

Abstract
Uniquely among malaria parasites, Plasmodium falciparum-infected erythrocytes (iRBCs) develop membrane protrusions, known as knobs, where the parasite adhesion receptor P. falciparum erythrocyte membrane protein 1 (PfEMP1) clusters. Knob formation and the associated iRBC adherence to host endothelium are directly linked to the severity of malaria and are functional manifestations of protein export from the parasite to the iRBC. A family of exported proteins featuring Plasmodium helical interspersed subtelomeric (PHIST) domains has attracted attention, with members being implicated in host-parasite protein interactions and differentially regulated in severe disease and among parasite isolates. Here, we show that PHIST member PFE1605w binds the PfEMP1 intracellular segment directly with Kd = 5 ± 0.6 μM, comigrates with PfEMP1 during export, and locates in knobs. PHIST variants that do not locate in knobs (MAL8P1.4) or bind PfEMP1 30 times more weakly (PFI1780w) used as controls did not display the same pattern. We resolved the first crystallographic structure of a PHIST protein and derived a partial model of the PHIST-PfEMP1 interaction from nuclear magnetic resonance. We propose that PFE1605w reinforces the PfEMP1-cytoskeletal connection in knobs and discuss the possible role of PHIST proteins as interaction hubs in the parasite exportome.—Oberli, A., Slater, L. M., Cutts, E., Brand, F., Mundwiler-Pachlatko, E., Rusch, S., Masik, M. F. G., Erat, M. C., Beck, H.-P., Vakonakis, I. A Plasmodium falciparum PHIST protein binds the virulence factor PfEMP1 and comigrates to knobs on the host cell surface.
Keywords: cytoadherence, exported proteins, interactions, malaria, protein structure
During the intraerythrocytic cycle, Plasmodium falciparum completely refurbishes the human erythrocyte by establishing membranous networks and new permeation pathways (1). This refurbishment involves export of hundreds of proteins into the cytosol of the infected red blood cell (iRBC) and dramatic changes in the host cell membrane. The infected cell increases its permeability (2) and becomes more rigid (3, 4), and electron-dense surface protrusions called knobs form, conveying cytoadherence of mature iRBCs to the endothelial lining (5). The major parasite virulence factor P. falciparum erythrocyte membrane protein 1 (PfEMP1) is embedded in these knob structures through a transmembrane helix and comprises a highly variable ectodomain and a semiconserved intracellular segment [acidic terminal segment (ATS)] anchoring the molecule to the host cell (6). The presentation of PfEMP1 on the host cell surface is thought to be a major cause of pathological changes (7).
The importance of parasite-exported proteins in these host cell modifications has been acknowledged, but little is known about their function and interactions. Transport of proteins through the parasitophorous vacuole into the host cell has been shown to be facilitated by a short amino-terminal sequence termed PEXEL or VTS (8, 9). The majority of exported proteins carry this PEXEL motif, which allowed the establishment of the P. falciparum exportome with ∼400 members (8). A smaller but unknown number of parasite proteins are exported despite lacking an identifiable motif (10). In both groups, only a few proteins have been studied in detail; these include some constituents of the translocon (11) and, in particular, knob components such as PfEMP1 (12) and others (13–16). More recently, PEXEL-negative proteins localizing in membrane structures formed in the host erythrocyte (Maurer's clefts), such as SBP1 (17) and MAHRP1 (18), have also been studied in greater detail. Maier et al. (4) used these predictions and identified through a knockout strategy a number of proteins that were essential for the transport of PfEMP1 to the surface, including members of the Plasmodium helical interspersed subtelomeric (PHIST) family. PHIST proteins comprise 72 variants in the P. falciparum 3D7 reference genome (19) and are organized into 3 subfamilies according to their species distribution: PHISTa proteins are entirely P. falciparum specific, PHISTb proteins are present in Plasmodium vivax and Plasmodium knowlesi but have extensively expanded in P. falciparum, and PHISTc proteins are shared between P. falciparum and P. vivax and appear as single-copy genes in the Plasmodium berghei lineage (19).
To date, no molecular function has been assigned to any of the PHIST proteins despite their wide distribution within infected cells. Yeast 2-hybrid analysis identified PHIST proteins as putative interactors with SBP1 (20) and erythrocyte band 4.1 (21). Transcriptome data suggest differences in PHIST expression during the parasite life cycle (19, 22–24) and among parasite isolates (25) and up-regulation of a specific PHIST member in parasites targeting the brain endothelium (26). Proteome data show a consistent presence of PHIST proteins in iRBC membrane fractions (27–29). Recently, a member of the PHIST family was identified within the Maurer's clefts (4); there is also evidence for a PHIST protein in J dots, and very recently PfPTP2, a PHISTb protein, was shown to be present in exosomes, a newly discovered means of P. falciparum communication (30). The PHISTc protein PFI1780w (PF3D7_0936800) has been detected in detergent-resistant membrane fractions (31), and we identified the same variant as an interaction partner with the ATS of PfEMP1, demonstrating the first direct association for a PHIST protein (32).
In this study, we show that another PHIST variant, PFE1605w (PF3D7_0532400), directly binds ATSs with higher affinity than PFI1780w. Both PFI1780w and PFE1605w were shown to localize to the iRBC membrane, but only PFE1605w is transported similar to PfEMP1 in time and space, and it locates specifically to knobs. Finally, we elucidate the first PHIST crystallographic structure from PFI1780w and suggest a partial model for the PHIST-ATS complex. This is the first functional information for any PHIST protein and provides evidence that PHIST proteins might be involved in PfEMP1 function and certainly are constituents of knobs.
MATERIALS AND METHODS
Cloning and protein production
PHIST domains
P. falciparum PFI1780w residues 85–247 or 98–247, PFD1170c residues 132–309, and MAL8P1.163 residues 131–284 were cloned in vector pGEX-6P-2 (GE Healthcare Life Sciences, Glattbrugg, Switzerland), and transformed into Escherichia coli BL21(DE3). PFE1605w residues 122–335 and MAL8P1.4 residues 310–456 were cloned in pOPINF vector (Oxford Protein Production Facility, Harwell, UK), and transformed into E. coli Rosetta2pLacI. Recombinant MAL8P1.4 for antibody production was derived from a full-length codon-optimized gene cloned into psCodon (Eurogentec, Seraing, Belgium) and expressed in the CherryCodon system (Eurogentec). Cells were grown in Luria-Bertani medium, supplemented with 1% (w/v) glucose for PFE1605w and MAL8P1.4. For nuclear magnetic resonance (NMR) samples, cells were grown in M9 medium supplemented with 15NH4Cl and 13C6 d-glucose, and after protein induction were grown for 16 h at 18°C .
Cells were resuspended in phosphate-buffered saline (PBS; 150 mM NaCl and 20 mM Na2HPO4, pH 7.4) and lysed by sonication; lysates were spun at 24,000 g for 30 min. Lysate supernatants of PHIST domains cloned in pGEX-6P-2 were incubated with glutathione Sepharose resin (GE Healthcare LifeSciences) equilibrated in PBS, and proteins were eluted in a 50 mM Tris-Cl (pH 7.8), 12 mM reduced glutathione buffer. The glutathione S-transferase (GST) tag was removed by 3C protease cleavage, followed by buffer exchange to PBS using a Sephadex G-20 column (GE Healthcare LifeSciences). GST was retained by reverse glutathione Sepharose affinity.
Lysate supernatants of pOPNIF-cloned PHIST domains were applied to a Talon HiTrap column (GE Healthcare LifeSciences) equilibrated in 20 mM Na2HPO4 (pH 7.4) and 300 mM NaCl and eluted with a gradient to an imidazole-containing buffer (20 mM Na2HPO4, pH 7.4; 300 mM NaCl; and 500 mM imidazole). For PFE1605w, the His6 tag was removed by 3C protease cleavage during dialysis in 50 mM Tris-Cl (pH 7.5), 150 mM NaCl, and 2 mM dithiothreitol (DTT), followed by dialysis in 20 mM 2-(N-morpholino)ethanesulfonic acid (MES; pH 6.0), 50 mM NaCl, and 2 mM DTT. PFE1605w was then applied to an SP ion exchange column (GE Healthcare LifeSciences) equilibrated in the dialysis buffer and eluted with a NaCl gradient (20 mM MES, pH 6.0; 1 M NaCl, and 2 mM DTT). For MAL8P1.4, the His6 tag was removed by 3C protease cleavage, followed by dialysis in 20 mM Na2HPO4 (pH 7.0), 150 mM NaCl, and 2 mM DTT.
Final purification of all PHIST domains was performed by size-exclusion chromatography over a Superdex S75 column (GE Healthcare LifeSciences) equilibrated in PBS supplemented with 1 mM DTT or buffer A (50 mM NaCl; 20 mM Na2HPO4, pH 7.0; and 1 mM DTT).
PfEMP1 intracellular segments
The cloning and protein production of PfEMP1 intracellular segments, both full-length segments and fragments, and fluorescent labeling of the PF08_0141 ATS were described earlier (32). A further 5 fluorescent-labeled full-length ATS variants were produced in an analogous manner by substituting single amino acids for cysteines; these are PFF0010 (G156C), PFB1055c (Q161C), PFC1120c (Q182C), PF08_0103 (H154C), and PFF0845c (H159C).
Biophysical characterization
Protein identity was confirmed by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Unless otherwise noted, all biophysical experiments were performed in buffer A, except for analytical ultracentrifugation (AUC) experiments in which 1 mM tris(2-carboxyethyl)phosphine was used instead of DTT. Fluorescence polarization measurements were recorded at 20°C with a 5-FAM label using a PHERAstar FS fluorimeter (λex=485 nm, λem=520 nm; BMG Labtech, Ortenberg, Germany). Differences in fluorescence polarization were fit using a single binding model in the program Origin (OriginLab, Northampton, MA, USA). AUC velocity experiments were performed on 25 μM protein samples using an Optima XL-I analytical ultracentrifuge (Beckman Coulter, Fullerton, CA, USA). Sedimentation velocities were recorded by measuring absorbance at 280 nm, with 200 scans every 4 min at 10°C and 35,000 rpm. Data were processed using SEDPHIT (33). The protein partial specific volume was calculated from the amino acid sequence.
Crystallization and structure determination
Crystals were obtained using the sitting drop vapor-diffusion technique at 20°C. A Mosquito robot (TTP LabTech, Melbourn, UK) was used to set up 200-nl sized drops with a 1:1 ratio of protein to mother liquor. PFI1780w residues 85–247 at a concentration of 4.0 mg/ml were mixed with 0.1 M sodium acetate (pH 4.6) and 2.0 M NaCl buffer. Crystals developed in 7 d were cryoprotected by a brief incubation in mother liquor supplemented with 22.5% (v/v) glycerol, flash-cooled in liquid nitrogen, and diffracted up to 2.35 Å at the Diamond Light Source (Harwell, UK), beamline I04. The space group was determined to be P3121 with 2 molecules/asymmetric unit. For phasing experiments, the crystals were incubated with 250 mM 5-amino-2,4,6-triiodoisophthalic acid (Hampton Research, Aliso Viejo, CA, USA) for 5 min before cooling.
Crystallographic data were integrated in XDS (34) and scaled in SCALA (35). Phase information for PFI1780w was obtained from a 2.44-Å resolution data set collected at a wavelength of 1.6531 Å using the Diamond Light Source, beamline I04. Phasing by single-wavelength anomalous diffraction was performed using PHENIX.autosol (36), which located and refined 27 iodine atoms to produce a density map with initial figure of merit of 0.51. Initial model building was done with PHENIX.autosol (247 residues built and 184 identified). Iterative model building was performed with COOT (37) and refinement against the native 2.35 Å data was performed with BUSTER 2.10 (38).
Crystallographic data processing and refinement statistics are provided in Supplemental Table S1. Model quality was assessed by MolProbity (39). For graphical representation, we used PyMOL (40). The model and associated data have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (http://www.rcsb.org) under accession number 4JLE.
ATS structure prediction
CS-Rosetta (41) structure prediction was performed on residues 306–346 of ATSs using backbone 15N, 1HN, and 13C′ chemical shifts recorded in the presence of the PFI1780w PHIST domain and extrapolated to complex saturation. Data were processed by TALOS+ (42) before input in CS-Rosetta for fragment selection and generation of 12,800 models. Model superposition with a 3-Å root mean square deviation (RMSD) cutoff yielded large clusters of which the top 3 had 1313, 943, and 719 members. These clusters showed characteristic β-sheet structures but incomplete convergence due to flexible loops between β strands. Superposition using the most stable secondary structure elements (residues 311–314, 320–324, and 329–333) and 1-Å RMSD cutoff resulted in model convergence as judged by the funnel-shaped plot of model score vs. pairwise RMSD.
NMR
Sequence-specific resonance assignments of ATSs have been reported previously (32). Assignments of PFI1780w were performed at 37°C using triple-resonance experiments on a 600-MHz Avance III spectrometer (Bruker, Newark, DE, USA) with a cryogenic probehead. NMR samples for assignments consisted of 0.2 mM 13C/15N-enriched PFI1780w residues 98–247 in buffer A (pH 6.5) supplemented with 0.1 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid, 0.02% (w/v) NaN3, and 5% (v/v) D2O. The limited sample concentration and stability under these conditions (typical lifetime of ∼2 d) necessitated the use of multiple samples to obtain a sufficient signal/noise ratio. Assignments have been deposited in BioMagResBank (University of Wisconsin, Madison, WI, USA; http://www.bmrb.wisc.edu/) under accession number 19719.
Perturbations in 1H and 15N chemical shifts were combined as Δδ(1H,15N) using the formula
Similar perturbations of carbonyl 13C resonances were expressed as Δδ(13C) = δ(13C)free − δ(13C)complex.
Plasmodium culture, transfection, and protein analysis
Full-length PFI1780w and PFE1605w inserts were C-terminally fused either to green fluorescent protein (GFP) into pARL1a-GFP (kindly provided by T. Spielmann, Bernhard Nocht Institut, Hamburg, Germany; ref. 43) or C-terminally to hemagglutinin (HA) into pBcamR_3xHA (44). P. falciparum strain 3D7 was cultured in human 0+ erythrocytes according to standard procedures (45). Transfected parasites were drug selected with either 10 nM WR99210 (Jacobs Pharmaceuticals, Cologne, Germany) or 2.5 mg/ml blasticidin (Life Technologies, Zug, Switzerland).
Parasite proteins were obtained through saponin lysis of synchronized parasites (5–10% parasitemia) after 2 sorbitol treatments within 4 h and Percoll purified after 30 h. Parasite aliquots were taken every 8 h. Samples were run on a 12.5% (w/v) SDS-PAGE column with complete protease inhibitor cocktail (Roche, Rozkreuz, Switzerland) and transferred to a nitrocellulose membrane (Hybond-C Extra; GE Healthcare LifeSciences). Antibodies were diluted in 5% (v/v) milk-PBS: mouse monoclonal anti-GFP (1:1000; Roche), rabbit anti-HA (1:20; Invitrogen, Zug, Switzerland), rabbit anti-MAHRP1 (1:5000), rabbit anti-MAHRP2 (1:1000), mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:20,000), mouse anti-PFE1605w (1:500), mouse anti-PFI1780w (1:500), and mouse anti-MAL8P1.4 (1:500). Binding was made visible by chemiluminescence (SuperSignal West Pico, Thermo Scientific, Reinach, Switzerland). Parasite protein solubility was analyzed as described previously (46).
Immunofluorescence assay (IFA) and live cell imaging
Blood smears of infected parasite cultures were fixed in 100% acetone for 30 min (47) and blocked with 3% (v/w) bovine serum albumin. Primary antibodies, rabbit anti-MAHRP1 (1:500), mouse anti-PFI1780w (1:100), mouse anti-PFE1605w (1:100), mouse anti-MAL8P1.4 (1:200), mouse anti-GFP (1:100, Roche), and mouse anti-ATS (1:50), were incubated for 1 h (48). Secondary antibodies (goat anti-mouse Alexa 488, goat anti-mouse Alexa 594, and goat anti-rabbit Alexa 594; Invitrogen) were incubated with 1 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI; Roche) for 1 h at 1:200 dilution. Alternatively, iRBCs were fixed with 4% paraformaldehyde (PFA)-0.01% glutaraldehyde and permeabilized with 0.1% Triton X-100. Slides were viewed with a Zeiss LSM 700 confocal microscope (Carl Zeiss GmbH, Jena, Germany), with a ×63 oil-immersion lens (1.4 numerical aperture).
Transgenic parasites expressing GFP fusion proteins were imaged as described previously (49). Live parasites were imaged with a Leica DM 5000B fluorescence microscope using a ×100 oil immersion lens (1.4 numerical aperture) with an attached Leica DFC300FX camera and Leica Application Suite software (Leica Microsystems, Heerbrugg, Switzerland).
Immunoelectron microscopy
PFE1605w-HA and PFI1780w-GFP transfected mature parasites were purified by Percoll density gradient, fixed in 2% PFA-0.2% glutaraldehyde in phosphate buffer, and prepared according to Tokuyasu (50). Ultrathin sections (70−90 nm) prepared on an FC7/UC7-ultramicrotome (Leica Microsystems) at −120°C were immunogold-labeled with rabbit anti-HA (1:20; Invitrogen) or rabbit anti-GFP (1:20; Abcam, Cambridge, UK) antibodies and 5 nM protein A-gold (1:70; UMC, Utrecht, The Netherlands). Sections were stained with 4% uranyl acetate-methylcellulose (1:9) and examined with a transmission electron microscope (CM10 or CM100; Philips, Eindhoven, The Netherlands) at 80 kV.
RESULTS
PFE1605w interacts with the PfEMP1 intracellular domain
Previously, we showed that intracellular segments (referred to as ATSs) from members of the PfEMP1 family comprise a stably folded core and 3 flexible regions (32). We demonstrated that the PHISTc protein PFI1780w interacts with moderate strength (Kd∼150 μM) with the ATS of PfEMP1 variant PF08_0141. However, it was not clear whether PFI1780w is a physiological partner of PfEMP1. Thus, we produced PHIST domains from proteins that had been reported as important for cytoadherence of iRBCs (PFD1170c; ref. 4), that are adjacent on the genome to the locus of PfEMP1 variant PF08_0141 (MAL8P1.163), or, alternatively, that were identified in proteomic studies on tethers (PFE1605w) and tested them for interactions with the ATS. As a negative control, we produced a PHIST member that was shown not to localize to the iRBC membrane and thus would not be expected to associate with the ATS (MAL8P1.4). Polarization experiments showed that PFE1605w, a PHISTb member, bound fluorescently labeled ATS PF08_0141 with ∼30-fold higher affinity (Kd=5±0.6 μM) than PFI1780w (Fig. 1A). NMR experiments suggest that the PHIST interaction occurs at the C terminus of the ATS (see below), and polarization experiments using a fluorescently labeled ATS C-terminal construct (residues 293–392) showed that this fragment is sufficient for strong PFE1605w binding (Fig. 1D).
Figure 1.

PHIST domains directly interact with ATS. A) Fluorescence polarization titrations of labeled ATS PF08_0141 with PHIST domains. Error bars derive from 5 measurements. Solid lines correspond to fits to single-site association models when possible. B, C) Similar titrations with different labeled PfEMP1 intracellular domains and PFE1605w (B) or PFI1780w (C). Binding by PfEMP1 variant PFF0845c was weak and could not be fit. In panel B, the fits of ATS variants PFF0010w and PF08_0103 overlap closely. D) Titrations of ATS PF08_0141 full-length or C-terminal fragment (residues 293–392) with PFE1605w.
To test whether different PfEMP1 members retain the PFE1605w interaction, we produced fluorescently labeled ATSs from PfEMP1 variants PFF0010, PFB1055c, PFC1120c, PF08_0103, and PFF0845c, which were selected on the basis of sequence divergence (32). Intriguingly, polarization experiments with PFE1605w showed up to 25-fold difference in affinities (Kd range, 2.7–67 μM; Fig. 1B) across ATS variants. Similar experiments with PFI1780w showed only ∼2.5-fold different affinities between ATS members (Fig. 1C). PFF0845c, a C-terminally truncated ATS variant (Supplemental Fig. S1A), showed little affinity to either PFE1605w or PFI1780w (Fig. 1B, C). Whether these differences reflect closer associations of PFE1605w with particular PfEMP1 members in vivo remains to be studied; however, time course experiments did suggest a closer association of PFE1605w with PfEMP1 than PFI1780w during transport.
PFI1780w and PFE1605w are membrane associated and exported to the iRBC membrane
To test whether these PHIST proteins are exported and to determine their subcellular localizations, they were C-terminally GFP tagged and episomally expressed in 3D7 parasites under the control of the crt promoter. The integrity of the GFP-fusion proteins was shown on Western blots (Fig. 2A). The full-length PFI1780w-GFP was exported to the iRBC cytosol similarly to the previously reported GFP-tagged N terminus (19), but full-length PFI1780w-GFP additionally revealed fluorescence at the periphery of iRBCs (Fig. 2B; top panel), suggesting a localization close or adjacent to the erythrocyte membrane (Fig. 2B; panel 2). Immunofluorescent 3-dimensional reconstructions of fixed iRBCs with PFI1780w-GFP-expressing parasites showed focal fluorescence in parasite cytosol and uniform fluorescence around the biconcave rim of the iRBC (Fig. 2C), confirming this location. From all available GFP-tagged PHIST proteins only PFE1605w-GFP gave a similar fluorescent pattern except that the rim-like fluorescence was observed at discrete foci instead of a uniform signal (Fig. 2B, panels 4 and 5), indicating that both PFI1780w and PFE1605w were transported close to the iRBC membrane.
Figure 2.

PHIST domains are exported to the iRBC membrane. A) Western blots of extracts from cell lines used in this study. All extracts are derived from saponin-released parasites, and the origin is indicated on top. Decoration of the same Western blot with GAPDH as loading control is shown at bottom. B) Live cell imaging of 3D7 parasites expressing PFI1780w-GFP and PFE1605w-GFP. For panels 2 and 5, the focal plane of the GFP signal was set on the surface of the iRBC, and the DAPI/differential interference contrast (DIC) microscopy signal was kept on the previous focal plane. Nuclei were stained with DAPI. Scale bar = 2 μm. Panel 3 shows a rare schizont with already ruptured erythrocyte membrane. C) Confocal immunofluorescence analysis of 3D7 parasite expressing PFI1780w-GFP. Merge image of GFP/DAPI/DIC channels representing stack 32 of 69 in total. Scale bar = 1 μm. D) Western blot of protein fractions of solubility assays of 3D7 parasites expressing PFI1780w-GFP and PFE1605w-GFP. Lane 1, soluble proteins; lane 2, peripheral membrane proteins; lane 3, Triton X-100 extract; lane 4, insoluble pellet.
Next, we tested the solubility of these 2 PHIST proteins (ref. 46 and Fig. 2D). The soluble parasite protein GAPDH was found as expected in the supernatant after hypotonic lysis, and MAHRP1, an integral Maurer's cleft membrane protein (51), was detected in the Triton X-100 supernatant. Both PFI1780w-GFP and PFE1605w-GFP lack a predicted transmembrane domain and were shown to be membrane-associated proteins by solubilization in sodium carbonate similar to MAHRP2, a membrane-associated protein localizing to Maurer's cleft tethers (46). This result was further confirmed by the fluorescent labeling of the iRBC membrane of a ruptured schizont expressing PFI1780w-GFP (Fig. 2B, panel 3).
PFI1780w localizes underneath the iRBC membrane and PFE1605w to knob structures
The subcellular localization of both PHIST proteins was shown by postembedding immunoelectron microscopy. Gold labeling of PFI1780w-GFP was found in proximity to the iRBC membrane but was clearly absent from knobs (Fig. 3A) and frequently at Maurer's clefts (Fig. 3B). In contrast, PFE1605w-3xHA (Fig. 3C) and PFE1605w-GFP were clearly localized in knobs, and in trophozoite stage parasites, PFE1605w-GFP was also frequently found in Maurer's clefts (Fig. 3D), suggesting that it localizes underneath the iRBC membrane, whereas PFE1605w localizes to knobs, but both proteins seem to be transiently transported through the Maurer's clefts.
Figure 3.

PFE1605w localizes to knobs. Shown here are postembedding immunoelectron microscopy images of iRBCs expressing PFI1780w-GFP (A, B), PFE1605w-HA (C), and PFE1605w-GFP (D). Insets: enlarged views of boxed sections. White arrows, 5 nM gold. Knobs (K) and Maurer's clefts (MC) are labeled. Scale bars = 200 nm (A); 500 nm (C); 250 nm (B, D).
Immunofluorescence reveals a similar trafficking pathway and timing of export for PFE1605w and PfEMP1
Specific antisera against PFI1780w (aa 80–280) recognized a single band of ∼45 kDa in agreement with its predicted mass of 45.5 kDa (Fig. 4A), and antisera against PFE1605w (aa 122–335) recognized a band of ∼55 kDa and a faint band of 50 kDa. Western blotting using samples from 6 time points of the intraerythrocytic cycle showed that both proteins were present throughout the cycle, which is in agreement with their transcription profile (52, 53). Both proteins were maximally expressed in trophozoite stages (Fig. 4A).
Figure 4.

PFE1605w is cotranslocated with PfEMP1. A) Western blot with extracts from stage-specific parasites (hpi indicated for each lane). GAPDH was used as loading control. B−D) Confocal immunofluorescence analysis of tightly synchronized 3D7 parasites. Samples were collected at 8-h intervals and stained with antibodies recognizing PFE1605w (B), PFI1780w (C), or ATS (D) and costained with DAPI. Scale bars = 2 μm. DIC, differential interference contrast microscopy.
We performed IFAs to visualize expression and export of PFE1605w/PFI1780w throughout the intraerythrocytic cycle using tightly synchronized parasites at 6 time points (Fig. 4B, C) and compared the timing of export of PfEMP1 using anti-ATS antibodies (Fig. 4D and ref. 48). PFE1605w was first observed at ∼0–8 h postinfection (hpi) in a “necklace of beads” pattern at the parasite surface (Fig. 4B; top panel) and in young trophozoites (16–24 hpi) most fluorescence was observed in the parasite with few fluorescent foci beyond the parasite's confines. After 24–32 hpi, the number of fluorescent foci in the iRBC cytosol increased, and faint fluorescence was visible at the iRBC membrane. Schizont stage parasites showed faint fluorescent dots at the iRBC membrane, similar to those seen with live cell images of PFE1605w-GFP (Fig. 2B). Bright fluorescent foci indicated that PFE1605w is exported via Maurer's clefts to the iRBC membrane and knobs.
PfEMP1 is known to display a necklace of beads pattern at the parasite surface at ∼8–11 hpi (54), which was confirmed (Fig. 4D) and was similar to the pattern observed for PFE1605w (Fig. 4B). Both proteins transiently associated with Maurer's clefts at ∼16–24 hpi (ref. 54 and Fig. 4B, D) before being transferred to the iRBC membrane, suggesting cotransport of PFE1605w with PfEMP1.
In contrast to PFE1605w, PFI1780w was found within the parasite cytosol until ∼24–32 hpi with limited focal fluorescence in the iRBC cytosol (Fig. 4C). In schizonts, PFI1780w was exported to the iRBC surface as shown in live cell imaging (Fig. 2B) with no distinct intermediate locations, although a few fluorescent foci and immunoelectron microscopy data (Fig. 3B) suggest that Maurer's clefts are intermediate transport compartments.
To confirm the transient location of PFE1605w at Maurer's clefts, we colocalized the protein with MAHRP1 in ring, trophozoite, and schizont stage parasites. While in ring stages, MAHRP1 already appeared in Maurer's clefts and PFE1605w exclusively localized within the parasite cytosol (Supplemental Fig. S2A, top panel). In young trophozoites PFE1605w and MAHRP1 colocalized to Maurer's clefts with a partial overlap of signals, suggesting subdomains in Maurer's clefts as described previously for MAHRP1 and PfEMP1 (54). This is also evidence for PFE1605w transport to already formed Maurer's clefts and proof that cargo arrives at clefts independent of their formation as reported previously (54, 55). At later stages, PFE1605w dissociates from Maurer's clefts and is transported to the iRBC membrane or knobs (Supplemental Fig. S2A, panel 3).
In contrast to PFE1605w and PFI1780w, MAL8P1.4, which does not interact with the ATS domain (Fig. 1A), showed persistent Maurer's cleft localization in IFAs (Supplemental Fig. S2B).
PHIST domains compete for a conserved epitope at the ATS C terminus
Despite our efforts, the PFE1605w PHIST domain evaded crystallization alone or in complex with ATS fragments and was unsuitable for extensive NMR analysis due to limited solubility. Thus, we turned to PFI1780w, which also interacts with ATS, to gain information on the PHIST-ATS complex. To delineate the specific PHIST-ATS interaction site, we performed NMR titrations of 13C/15N-enriched ATS PF08_0141 (32) with the unenriched PFI1780w PHIST domain (residues 98–247). As shown in Fig. 5A−C, the largest chemical shift perturbations, indicative of complex formation, span a wide region at the flexible C terminus of the ATS. Although sequence similarity across the C terminus of PfEMP1 intracellular segments is relatively low (32), ∼75% of the amino acids most perturbed on PFI1780w binding are conserved (I311, I313, I330, L331, D336, I338, Y339, Y340, and W391) or conservatively substituted (T329, D332, E335, I372, and V390) in the majority of members of the PfEMP1 family (Supplemental Fig. S1A). We were able to perform similar experiments on 13C/15N-enriched ATS with unenriched PFE1605w under dilute conditions and recorded the loss of ATS resonance intensity in the presence of PFE1605w (Supplemental Fig. S3A). Mapping interaction interfaces using this type of information is less sequence specific than that with the chemical shift perturbations observed with PFI1780w, as loss of resonance intensity may reflect interactions by a specific residue or its neighbors. Nonetheless, the data (Supplemental Fig. S3B) suggest that PFE1605w affects a wide span of the ATS C terminus that includes the PFI1780w interaction epitope (Fig. 5B, C). In addition, PFE1605w makes additional contacts with the ATS C terminus that span residues 303–309 and 353–362; these additional contacts may contribute to the higher ATS affinity of this PHIST variant.
Figure 5.

NMR analysis of the PFI1780w-ATS interaction. A) Combined perturbations of backbone amide nuclei of 0.1 mM 13C/15N-enriched ATS on titration with 0.2 mM unenriched PFI1780w PHIST domain. Data were recorded at 10°C and pH 7.0. Schematic representation of ATS structure (32) is shown at top. B) Expansion of panel A for the ATS C-terminal section. Dashed line denotes the level of average perturbation plus 1 sd; residues with perturbations above this line are considered as significantly affected. C) Carbonyl perturbations of the same ATS region as in panel B. Residues judged as significantly affected in either amide or carbonyl perturbations are colored based on their conservation: red if identical or orange if conservatively substituted in ≥75% of PfEMP1 members in P. falciparum isolate 3D7. D) 15N-heteronuclear single quantum coherence spectra overlay from 0.1 mM 15N-enriched PFI1780w PHIST domain alone (red) or in the presence of 0.2 mM unenriched ATS C-terminal fragment (residues 309–392, green). Spectra were recorded at 37°C and pH 6.5. Perturbations in peak positions show formation of the PFI1780w-ATS complex. E) Expansion of overlaid spectra from 50 μM PFI1780w PHIST domain alone (red), in the presence of 50 μM ATS residues 309–392 (black), after addition of 0.6× stoichiometric ratio of unenriched PFE1605w PHIST domain (blue), or after addition of 1.2× stoichiometric ratio of PFE1605w (green). PFE1605w addition reverses PFI1780w perturbations caused by ATS binding, suggesting that the 2 PHIST proteins compete for the same ATS binding site. F) Per-residue combined perturbations of backbone amide 1H and 15N nuclei from the PFI1780w spectra shown in panel D. Dashed line identifies significantly perturbed residues as in panel B.
To confirm that the 2 PHIST proteins compete for the same ATS interaction epitope, we performed NMR experiments using 15N-enriched PFI1780w and unenriched ATS C terminus and PFE1605w (Fig. 5D). Perturbations on PFI1780w resonances caused by ATS are reversed by PFE1605w (Fig. 5E), which supports the competition between these PHIST domains.
Structure of the PFI1780w PHIST domain
We obtained crystals of a PFI1780w fragment spanning residues 85–247 and solved the resulting structure to 2.35-Å resolution (Supplemental Table S1). The final model showed 2 protein chains forming a tight helix-swapped dimer in the crystal (Fig. 6A); however, AUC velocity experiments showed that PFI1780w is monomeric in solution under physiological conditions (Fig. 6B). Thus, we concluded that the dimer is a crystallization artifact and consider only the monomeric unit of the PHIST domain.
Figure 6.

Structure of PFI1780w. A) Crystallographic model of the PFI1780w PHIST domain, showing the dimer (blue and red chains). Residues 85–97 (purple) are visible in only one polypeptide chain. B) AUC velocity experiment of PFI1780w residues 85–247. Data analysis yielded a narrow distribution at 1.65 S, with best friction ratio of 1.61 and 19,919 Da corresponding molecular mass. C) Monomeric model of the PFI1780w PHIST domain. D) Twist of the triple-helix bundle for PFI1780w and spectrin (RCSB 3KBT). Both models are visualized with their C termini below the page.
PFI1780w residues 98–247 adopted a highly α-helical configuration (Fig. 6C), in agreement with structure predictions (19) and circular dichroism data (32). A short initial helix (α1) is followed by an antiparallel bundle of 3 long helices (α2−α4), which are up to 51 aa in length. The PFI1780w triple-helical bundle is of length (∼6.5 nm) comparable to that of a spectrin repeat; however, in contrast to spectrin, it adopts a right-handed twist (Fig. 6D). PHIST domains show remarkable sequence divergence, and PFI1780w belongs to the most variable PHISTc subtype of this family (19). Alignment of the PFI1780w PHIST domain with its 5 closest relatives showed just 4% identical and 20.7% conservatively substituted residues (Supplemental Fig. S1B). The vast majority of these conserved amino acids, including all tryptophans characteristic of the PHIST family (19), are involved in the protein hydrophobic core and do not form a continuous surface area. Nonetheless, the PFI1780w structural model is sufficient to model with 98–100% confidence the domain structures for all members of this family using automated prediction pipelines.
Structural analysis of the PHIST-ATS interaction
The chemical shift perturbations of PFI1780w on ATS binding allowed us to map the binding site on the PHIST structure (Fig. 5F). The most significant changes localized at the middle of the PFI1780w triple-helical bundle, and, in particular, over a continuous surface on helices α2 and α3 (Fig. 7C and Supplemental Fig. S1B). The amino acids forming this ATS-binding interface (K136, E138, E139, H167, F168, and Q171) are not conserved between PFI1780w and PFE1605w, which probably contributes to the different ATS affinities of these proteins. Nonetheless, because PFI1780w and PFE1605w compete for the same ATS epitope, we reasoned that an analysis of the PFI1780w-ATS interaction may provide a partial model of the high-affinity complex.
Figure 7.

Structure prediction for the ATS C terminus and analysis of the PFI1780w-ATS interaction. A) Plot of CS-Rosetta model score vs. backbone RMSD. Models were predicted for a C-terminal fragment of ATS spanning residues 306–346, using chemical shift restraints that correspond to the PFI1780w-complexed state of ATS. A characteristic funnel shape in this plot suggests that the calculation converged to a single subset of solutions. B) Schematic representation of a low-energy ATS model from panel A. C) Opposite views of the PFI1780w PHIST domain surface, with areas significantly affected by ATS binding in NMR titrations shown in red. D) Proposed mode of PFI1780w-ATS binding based on the predicted structure of ATS residues 306–346.
The number of ATS-binding residues on PFI1780w is remarkably small compared with the broad span of perturbations observed on the ATS C terminus (Fig. 5A). In particular, the continuous surface identified on PFI1780w α2–α3 can only accommodate approximately 6 aa in an extended conformation. To understand how these observations relate, we predicted the transient structure adopted by the ATS C-terminal epitope (residues 306–346) on PFI1780w binding using CS-Rosetta (41). As shown in Fig. 7A, B, the structure prediction converged to a single β sheet formed by ATS residues 311–314 (β1), 320–324 (β2), 328–334 (β3), and 337–343 (β4). Formation of a single β sheet by ATS residues 306–346 may explain the widespread chemical shift changes seen in this construct on PFI1780w binding, as nearly all residues would transit from a random coil to an extended conformation. Yet, at the same time, the single ATS β sheet could provide a relatively narrow interaction interface with PFI1780w along the β-sheet edge. Thus, we propose that an ATS associates along one β-sheet edge with PFI1780w (Fig. 7D).
DISCUSSION
The extensive remodeling of host erythrocytes by invading Plasmodia is an impressive feat of biological engineering necessary for the parasites to grow, replicate, and evade the immune system; thus, it has clear implications for the human host. The unique alterations induced by P. falciparum resulting in iRBC cytoadherence are linked to disease severity (7). Yet our understanding of this process is largely incomplete, especially at a mechanistic level. Although iRBC remodeling involves hundreds of proteins, few interactions and even fewer structures have been examined in detail. Here, we present the first structural-functional insights on PHIST proteins, which comprise a large subset of the P. falciparum exportome (4, 19). Notably, our study suggests strong links between a specific PHIST member and PfEMP1, the parasite receptor responsible for cytoadherence.
PHIST domains, as demonstrated here by PFI1780w (Fig. 6), are characterized by a relatively simple α-helical bundle structure, a type of protein fold that can accommodate divergent sequences and, thus, present variable surface residues to accommodate different binding requirements. The PHIST family in Plasmodia probably serves precisely the role of a simple, easy-to-manipulate type of protein that can act to bridge or structurally support other components. PHIST proteins seem to have a variety of functions in the parasite (4, 20, 21, 26, 30), but this might reflect their role as interaction hubs that can bind flexible protein segments, as shown here for ATS (Fig. 7). Thus far, the molecular evidence of PHIST-mediated interactions presented here and elsewhere (20, 21, 56) involves only ∼10% of all family members, and high-throughput molecular methods may provide a clearer picture of the PHIST interactome.
What is the relationship of PHIST proteins with PfEMP1? In a limited set of PHIST variants, we identified 2 members, PFI1780w and PFE1605w, that bound the PfEMP1 intracellular segment (Fig. 1A); it is likely that additional PfEMP1-binding PHIST domains can be found among the 72 members of this family. Both PHIST proteins described here interact with the same PfEMP1 epitope but with significantly different affinities. Furthermore, substantial variation in binding strength was observed, depending on specific PHIST/PfEMP1 combinations (Fig. 1B), which leads us to propose that individual PHIST variants might have been optimized for different groups of PfEMP1 molecules. This proposal is strongly supported by transcriptomic evidence of a specific PHISTa member (PF14_0472) up-regulated in parasites expressing PfEMP1 variants binding human brain endothelial cells (26). Similarly, we observed that both PfEMP1-binding PHIST proteins localized to the iRBC membrane (Fig. 2B), but only PFE1605w was coexported with PfEMP1 and localized in knobs (Fig. 3).
Very recently Proellocks et al. (56) showed that disruption of PFE1605w, termed LyMP in their study, reduced iRBC cytoadherence to CD36 by 55%, while retaining knob formation and PfEMP1 surface localization. Further, they identified an interaction between inside-out vesicles prepared from uninfected human erythrocytes and the PFE1605w positively charged C terminus, but not its PHIST domain. Together with the data presented here, we can now suggest at least one mechanistic role for PFE1605w and potentially for other PHIST proteins in cytoadherence (Fig. 8). We propose that PFE1605w tightly binds to the PfEMP1 intracellular segment (Fig. 1) soon after export from the parasite and remains bound during trafficking to Maurer's clefts (Fig. 4) and eventually knobs (Fig. 3). When in knobs, the C-terminal segment of PFE1605w links PfEMP1 with the host cytoskeleton. The PFE1605w-ATS interaction (Fig. 7) probably complements direct ATS-cytoskeletal binding (57) and ensures mechanical robustness against shear forces exerted by blood flow, thereby allowing strong cytoadherence.
Figure 8.
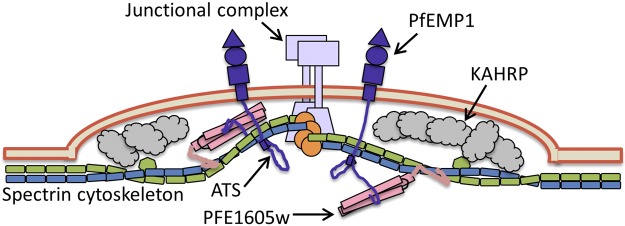
Schematic representation of an iRBC knob. Our proposed mechanism of PFE1605w function, connecting the C terminus of PfEMP1 ATS to the cytoskeleton, is illustrated.
In summary, we present here the first structural insights on members of the PHIST family and how they function as protein interaction modules. The interaction properties of specific members could be linked to their cellular localization and trafficking pattern alongside the well characterized PfEMP1, allowing the proposition of a mechanistic model of the function of PHIST molecules in cytoadherence.
Supplementary Material
Acknowledgments
The authors are grateful to David Staunton, Nick Soffe, Edward Lowe, and Philip Fowler for the upkeep of the supporting facilities at Oxford Biochemistry and to Henning Stahlberg and his team at the Center for Cellular Imaging and NanoAnalytics (C-CINA) and the Image Core Facility, Biozentrum, University of Basel, for the access and support. The authors acknowledge the Diamond Light Source (Harwell, UK) for access to synchrotron beamlines and the Oxford Protein Production Facility (Harwell, UK) for assistance with expression of PHIST domains.
This work received support from the Wellcome Trust for the Oxford Biochemistry NMR facility (grant 094872/Z/10/Z, grant 088497/Z/09/Z to I.V., and a Ph.D. studentship to E.C.). The cell biological part of this study was supported by the Swiss National Science Foundation (grants 31003A_132709/1 and 31003A_149297/1).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ATS
- acidic terminal segment
- AUC
- analytical ultracentrifugation
- DAPI
- 4′,6′-diamidino-2-phenylindole
- DTT
- dithiothreitol
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- GST
- glutathione S-transferase
- HA
- hemagglutinin
- hpi
- hours postinfection
- IFA
- immunofluorescence assay
- iRBC
- infected red blood cell
- MES
- 2-(N-morpholino)ethanesulfonic acid
- NMR
- nuclear magnetic resonance
- PFA
- paraformaldehyde
- PfEMP1
- P. falciparum erythrocyte membrane protein 1
- PHIST
- Plasmodium helical interspersed subtelomeric
- PBS
- phosphate-buffered saline
- RMSD
- root mean square deviation
REFERENCES
- 1. Mundwiler-Pachlatko E., Beck H. P. (2013) Maurer's clefts, the enigma of Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 110, 19987–19994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nguitragool W., Bokhari A. A., Pillai A. D., Rayavara K., Sharma P., Turpin B., Aravind L., Desai S. A. (2011) Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 145, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cooke B. M., Mohandas N., Coppel R. L. (2001) The malaria-infected red blood cell: structural and functional changes. Adv. Parasitol. 50, 1–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maier A. G., Rug M., O'Neill M. T., Brown M., Chakravorty S., Szestak T., Chesson J., Wu Y., Hughes K., Coppel R. L., Newbold C., Beeson J. G., Craig A., Crabb B. S., Cowman A. F. (2008) Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell 134, 48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kyes S., Horrocks P., Newbold C. (2001) Antigenic variation at the infected red cell surface in malaria. Annu. Rev. Microbiol. 55, 673–707 [DOI] [PubMed] [Google Scholar]
- 6. Baruch D. I., Pasloske B. L., Singh H. B., Bi X., Ma X. C., Feldman M., Taraschi T. F., Howard R. J. (1995) Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 82, 77–87 [DOI] [PubMed] [Google Scholar]
- 7. Chen Q., Schlichtherle M., Wahlgren M. (2000) Molecular aspects of severe malaria. Clin. Microbiol. Rev. 13, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marti M., Good R. T., Rug M., Knuepfer E., Cowman A. F. (2004) Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science 306, 1930–1933 [DOI] [PubMed] [Google Scholar]
- 9. Hiller N. L., Bhattacharjee S., van Ooij C., Liolios K., Harrison T., Lopez-Estrano C., Haldar K. (2004) A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 306, 1934–1937 [DOI] [PubMed] [Google Scholar]
- 10. Heiber A., Kruse F., Pick C., Gruring C., Flemming S., Oberli A., Schoeler H., Retzlaff S., Mesen-Ramirez P., Hiss J. A., Kadekoppala M., Hecht L., Holder A. A., Gilberger T. W., Spielmann T. (2013) Identification of new PNEPs indicates a substantial non-PEXEL exportome and underpins common features in Plasmodium falciparum protein export. PLoS Pathog. 9, e1003546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Koning-Ward T. F., Gilson P. R., Boddey J. A., Rug M., Smith B. J., Papenfuss A. T., Sanders P. R., Lundie R. J., Maier A. G., Cowman A. F., Crabb B. S. (2009) A newly discovered protein export machine in malaria parasites. Nature 459, 945–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wickham M. E., Rug M., Ralph S. A., Klonis N., McFadden G. I., Tilley L., Cowman A. F. (2001) Trafficking and assembly of the cytoadherence complex in Plasmodium falciparum-infected human erythrocytes. EMBO J. 20, 5636–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knuepfer E., Rug M., Klonis N., Tilley L., Cowman A. F. (2005) Trafficking determinants for PfEMP3 export and assembly under the Plasmodium falciparum-infected red blood cell membrane. Mol. Microbiol. 58, 1039–1053 [DOI] [PubMed] [Google Scholar]
- 14. Magowan C., Nunomura W., Waller K. L., Yeung J., Liang J., Van Dort H., Low P. S., Coppel R. L., Mohandas N. (2000) Plasmodium falciparum histidine-rich protein 1 associates with the band 3 binding domain of ankyrin in the infected red cell membrane. Biochim. Biophys. Acta 1502, 461–470 [DOI] [PubMed] [Google Scholar]
- 15. Taylor D. W., Parra M., Chapman G. B., Stearns M. E., Rener J., Aikawa M., Uni S., Aley S. B., Panton L. J., Howard R. J. (1987) Localization of Plasmodium falciparum histidine-rich protein 1 in the erythrocyte skeleton under knobs. Mol. Biochem. Parasitol. 25, 165–174 [DOI] [PubMed] [Google Scholar]
- 16. Weng H., Guo X., Papoin J., Wang J., Coppel R., Mohandas N., An X. (2014) Interaction of Plasmodium falciparum knob-associated histidine-rich protein (KAHRP) with erythrocyte ankyrin R is required for its attachment to the erythrocyte membrane. Biochim. Biophys. Acta 1838, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooke B. M., Buckingham D. W., Glenister F. K., Fernandez K. M., Bannister L. H., Marti M., Mohandas N., Coppel R. L. (2006) A Maurer's cleft-associated protein is essential for expression of the major malaria virulence antigen on the surface of infected red blood cells. J. Cell Biol. 172, 899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spycher C., Rug M., Pachlatko E., Hanssen E., Ferguson D., Cowman A. F., Tilley L., Beck H. P. (2008) The Maurer's cleft protein MAHRP1 is essential for trafficking of PfEMP1 to the surface of Plasmodium falciparum-infected erythrocytes. Mol. Microbiol. 68, 1300–1314 [DOI] [PubMed] [Google Scholar]
- 19. Sargeant T. J., Marti M., Caler E., Carlton J. M., Simpson K., Speed T. P., Cowman A. F. (2006) Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 7, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. LaCount D. J., Vignali M., Chettier R., Phansalkar A., Bell R., Hesselberth J. R., Schoenfeld L. W., Ota I., Sahasrabudhe S., Kurschner C., Fields S., Hughes R. E. (2005) A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438, 103–107 [DOI] [PubMed] [Google Scholar]
- 21. Parish L. A., Mai D. W., Jones M. L., Kitson E. L., Rayner J. C. (2013) A member of the Plasmodium falciparum PHIST family binds to the erythrocyte cytoskeleton component band 4.1. Malar. J. 12, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Young J. A., Fivelman Q. L., Blair P. L., de la Vega P., Le Roch K. G., Zhou Y., Carucci D. J., Baker D. A., Winzeler E. A. (2005) The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Mol. Biochem. Parasitol. 143, 67–79 [DOI] [PubMed] [Google Scholar]
- 23. Silvestrini F., Bozdech Z., Lanfrancotti A., Di Giulio E., Bultrini E., Picci L., Derisi J. L., Pizzi E., Alano P. (2005) Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol. Biochem. Parasitol. 143, 100–110 [DOI] [PubMed] [Google Scholar]
- 24. Eksi S., Haile Y., Furuya T., Ma L., Su X., Williamson K. C. (2005) Identification of a subtelomeric gene family expressed during the asexual-sexual stage transition in Plasmodium falciparum. Mol. Biochem. Parasitol. 143, 90–99 [DOI] [PubMed] [Google Scholar]
- 25. Rovira-Graells N., Gupta A. P., Planet E., Crowley V. M., Mok S., Ribas de Pouplana L., Preiser P. R., Bozdech Z., Cortes A. (2012) Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res. 22, 925–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Claessens A., Adams Y., Ghumra A., Lindergard G., Buchan C. C., Andisi C., Bull P. C., Mok S., Gupta A. P., Wang C. W., Turner L., Arman M., Raza A., Bozdech Z., Rowe J. A. (2012) A subset of group A-like var genes encodes the malaria parasite ligands for binding to human brain endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 109, E1772–E1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Florens L., Liu X., Wang Y., Yang S., Schwartz O., Peglar M., Carucci D. J., Yates J. R., 3rd, Wub Y. (2004) Proteomics approach reveals novel proteins on the surface of malaria-infected erythrocytes. Mol. Biochem. Parasitol. 135, 1–11 [DOI] [PubMed] [Google Scholar]
- 28. Florens L., Washburn M. P., Raine J. D., Anthony R. M., Grainger M., Haynes J. D., Moch J. K., Muster N., Sacci J. B., Tabb D. L., Witney A. A., Wolters D., Wu Y., Gardner M. J., Holder A. A., Sinden R. E., Yates J. R., Carucci D. J. (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419, 520–526 [DOI] [PubMed] [Google Scholar]
- 29. Lasonder E., Ishihama Y., Andersen J. S., Vermunt A. M., Pain A., Sauerwein R. W., Eling W. M., Hall N., Waters A. P., Stunnenberg H. G., Mann M. (2002) Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature 419, 537–542 [DOI] [PubMed] [Google Scholar]
- 30. Regev-Rudzki N., Wilson D. W., Carvalho T. G., Sisquella X., Coleman B. M., Rug M., Bursac D., Angrisano F., Gee M., Hill A. F., Baum J., Cowman A. F. (2013) Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 153, 1120–1133. [DOI] [PubMed] [Google Scholar]
- 31. Sanders P. R., Gilson P. R., Cantin G. T., Greenbaum D. C., Nebl T., Carucci D. J., McConville M. J., Schofield L., Hodder A. N., Yates J. R., 3rd, Crabb B. S. (2005) Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J. Biol. Chem. 280, 40169–40176 [DOI] [PubMed] [Google Scholar]
- 32. Mayer C., Slater L., Erat M. C., Konrat R., Vakonakis I. (2012) Structural analysis of the plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) intracellular domain reveals a conserved interaction epitope. J. Biol. Chem. 287, 7182–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 36. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Bricogne G., Blanc E., Brandl M., C., F., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER version 2.10. Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 39. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA, USA [Google Scholar]
- 41. Shen Y., Lange O., Delaglio F., Rossi P., Aramini J. M., Liu G., Eletsky A., Wu Y., Singarapu K. K., Lemak A., Ignatchenko A., Arrowsmith C. H., Szyperski T., Montelione G. T., Baker D., Bax A. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U. S. A. 105, 4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crabb B. S., Rug M., Gilberger T. W., Thompson J. K., Triglia T., Maier A. G., Cowman A. F. (2004) Transfection of the human malaria parasite Plasmodium falciparum. Methods Mol. Biol. 270, 263–276 [DOI] [PubMed] [Google Scholar]
- 44. Flueck C., Bartfai R., Volz J., Niederwieser I., Salcedo-Amaya A. M., Alako B. T., Ehlgen F., Ralph S. A., Cowman A. F., Bozdech Z., Stunnenberg H. G., Voss T. S. (2009) Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog. 5, e1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trager W., Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675 [DOI] [PubMed] [Google Scholar]
- 46. Pachlatko E., Rusch S., Muller A., Hemphill A., Tilley L., Hanssen E., Beck H. P. (2010) MAHRP2, an exported protein of Plasmodium falciparum, is an essential component of Maurer's cleft tethers. Mol. Microbiol. 77, 1136–1152 [DOI] [PubMed] [Google Scholar]
- 47. Spielmann T., Fergusen D. J., Beck H. P. (2003) etramps, a new Plasmodium falciparum gene family coding for developmentally regulated and highly charged membrane proteins located at the parasite-host cell interface. Mol. Biol. Cell 14, 1529–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maier A. G., Rug M., O'Neill M. T., Beeson J. G., Marti M., Reeder J., Cowman A. F. (2007) Skeleton-binding protein 1 functions at the parasitophorous vacuole membrane to traffic PfEMP1 to the Plasmodium falciparum-infected erythrocyte surface. Blood 109, 1289–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gruring C., Spielmann T. (2012) Imaging of live malaria blood stage parasites. Methods Enzymol. 506, 81–92 [DOI] [PubMed] [Google Scholar]
- 50. Tokuyasu K. T. (1973) A technique for ultracryotomy of cell suspensions and tissues. J. Cell Biol. 57, 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spycher C., Klonis N., Spielmann T., Kump E., Steiger S., Tilley L., Beck H. P. (2003) MAHRP-1, a novel Plasmodium falciparum histidine-rich protein, binds ferriprotoporphyrin IX and localizes to the Maurer's clefts. J. Biol. Chem. 278, 35373–35383 [DOI] [PubMed] [Google Scholar]
- 52. Bozdech Z., Llinas M., Pulliam B. L., Wong E. D., Zhu J., DeRisi J. L. (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS. Biol. 1, E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Le Roch K. G., Zhou Y., Blair P. L., Grainger M., Moch J. K., Haynes J. D., De La Vega P., Holder A. A., Batalov S., Carucci D. J., Winzeler E. A. (2003) Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301, 1503–1508 [DOI] [PubMed] [Google Scholar]
- 54. McMillan P. J., Millet C., Batinovic S., Maiorca M., Hanssen E., Kenny S., Muhle R. A., Melcher M., Fidock D. A., Smith J. D., Dixon M. W., Tilley L. (2013) Spatial and temporal mapping of the PfEMP1 export pathway in Plasmodium falciparum. Cell. Microbiol. 15, 1401–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gruring C., Heiber A., Kruse F., Ungefehr J., Gilberger T. W., Spielmann T. (2011) Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nat. Commun. 2, 165. [DOI] [PubMed] [Google Scholar]
- 56. Proellocks N. I., Herrmann S., Buckingham D. W., Hanssen E., Hodges E. K., Elsworth B., Morahan B. J., Coppel R. L., Cooke B. M. (2014) A lysine-rich membrane-associated PHISTb protein involved in alteration of the cytoadhesive properties of Plasmodium falciparum-infected red blood cells. FASEB J. 28, 3103–3113 [DOI] [PubMed] [Google Scholar]
- 57. Oh S. S., Voigt S., Fisher D., Yi S. J., LeRoy P. J., Derick L. H., Liu S., Chishti A. H. (2000) Plasmodium falciparum erythrocyte membrane protein 1 is anchored to the actin-spectrin junction and knob-associated histidine-rich protein in the erythrocyte skeleton. Mol. Biochem. Parasitol. 108, 237–247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.