs (41). Moreover, intragastric sensitization to induce food allergy resulted in reduced OVA-specific IgE in SphK1−/− mice but not when immunized intraperitoneally (47).
Proliferation of B cells has been shown to correlate with isotype switching (48). Blockade of CD40-CD40L interactions or genetic ablation of the CD40L or CD40 genes prevented the clonal proliferation of antigen-responding B cells and averted GC formation, and antibody responses were mainly limited to low affinity IgM (4, 49). In addition, CD40 has been implicated in the formation of memory B cells, where the role of S1P and SphK1 is less clear. Here we provide evidence that blockade of SphK1 can selectively inhibit Ig class switching and thus the formation of long-lived memory plasma cells. This finding may have clinical implications for the treatment of humoral alterations seen in atopic or autoimmune disease. Similar observations were made when some signaling molecules downstream of CD40 were deleted. For example, IKKβ-deficient B cells are also impaired in mitogenic responses to anti-CD40 that correlate with a failure to mount effective antibody responses to T-cell-dependent and independent antigens (50). Likewise, MEKK1 activation is needed for CD40-induced B cell proliferation and thymus-dependent antigen-specific antibody production (32). Although inhibition of SphK1 decreased B-cell proliferation in a dose-dependent manner, in B cells lacking SphK1, there was a tendency for reduced proliferation, albeit not a statistically significant effect. Yet differentiation of these B cells to plasma cells was greatly reduced. This might be due to the short 3 d assay for proliferation, whereas plasma cell differentiation and antibody production were determined after longer incubations with agonistic anti-CD40 antibody.
S1P and its receptors are well known to regulate movement and localization of different B-cell subsets at distinct stages of development. S1PR1 directs the release of immature B cells from bone marrow into blood (51). Immature B cells also express S1PR3, which directs immature B cells to a bone marrow microenvironment important for both tolerance induction and maturation (52). S1PR1 expression in differentiating plasma cells is also required for S1P to guide IgG- or IgA-secreting B cells from secondary lymphoid tissues to the bone marrow or from Peyer's patches to mucosal sites, respectively (53, 54). S1P signaling via S1PR1 and S1PR3 also maintains proper positioning of marginal zone B cells within the spleen (55) and regulates movement of marginal zone B cells to and from the marginal zone and follicle, providing an efficient mechanism for systemic antigen capture and delivery to follicular dendritic cells (12). Germinal center B cells down-regulate S1PR1 and up-regulate S1PR2, a receptor that decreases B-cell movement and inhibits Akt activation and thus restricts GC B-cell survival and localization to the follicle center (7). It has been suggested that the local S1P concentration is lowest at the follicle center and highest at the follicle edge, where it signals S1PR2-expressing B cells to turn back toward the GC to prevent exit from the niche, thus controlling the number of B cells in the GC (7). Although it has been suggested that B cells express S1P-degrading enzymes at high levels (7), it is still not known how levels of S1P in the follicle center or interstitional concentrations are regulated. It is tempting to speculate that CD40-mediated activation of SphK1 regulates interstitial concentrations of S1P within lymphoid organs and influences residence time in the follicle. Although the identity of the cells that produce S1P in response to CD40 ligation is unknown, it has been suggested that follicular S1P is derived from radiation-resistant cells other than follicular dendritic cells and is rapidly degraded by B cells (7). Nevertheless, CD40 is also expressed on other antigen-presenting cells, such as monocytes, and can also be expressed on both endothelial and epithelial cells. Thus, the CD40-SphK1 axis could have a major role in maintaining lymph node homeostasis and formation of appropriate humoral immune responses. Interestingly, previous studies suggest that stimulation of B lymphocytes via CD40 ligation results in acid sphingomyelinase translocation from intracellular stores to the plasma membrane, leading to increased ceramide, which, in turn, mediates clustering of CD40 and CD40-initiated cell signaling (56, 57). Taken together with our results, it is possible that increased formation of bioactive sphingolipids at the plasma membrane plays an important role in CD40-mediated events.
Acknowledgments
The authors thank Dr. Michael Karin (University of California–San Diego, La Jolla, CA, USA) for kindly providing HEK-CD40 cells.
This study was supported by U.S. National Institutes of Health (NIH) grants R37GM043880 and R01 AI50094 (S.S.) and in part by U19AI077435 (S.S. and D.C.). E.Y.K. and S.L. were supported by NIH grant T32HL094290. Flow cytometry was supported in part by NIH grant P30CA16059 to the Virginia Commonwealth University (VCU) Massey Cancer Center, and microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, by NIH grant P30NS047463.
Footnotes
- ANOVA
- analysis of variance
- AP
- alkaline phosphatase
- CD40L
- CD40 ligand
- FBS
- fetal bovine serum
- GC
- germinal center
- HRP
- horseradish peroxidase
- Ig
- immunoglobulin
- LC-ESI-MS/MS
- electrospray ionization-tandem mass spectrometry
- OVA
- ovalbumin
- PI
- propidium iodide
- S1P
- sphingosine-1-phosphate
- SphK1
- sphingosine kinase 1
- TNF
- tumor necrosis factor
- TRAF2
- tumor necrosis factor receptor-associated factor 2
REFERENCES
- 1. Van Kooten C., Banchereau J. (2000) CD40-CD40 ligand. J. Leukoc. Biol. 67, 2–17 [DOI] [PubMed] [Google Scholar]
- 2. Bishop G. A. (2004) The multifaceted roles of TRAFs in the regulation of B-cell function. Nat. Rev. Immunol. 4, 775–786 [DOI] [PubMed] [Google Scholar]
- 3. Rickert R. C., Jellusova J., Miletic A. V. (2011) Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol. Rev. 244, 115–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawabe T., Naka T., Yoshida K., Tanaka T., Fujiwara H., Suematsu S., Yoshida N., Kishimoto T., Kikutani H. (1994) The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1, 167–178 [DOI] [PubMed] [Google Scholar]
- 5. Jabara H. H., Fu S. M., Geha R. S., Vercelli D. (1990) CD40 and IgE: synergism between anti-CD40 monoclonal antibody and interleukin 4 in the induction of IgE synthesis by highly purified human B cells. J. Exp. Med. 172, 1861–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fuleihan R., Ramesh N., Loh R., Jabara H., Rosen R. S., Chatila T., Fu S. M., Stamenkovic I., Geha R. S. (1993) Defective expression of the CD40 ligand in X chromosome-linked immunoglobulin deficiency with normal or elevated IgM. Proc. Natl. Acad. Sci. U. S. A. 90, 2170–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Green J. A., Suzuki K., Cho B., Willison L. D., Palmer D., Allen C. D., Schmidt T. H., Xu Y., Proia R. L., Coughlin S. R., Cyster J. G. (2011) The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 12, 672–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spiegel S., Milstien S. (2011) The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cyster J. G., Schwab S. R. (2012) Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 30, 69–94 [DOI] [PubMed] [Google Scholar]
- 10. Pappu R., Schwab S. R., Cornelissen I., Pereira J. P., Regard J. B., Xu Y., Camerer E., Zheng Y. W., Huang Y., Cyster J. G., Coughlin S. R. (2007) Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316, 295–298 [DOI] [PubMed] [Google Scholar]
- 11. Grigorova I. L., Schwab S. R., Phan T. G., Pham T. H., Okada T., Cyster J. G. (2009) Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat. Immunol. 10, 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cinamon G., Zachariah M. A., Lam O. M., Foss F. W., Jr., Cyster J. G. (2008) Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 9, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xia P., Wang L., Moretti P. A., Albanese N., Chai F., Pitson S. M., D'Andrea R. J., Gamble J. R., Vadas M. A. (2002) Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 277, 7996–8003 [DOI] [PubMed] [Google Scholar]
- 14. Pitson S. M., Moretti P. A., Zebol J. R., Lynn H. E., Xia P., Vadas M. A., Wattenberg B. W. (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 22, 5491–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baker D. A., Barth J., Chang R., Obeid L. M., Gilkeson G. S. (2010) Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNF-alpha-induced arthritis. J. Immunol. 185, 2570–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alvarez S. E., Harikumar K. B., Hait N. C., Allegood J., Strub G. M., Kim E. Y., Maceyka M., Jiang H., Luo C., Kordula T., Milstien S., Spiegel S. (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harikumar K. B., Yester J. W., Surace M. J., Oyeniran C., Price M. M., Huang W.-C., Hait N. C., Allegood J. C., Yamada A., Kong X., Lazear H. M., Bhardwaj R., Takabe K., Diamond M. S., Luo C., Milstien S., Spiegel S., Kordula T. (2014) K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat. Immunol. 15, 231–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., Karin M. (2008) Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 9, 1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsuzawa A., Tseng P. H., Vallabhapurapu S., Luo J. L., Zhang W., Wang H., Vignali D. A., Gallagher E., Karin M. (2008) Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 321, 663–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sturgill J. L., Mathews J., Scherle P., Conrad D. H. (2011) Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J. Neuroimmunol. 233, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rabah D., Conrad D. H. (2002) Effect of cell density on in vitro mouse immunoglobulin E production. Immunology 106, 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho S. W., Kilmon M. A., Studer E. J., van der Putten H., Conrad D. H. (1997) B cell activation and Ig, especially IgE, production is inhibited by high CD23 levels in vivo and in vitro. Cell. Immunol. 180, 36–46 [DOI] [PubMed] [Google Scholar]
- 23. Mills D. M., Bonizzi G., Karin M., Rickert R. C. (2007) Regulation of late B cell differentiation by intrinsic IKKalpha-dependent signals. Proc. Natl. Acad. Sci. U. S. A. 104, 6359–6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maceyka M., Alvarez S. E., Milstien S., Spiegel S. (2008) Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol. Cell. Biol. 28, 5687–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hait N. C., Sarkar S., Le Stunff H., Mikami A., Maceyka M., Milstien S., Spiegel S. (2005) Role of sphingosine kinase 2 in cell migration towards epidermal growth factor. J. Biol. Chem. 280, 29462–29469 [DOI] [PubMed] [Google Scholar]
- 26. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pettus B. J., Bielawski J., Porcelli A. M., Reames D. L., Johnson K. R., Morrow J., Chalfant C. E., Obeid L. M., Hannun Y. A. (2003) The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 17, 1411–1421 [DOI] [PubMed] [Google Scholar]
- 28. Billich A., Bornancin F., Mechtcheriakova D., Natt F., Huesken D., Baumruker T. (2005) Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell. Signal. 17, 1203–1217 [DOI] [PubMed] [Google Scholar]
- 29. De Palma C., Meacci E., Perrotta C., Bruni P., Clementi E. (2006) Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: a novel pathway relevant to the pathophysiology of endothelium. Arterioscler. Thromb. Vasc. Biol. 26, 99–105 [DOI] [PubMed] [Google Scholar]
- 30. Scherer E. Q., Yang J., Canis M., Reimann K., Ivanov K., Diehl C. D., Backx P. H., Wier W. G., Strieth S., Wangemann P., Voigtlaender-Bolz J., Lidington D., Bolz S. S. (2010) Tumor necrosis factor-alpha enhances microvascular tone and reduces blood flow in the cochlea via enhanced sphingosine-1-phosphate signaling. Stroke 41, 2618–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bishop G. A., Hostager B. S. (2003) The CD40-CD154 interaction in B cell-T cell liaisons. Cytokine Growth Factor Rev. 14, 297–309 [DOI] [PubMed] [Google Scholar]
- 32. Gallagher E., Enzler T., Matsuzawa A., Anzelon-Mills A., Otero D., Holzer R., Janssen E., Gao M., Karin M. (2007) Kinase MEKK1 is required for CD40-dependent activation of the kinases Jnk and p38, germinal center formation, B cell proliferation and antibody production. Nat. Immunol. 8, 57–63 [DOI] [PubMed] [Google Scholar]
- 33. Chan H., Pitson S. M. (2013) Post-translational regulation of sphingosine kinases. Biochim. Biophys. Acta 1831, 147–156 [DOI] [PubMed] [Google Scholar]
- 34. Spiegel S., Milstien S. (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407 [DOI] [PubMed] [Google Scholar]
- 35. Paugh S. W., Paugh B. S., Rahmani M., Kapitonov D., Almenara J. A., Kordula T., Milstien S., Adams J. K., Zipkin R. E., Grant S., Spiegel S. (2008) A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 112, 1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terstappen L. W., Johnsen S., Segers-Nolten I. M., Loken M. R. (1990) Identification and characterization of plasma cells in normal human bone marrow by high-resolution flow cytometry. Blood 76, 1739–1747 [PubMed] [Google Scholar]
- 37. Caven T. H., Sturgill J. L., Conrad D. H. (2007) BCR ligation antagonizes the IL-21 enhancement of anti-CD40/IL-4 plasma cell differentiation and IgE production found in low density human B cell cultures. Cell. Immunol. 247, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schnute M. E., McReynolds M. D., Kasten T., Yates M., Jerome G., Rains J. W., Hall T., Chrencik J., Kraus M., Cronin C. N., Saabye M., Highkin M. K., Broadus R., Ogawa S., Cukyne K., Zawadzke L. E., Peterkin V., Iyanar K., Scholten J. A., Wendling J., Fujiwara H., Nemirovskiy O., Wittwer A. J., Nagiec M. M. (2012) Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 444, 79–88 [DOI] [PubMed] [Google Scholar]
- 39. Banchereau J., Bazan F., Blanchard D., Briere F., Galizzi J. P., van Kooten C., Liu Y. J., Rousset F., Saeland S. (1994) The CD40 antigen and its ligand. Annu. Rev. Immunol. 12, 881–922 [DOI] [PubMed] [Google Scholar]
- 40. Nagahashi M., Ramachandran S., Kim E. Y., Allegood J. C., Rashid O. M., Yamada A., Zhao R., Milstien S., Zhou H., Spiegel S., Takabe K. (2012) Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 72, 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Price M. M., Oskeritzian C. A., Falanga Y. T., Harikumar K. B., Allegood J. C., Alvarez S. E., Conrad D., Ryan J. J., Milstien S., Spiegel S. (2013) A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J. Allergy Clin. Immunol. 131, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Allende M. L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C. A., Mandala S., Spiegel S., Proia R. L. (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 279, 52487–52492 [DOI] [PubMed] [Google Scholar]
- 43. Olivera A., Mizugishi K., Tikhonova A., Ciaccia L., Odom S., Proia R. L., Rivera J. (2007) The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 26, 287–297 [DOI] [PubMed] [Google Scholar]
- 44. Snider A. J., Kawamori T., Bradshaw S. G., Orr K. A., Gilkeson G. S., Hannun Y. A., Obeid L. M. (2009) A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 23, 143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishiuma T., Nishimura Y., Okada T., Kuramoto E., Kotani Y., Jahangeer S., Nakamura S. (2008) Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1085–L1093 [DOI] [PubMed] [Google Scholar]
- 46. Chiba Y., Takeuchi H., Sakai H., Misawa M. (2010) SKI-II, an inhibitor of sphingosine kinase, ameliorates antigen-induced bronchial smooth muscle hyperresponsiveness, but not airway inflammation, in mice. J. Pharmacol. Sci. 114, 304–310 [DOI] [PubMed] [Google Scholar]
- 47. Diesner S. C., Olivera A., Dillahunt S., Schultz C., Watzlawek T., Forster-Waldl E., Pollak A., Jensen-Jarolim E., Untersmayr E., Rivera J. (2012) Sphingosine-kinase 1 and 2 contribute to oral sensitization and effector phase in a mouse model of food allergy. Immunol. Lett. 141, 210–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hasbold J., Lyons A. B., Kehry M. R., Hodgkin P. D. (1998) Cell division number regulates IgG1 and IgE switching of B cells following stimulation by CD40 ligand and IL-4. Eur. J. Immunol. 28, 1040–1051 [DOI] [PubMed] [Google Scholar]
- 49. Elgueta R., Benson M. J., de Vries V. C., Wasiuk A., Guo Y., Noelle R. J. (2009) Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 229, 152–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li Z. W., Omori S. A., Labuda T., Karin M., Rickert R. C. (2003) IKK beta is required for peripheral B cell survival and proliferation. J. Immunol. 170, 4630–4637 [DOI] [PubMed] [Google Scholar]
- 51. Allende M. L., Tuymetova G., Lee B. G., Bonifacino E., Wu Y. P., Proia R. L. (2010) S1P1 receptor directs the release of immature B cells from bone marrow into blood. J. Exp. Med. 207, 1113–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Donovan E. E., Pelanda R., Torres R. M. (2010) S1P3 confers differential S1P-induced migration by autoreactive and non-autoreactive immature B cells and is required for normal B-cell development. Eur. J. Immunol. 40, 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gohda M., Kunisawa J., Miura F., Kagiyama Y., Kurashima Y., Higuchi M., Ishikawa I., Ogahara I., Kiyono H. (2008) Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer's patches for intestinal IgA responses. J. Immunol. 180, 5335–5343 [DOI] [PubMed] [Google Scholar]
- 54. Kabashima K., Haynes N. M., Xu Y., Nutt S. L., Allende M. L., Proia R. L., Cyster J. G. (2006) Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J. Exp. Med. 203, 2683–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cinamon G., Matloubian M., Lesneski M. J., Xu Y., Low C., Lu T., Proia R. L., Cyster J. G. (2004) Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat. Immunol. 5, 713–720 [DOI] [PubMed] [Google Scholar]
- 56. Grassme H., Jendrossek V., Bock J., Riehle A., Gulbins E. (2002) Ceramide-rich membrane rafts mediate CD40 clustering. J. Immunol. 168, 298–307 [DOI] [PubMed] [Google Scholar]
- 57. Henry B., Ziobro R., Becker K. A., Kolesnick R., Gulbins E. (2013) Acid sphingomyelinase. Handb. Exp. Pharmacol. 2013, 77–88 [DOI] [PubMed] [Google Scholar]