

Abstract
Neuronal apoptosis is one of the major causes of poststroke neurological deficits. Inflammation during the acute phase of stroke results in nuclear translocation of NFκB in affected cells in the infarct area. Macrophage migration inhibitory factor (MIF) promotes cardiomyocyte survival in mice following heart ischemia. However, the role of MIF during stroke remains limited. In this study, we showed that MIF expression is down-regulated by 0.75 ± 0.10-fold of the control in the infarct area in the mouse brains. Two functional cis-acing NFκB response elements were identified in the human MIF promoter. Dual activation of hypoxia and NFκB signaling resulted in significant reduction of MIF promoter activity to 0.86 ± 0.01-fold of the control. Furthermore, MIF reduced caspase-3 activation and protected neurons from oxidative stress- and in vitro ischemia/reperfusion-induced apoptosis. H2O2 significantly induced cell death with 12.81 ± 0.58-fold increase of TUNEL-positive cells, and overexpression of MIF blocked the H2O2-induced cell death. Disruption of the MIF gene in MIF-knockout mice resulted in caspase-3 activation, neuronal loss, and increased infarct development during stroke in vivo. The infarct volume was increased from 6.51 ± 0.74% in the wild-type mice to 9.07 ± 0.66% in the MIF-knockout mice. Our study demonstrates that MIF exerts a neuronal protective effect and that down-regulation of MIF by NFκB-mediated signaling under hypoxia accelerates neuronal loss during stroke. Our results suggest that MIF is an important molecule for preserving a longer time window for stroke treatment, and strategies to maintain MIF expression at physiological level could have beneficial effects for stroke patients.—Zhang, S., Zis, O., Ly, P. T. T., Wu, Y., Zhang, S., Zhang, M., Cai, F., Bucala, R., Shyu, W.-C., Song, W. Down-regulation of MIF by NFκB under hypoxia accelerated neuronal loss during stroke.
Keywords: macrophage migration inhibitory factor, caspase-3, nuclear factor κB
Ischemic stroke that results from disturbance of blood supply to the brain accounts for 80% of stroke cases and significantly causes poststroke neurological deficits and death (1). Among a number of mechanisms involved in stroke pathogenesis, activation of the inflammatory response and the apoptotic pathways have been shown to play pivotal roles in stroke progression (2–4).
Ischemic stroke differentially affects brain regions and results in an ischemic core and penumbra. Neurons in the ischemic core are rarely salvageable due to their immediate necrosis. In contrast, neurons in the penumbra experience a relatively milder ischemic insult and undergo delayed cell death and are the targets for therapeutic intervention (4). In the penumbra, neurons can undergo both necrosis and apoptosis. Necrosis induced by glutamate diffusion from the necrotic core can be diminished by blocking a specific subtype of NMDA receptors (5). Most of the neurons in the penumbra undergo apoptosis. Antiapoptotic strategies, including inhibition of Apaf-1 (involved in intrinsic apoptosis), expressing dysfunctional FasL (involved in extrinsic apoptosis) and knockdown of AIF (involved in caspase-independent apoptosis), were shown to successfully reduce neuronal loss and infarct volume (2).
Ischemic stroke-induced acute brain damage rapidly activates inflammatory responses, which lead to up-regulation and release of pro- and anti-inflammatory factors, such as tumor necrosis factor α (TNF-α), IL-1β, IL-6, IL-10, IL-20 and TGF-β (3). In the brain, the cytokines are produced not only by infiltrating immune cells but also by resident brain cells including neurons and glial cells (6). As one of the major consequences of ischemic stroke, inflammation amplifies stroke outcome regardless of the triggering stimuli (7, 8), and anti-inflammatory approaches are beneficial in reducing infarct volume and promoting cell survival (9, 10). This highly finding suggests that inflammation plays a key role in stroke-induced progressive brain damage.
Macrophage migration inhibitory factor (MIF), a 114 aa protein, was first identified as a proinflammatory cytokine (11, 12) and later was found to function as a pleiotropic protein involved in many cellular activities, such as regulating cell death and survival (13–15). The role of MIF during ischemia, either being protective by suppressing apoptosis or detrimental by promoting inflammation, is not well characterized. A study on intestinal ischemia demonstrated that lack of MIF gene expression significantly suppressed circulating inflammatory cytokines and reduced lethality in mice (16). MIF has been shown to promote cell survival by modulating glucose uptake and metabolism during energy deprivation in mouse model of myocardial infarction (17, 18). MIF has also been shown to inhibit the transcription of tumor suppressor p53 and negatively regulate JNK signaling, thereby preventing immune cells, such as macrophages, to undergo apoptosis, growth arrest, and cellular senescence (13–15). In addition, MIF possesses redox activity through its Cys-Xaa-Xaa-Cys group, suppressing oxidative stress induced apoptosis (19, 20). These studies suggest that MIF could be protective during ischemia. Limited studies have focused on the role of MIF in brain disorders. We recently reported that MIF is dysregulated during ischemic stroke in both patients and a rat stroke model in a temporal manner (21). MIF was found to contribute to stroke pathology in mice and neuronal death in vitro following oxygen-glucose deprivation (22) and knockout of the Mif gene results in smaller infarct volume at 7 d after stroke by promoting the macrophage and microglia response (23), which suggests a detrimental role of MIF in cerebral ischemia. The same group also concluded that MIF was not involved in promoting inflammatory response during these 7 d (22), leaving an open debate regarding the role of MIF in regulating neuronal death/survival in the central nervous system.
The human MIF gene, located on chromosome 22q11.23, spans 855 bp and contains 3 exons and 2 introns. Human MIF is expressed in the heart, brain, lung, liver and kidney (24–26). However, the expression of MIF in the central nervous system has not been well characterized. The TATA-less promoter of MIF containing a GC box controls its constitutive expression in many types of cells including neurons and glial cells (11, 25, 26). The MIF promoter also contains cyclic-AMP response elements (CREs), which directly interact with CRE-binding protein (CREB) for transcriptional regulation of MIF gene expression in pituitary cells (27). Our laboratory and others identified several hypoxia responsive elements (HREs) in the MIF promoter (21, 28, 29). The binding of hypoxia-inducible factor 1α (HIF-1α) to the HREs in MIF promoter resulted in transcriptional up-regulation in vitro and in the ischemic hemisphere of rats during stroke (21).
Nuclear factor κB (NFκB) signaling plays an important role in gene regulation and is implicated in inflammation, oxidative stress, apoptosis, and neurodegenerative disorders (30–34). Activation of NFκB signaling also has been observed during stroke, despite appearing to have dual roles with respect to neuronal survival and stroke outcomes (35, 36). NFκB regulates genes involved in both pro- and antiapoptotic processes and plays a complex role in neuronal survival/death (36, 37). The active NFκB is a dimer composed of 2 of the 5 subunits in the NFκB family, RelA (p65), c-Rel, RelB, p105/p50, and p100/p52. The most abundant dimer in mammalian cells consists of p65 and p50 and specifically recognizes a DNA sequence of 5′-GGGRNNYYCC (where N and Y represent any base and pyrimidine, respectively; refs. 38, 39). Active NFκB modulates gene transcription by direct binding to the cis-acting elements on DNA sequences. On stimulation, the IκB kinase (IKK) phosphorylates IκB, resulting in the degradation of IκB by the ubiquitin proteasome pathway. Degradation of IκB releases its binding partner NFκB, and allows it to be translocated from the cytoplasm to the nucleus, where it activates the transcription of its target genes. During stroke, nuclear translocation of NFκB p65 is observed in neurons located in the infarct, indicating an active role of NFκB in ischemic neurons (35). Despite the fact that both NFκB activation and alteration of MIF expression are observed during the early stages of cerebral ischemia (21, 40), the interaction between NFκB and MIF and its role in cerebral ischemia remain undefined. In the present study, we report two functional cis-acting NFκB binding elements on the MIF gene promoter and demonstrate that NFκB regulates MIF expression by transcriptional activation of the MIF gene promoter via these two sites. Futhermore, we demonstrate that MIF gene transcription is reduced by activation of NFκB signaling under hypoxic condition. We show that NFκB plays an important role in the temporospatial expression pattern of MIF in the brain following focal ischemia. We further show that MIF reduces caspase-3 activation and protects neurons from oxidative stress-induced and in vitro ischemia/reperfusion-induced apoptosis. Finally, we demonstrate that disruption of MIF gene expression results in elevated caspase-3 activation and exacerbates neuronal death as well as increased infarct development during stroke in Mif-knockout mice in vivo. Our results suggest that MIF exhibits neuroprotective qualities following stroke and may be an important molecule for preserving a longer time window for stroke treatment.
MATERIALS AND METHODS
Generation of human MIF expression plasmid and promoter constructs
Human MIF cDNA was amplified from human embryonic kidney 293 (HEK293) cells and cloned into pcDNA4-mycHis vector to generate pMIF-mycHis mammalian expression plasmid. pMIF-Stop, expressing human MIF protein without any tag, was generated from pMIF-mycHis. Previously, we have constructed a 5′ upstream fragment of the human MIF gene promoter regions −2634 to +35 and −553 to +8 bp into pGL3-basic vector to generate MIF promoter luciferase report plasmid phMIFluc and HRE-deleting plasmid phMIFδHluc, respectively (21). phMIFluc was digested by KpnI to remove a fragment between two KpnI sites and the new construct phMIF-Bluc plasmid containing promoter region from −2158 to +35 bp. Plasmids phMIF-Cluc and phMIF-Dluc, containing the human MIF promoter region from −553 to +35, and −194 to +35 bp, respectively, were generated by PCR amplification from phMIFluc and cloned into pGL3-basic vector at XhoI and HindIII sites. Human MIF promoter fragment of −5415 to −3090 bp was amplified by PCR with primers NheIHMIF1f (5′-ctagctagcgagcgaggccttgtttctacca) and BglIIHMIF1r (5′-ggaagctctctttggccatgcgagtccttaca), and cloned into pGL3-basic at NheI and BglII sites to generate phMIF1-luc. Plasmid phMIF2-luc contains human MIF promoter from −3358 to +35 bp inserted between NheI and HindIII, having an endogenous BglII site located at −3355. To generate phMIF5k-luc, phMIF1-luc digested by NheI and BglII, and the fragment spanning −5415 to −3354 bp was inserted into phMIF2-luc between NheI and BglII site. Plasmid pMIF-NFκBluc was generated by cloning a double-strand oligonucleotide (5′-ccgGGGGCTTTCCcaatGGGGCCTCCCagcaGGGAAGTTCCctg) containing 3 NFκB cis-acting elements from human MIF promoter, namely MIF-NFκB, into the pGL-pl vector (41) at SacI and NheI sites.
Cell culture, transfection, and ELISA
The HEK293 cell line, mouse microglia BV-2 cell line, and human neuroblastoma N2A cell line were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1 mM of sodium pyruvate, 2 mM of l-glutamine (Invitrogen, Carlsbad, CA, USA). Cells were cultured at 37°C in an incubator supplemented with 5% CO2. Plasmids were transfected by either the calcium-phosphate method or Lipofectamine 2000 (Invitrogen) following the manufacturer's instruction. Plasmid (2 μg) was transfected into one 35 mm tissue culture plate. Fresh medium was changed 6 h after transfection. Culture medium was collected, centrifuged at 3000 g for 2 min at 4°C. Human MIF expression was measured by human MIF ELISA kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer's instructions.
Luciferase assays
Cells were plated onto 24-well plates 24 h prior to transfection and cultured to ∼50% confluency before transfection. In each well, cells were transfected with 0.5 μg of plasmids using calcium phosphate transfection methods. pCMV-Rluc plasmid (1 ng) expressing Renilla luciferase was cotransfected for normalization for the transfection efficiency. After 48 h transfection, cells were harvested and lysed with 100 μl 1× passive lysis buffer, and a dual luciferase assay was performed as described previously (42). Firefly luciferase activities and Renilla luciferase activities were measured using the dual-luciferase reporter assay system (Promega, Madison, WI, USA). The firefly luciferase activity was normalized to the Renilla luciferase activity and expressed as relative luciferase units (RLU) to reflect the promoter activity.
Gel shift assay (GSA)
GSA, or electrophoretic mobility shift assay (EMSA), was performed as described previously (43). Nuclear extract was fractionated from the HEK293 lysate with NFκB overexpression, as described previously (44). Oligonucleotide probes were labeled with IR700 dye (Integrated DNA Technologies, Coralville, IA, USA) and annealed to generate double-stranded probes at a final concentration of 0.1 μM. For competition experiments, nuclear extract was incubated with the 0.5 pmol of labeled probe and excess of unlabeled competition oligonucleotides for 20 min at 22°C. For the supershifting assay, mouse anti-NFκB p65 monoclonal antibody (Sigma, St. Louis, MO, USA) was added to the above reaction and incubated for an additional 20 min. The samples were analyzed on a 4% nondenaturing polyacrylamide gel, and the gel was scanned using the Odyssey scanner at a wavelength of 700 nm (LI-COR Biosciences, Lincoln, NE, USA). The sequences of the sense strand oligonucleotides to generate the double-strand probes or competitors are as follows: consensus wild-type NFκB (wt-NFκB), 5′-agttgagGGGACTTTCCcaggc; mutant NFκB (mu-NFκB), 5′-agttgagGCCACTTTCCcaggc; MIF-NFκB-A, 5′-gctccgGGGGCTTTCCcaagga; MIF-NFκB-B, 5′-ggccatGGGGCCTCCCagctgg; MIF-NFκB-C, 5′-ggttcaGGGAAGTTCCctggat.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed, as described previously (45). Cross-linking between protein and chromatin was achieved by adding formaldehyde to the final concentration of 1.42% in NFκB-enriched HEK293 cells for 15 min at 22°C and was quenched with glycine at final concentration of 125 mM for 5 min. Cells then were harvested in cold PBS and lysed with immunoprecipitation (IP) buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, Nonidet P-40 (0.5% v/v), and Triton X-100 (1.0% v/v), supplemented with protease inhibitor cocktail Complete (Roche Molecular Biochemicals, Pleasanton, CA, USA). The nuclear pellets were isolated by centrifugation at 12,000 g for 1 min and resuspended with the IP buffer. The chromatin then was sheared by sonication on ice. For isolation of NFκB binding complex, the chromatin solution was incubated with a monoclonal rabbit-anti-NFκB p65 antibody (cat no. 8242; Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C. The normal rabbit serum was used for the sham IP control. After IP, cross-link was reversed by boiling the sample with Chelex100 (Bio-Rad, Richmond, CA, USA) for 10 min. The supernatant containing DNA fragments was isolated by centrifugation at 12,000 g for 1 min and then used as template for PCR analysis of the chromatin fragments. The following pairs of primers, 5′-atagcctcgaaggacaggacagg and 5′-aatccagtttgcccacattttcc, 5′-cagctgcaggaaccaataccc and 5′-ctcagagatttccagggagg, and 5′-agctgagcacgtttgaaccac and 5′-tagtcatcgcggcaggtgaga, were used to specifically amplify fragments containing MIF-NFκB-A, -B, and -C, respectively. β-Actin was amplified by 5′-gacaggatgcagaaggagat and 5′-ttgctgatccacatctgctg as the internal control.
Semiquantitative RT-PCR
Cell samples were homogenized with the Tri reagent (Invitrogen), and total RNA was extracted following the manufacturer's instruction. The first-strand cDNA was generated from the total RNA (1 μg) by the ThermoScript reverse transcriptase with Oligo(dT)20 as the primer (Invitrogen). The resultant cDNA products were used as templates to quantify the expression of target genes by semiquantitative PCR with TaqDNA polymerase and specific primer sets. The human MIF gene specific primers were 5′-gtagtctgacgtcagcggaggc (mMIFqRT-57F) and 5′-ctaaaagtcatgagctggtccg (mMIFqRT234R). The primers specifically amplifying a mouse EST were 5′-atgccgatgttcatcgtaaacacc and 5′-ttaggcgaaggtggagttgttccagc. The GAPDH gene specific primers were 5′-ccagtgagcttcccgttcagc and 5′-cccatcaccatcttccaggagc. The PCR products were separated on a 2% agarose gel, and the band intensity was analyzed by ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA).
Middle cerebral artery ligation (MCAl)
Animal experiment protocols were approved by The University of British Columbia Animal Care and Use committee. Mif+/− mice on the BALB/c (generation N10) have been described previously (46). All the mice were allowed access to water and food ad libitum. Male mice, between 10 and 12 wk of age, were subjected to transient distal MCAl (tMCAl) surgery (47) with modifications. Briefly, following isolation of the right common carotid artery (rCCA), the mouse was put on a stereotactic frame (Kopf, Tujunga, CA, USA) and positioned on its right side for MCAl. A skin incision was made between the right eye and ear, and the muscle fibers were separated and retracted to expose the skull surface. The right MCA (rMCA) was easily visualized through the skull, and a 1 mm burr hole was opened by a dental drill around the rMCA to expose the rMCA for ligation with a 10–0 suture. Quickly after ligation of rMCA, the rCCA was occluded by a nontraumatic microclamp (S&T Microsurgical Instruments, Birmingham, AL, USA). Saline (1 ml) was injected subcutaneously before and after the surgery, and the mouse was laid on a warm blanket to recover. After transient rMCA and rCCA ligation for 2 h, the suture and microclamp were removed, and the mice were allowed reperfusion. Control mice underwent similar surgical procedures, except that the rMCA and rCCA were not ligated.
Sample collection and 2,3,5-triphenyl-tetrazolium chloride (TTC) staining
A pilot study was carried out to determine the sampling strategy. Following the tMCAl procedure, mice were euthanized at 0, 2, 8, or 22 h after reperfusion. The freshly dissected brains were placed on a brain slice matrix, and coronal sections were made to generate 1-mm-thick slices beginning from the anterior edge of the cerebral cortex. The brain slices were incubated in 1% TTC solution in PBS for 10 min at 37°C. After incubation, the slices were fixed in cold 4% paraformaldehyde (PFA) solution and scanned for subsequent analysis. ImageJ was used for infarct area determination. Focal ischemia resulted in tissue infarction and was illustrated by the complete loss of TTC staining, contrasting with the surrounding dark red-stained viable tissue. The infarcted area was restricted to the cerebral cortex supplied by MCA at 24 h postischemia. The rest of the cerebral cortex supplied by anterior/posterior cerebral arteries was not affected following the tMCAl procedure. Brain tissues from the ischemic and contralateral nonischemic area were dissected for protein expression analysis. For the sham surgery mice, equivalent areas were dissected. All samples were stored in −80°C before biochemical analysis.
Immunohistology, immunoblot analysis, and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
The 4% PFA-fixed brain samples were prepared for either cryosectioning at 20 μm thickness or paraffin-embedded sectioning at 5 μm thickness. TUNEL assay (Promega) was performed on the 5-μm-thick slices following the manufacturer's instruction. For immunostaining, the brain slices were permeabilized in 0.3% Triton X-100 in PBS (PBS-Tx) for 30 min and blocked with 5% BSA in PBS for 1 h at 22°C. Next, the slices were incubated with primary antibodies in PBS with 1% BSA at 4°C overnight. After rinsing with PBS-Tx 3 times, the slices were applied with secondary antibodies biotinylated swine anti-rabbit in PBS-Tx with 1% BSA for 1 h at 22°C, and visualized by the ABC and DBA method. For immunoblotting, brain tissue and cells were lysed with the RIPA-DOC buffer. The lysates were resolved by gel electrophoresis, and the protein was analyzed by Western blot. The primary antibodies were recognized by IRDye 800CW-labeled goat anti-mouse or IRDye 700CW-labled goat anti-rabbit, and were visualized and analyzed using the Odyssey system (LI-COR Biosciences). The primary antibodies were anti-cleaved caspase-3 (Cell Signaling Technology) for immunohistology, anti-NFκB p65 subunit (cat no. 8242; Cell Signaling Technology) for immunohistology and immunoblotting, anti-MIF antibody (Torrey Pines Biolabs, Secaucus, NJ, USA) for immunoblotting, and anti-β-actin antibody (AC-15; Sigma) for immunoblot, used as a loading control.
Human MIF purification, primary neuronal culture, oxygen-glucose deprivation (OGD) treatment, and MTS assay
To obtain human MIF protein for primary neuronal treatment, pMIF-mycHis was transfected into HEK293 cells, and cell lysate was prepared under undenatured conditions. MycHis-tagged MIF protein was purified using nickel-NTA magnetic agarose beads (Qiagen, Valencia, CA, USA) following the manufacturer's protocol. The identity of the purified protein was verified by Western blot, and the purity was assessed by Coomassie blue staining following gel electrophoresist (Supplemental Fig. S2). Primary neuronal cultures were derived from embryonic day 14 (E14) mouse embryos as described previously (48). Cultures of mouse neurons (10 d) were subjected to either 50 μM H2O2 for 16 h or “in vitro ischemia/reperfusion” treatment. The latter was achieved by incubating primary cultures under OGD condition for 2 h, followed by normal incubation conditions for an additional 16 h. For cells undergoing OGD, the cells were cultured with either fresh Neurobasal medium (Invitrogen) for control cells or glucose-free Neurobasal medium (Invitrogen) for OGD treatment under hypoxic condition (2% oxygen complemented with nitrogen). Both Neurobasal media were supplemented with B27 (Invitrogen) and L-glu (Invitrogen). The cells were treated with purified mycHis-tagged human MIF protein or vehicle solution during the entire experimental period. Cell viability was assessed using the MTS assay (Promega) following the manufacturer's instructions. Cell viability was calculated by dividing the absorbance of treated or untreated cells by the average absorbance of nontreated (control) cells and expressed as a survival ratio.
Statistics
All results are presented as means ± sd. Statistical analysis was performed by 1-way ANOVA with Tukey posttest, 2-way ANOVA with Bonferroni posttests, and Student's t test.
RESULTS
Down-regulation of MIF expression in infarct regions after cerebral ischemia in mice
Previously we showed that MIF gene expression was altered in patients with stroke and in the brains of stroke model rats (21). In the present study, we further studied the spatial alteration of MIF expression and focused on the brain region undergoing acute infarction within 24 h after focal ischemia induced by the tMCAl procedure. Since the rMCA was ligated, with additional ligation of the rCCA, the blood supply was impaired in the entire right hemisphere, and this stroke model precisely produced an infarct restricted to the MCA-supplied cortical region. Mice (10 to 12 wk of age) were subjected to the tMCAl procedure and reperfused for 4 or 22 h. At 22 h postreperfusion, a clear infarction in the MCA-supplied cortical region was obtained, as assessed by TTC staining (Fig. 1A). The MCA territory and cortical peri-MCA area were defined as described in Fig. 1A. The brain tissue in the respective areas, along with its counterpart in the unaffected hemisphere (as control), were dissected for MIF protein analysis. At 6 h after the onset of ischemia, MIF expression in the MCA territory was significantly reduced to 0.80 ± 0.07-fold on the ischemic hemisphere compared to the unaffected counterpart (P<0.05; Fig. 1B, C). At 24 h after the onset of ischemia, brain samples collected from the MCA-supplied territory were assessed for protein expression even though significant tissue decomposition had occurred. The results showed that the level of β-actin, serving as internal control, was significantly reduced (Fig. 1D), even though the same amount of protein was loaded for evaluation, confirming severe decomposition of the parenchyma. Significantly lower expression of MIF to 0.75 ± 0.10-fold (P<0.05) reduction in the ischemic hemisphere was also observed (Fig. 1D, E). MIF protein expression in the cortical peri-MCA territory was also examined. The results showed no apparent effect on protein expression in the cortical peri-MCA area at either 6 h (Fig. 1F, G) or 24 h (Fig. 1H, I) postischemia (P>0.05).
Figure 1.
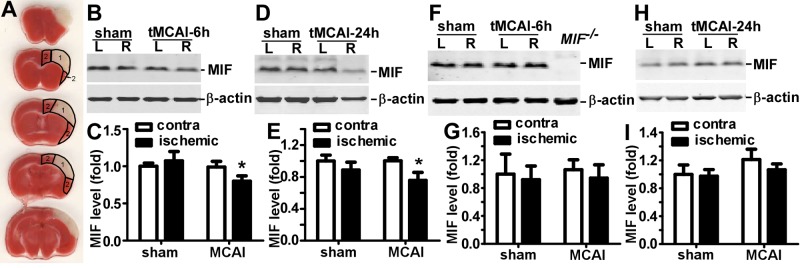
Reduced MIF expression in the infarct-targeted area after cerebral focal ischemia in mice. BALB/c mice were subjected to 2 h rMCAl, followed by 4 or 22 h of reperfusion or a sham procedure on the right hemisphere. The brain was freshly cut into 1 mm slices and subjected to TTC staining or protein expression analysis. A) At 22 h after reperfusion, infarcted tissue restricted to the MCA-supplied cerebral cortex was clearly distinguished by pale TTC staining and defined as the MCA territory (area 1) and the cortical peri-MCA area (area 2). B, D) At 6 h (B) or 24 h (D) after tMCAl, the MCA territory was lysed by RIPA-Doc buffer with 1% SDS. The lysate was resolved on a 12% Tris-tricine SDS-PAGE gel. MIF was detected by anti-MIF antibody, and β-actin was detected by β-actin antibody as control. C, E) Quantification of B and D, respectively. The ratio of MIF to β-actin was further normalized to unaffected hemisphere in sham-treated mice. Values are expressed as means ± sd, n = 6 for sham treatment and 7 for MCAl. *P < 0.05; 2-way ANOVA with Bonferroni posttest. F, H) At 6 h (F) or 24 h (H) after tMCAl, the lysates of peri-MCA territory were subjected to 12% Tris-tricine SDS-PAGE. MIF and β-actin were detected by their respective antibodies. G, I) Quantification of F and H, respectively. The ratio of MIF to β-actin was normalized to unaffected hemisphere in sham-treated mice. Values are expressed as means ± sd, n = 6 for sham treatment and 7 for MCAl. P > 0.05; 2-way ANOVA with Bonferroni posttest.
Identification of functional NFκB elements in the human MIF promoter
A significant increase in nuclear NFκB and the activation of the NFκB signaling pathway within the infarcted area following stroke suggest that NFκB may be involved in MIF dysregulation in the MCA territory (35, 49). To investigate this issue, we first examined the expression of NFκB p65 in the brain following stroke. Compared with the peri-MCA area, the increase of nuclear p65 in the MCA territory was confirmed by immunostaining (Fig. 2A), indicating that NFκB was activated in this region. To examine whether NFκB signaling modulates MIF gene expression following stroke, human MIF gene promoter function was analyzed. Sequence analysis revealed 3 putative NFκB cis-acting binding elements on the MIF promoter, located at −2536 to −2527, −1027 to −1018, and −513 to −504 bp (Fig. 2B), suggesting that MIF may be regulated by NFκB. To investigate whether these putative binding sites could bind to NFκB p65, GSA was performed. Double-strand oligonucleotides containing the core sequences of all 3 sites (MIF-NFκB) or an individual site (MIF-NFκB-A, -B, or -C) with flanking nucleotides were synthesized (Fig. 2B). Incubation of the wt-NFκB probe (wt-NFκB-IRDye 700) with NFκB p65-enriched HEK nuclear extracts resulted in a shifted band composed of DNA-protein complex (Fig. 2C, lane 2). Competition assays demonstrated that the intensity of the shifted band was significantly reduced or abolished by adding a 5- or 50-fold molar excess of unlabeled wt-NFκB oligonucleotide (Fig. 2C, lanes 3, 4), while the mu-NFκB oligonucleotide did not compete with the probe at either concentration (Fig. 2C, lanes 5, 6). MIF-NFκB significantly reduced the intensity, or completely abolished the shifted band at a 5- or 50-fold molar excess of the probe, respectively (Fig. 2C, lanes 7, 8). Addition of anti-NFκB p65 monoclonal antibody further retarded the migration of the DNA-protein complex (Fig. 2C, lane 9), resulting in a supershifted band, which was abolished by adding 50-fold molar excess of wt-NFκB (Fig. 2C, lanes 9, 10). This result confirmed the binding between the NFκB p65 and the probe. These data indicated that ≥1 of 3 putative binding sites could bind with NFκB p65.
Figure 2.

Identification of the functional NFκB binding sites on the human MIF promoter. A) Brain slices from BALB/c mice subjected to 2 h tMCAl followed by 2 h reperfusion were stained for NFκB p65. In the MCA territory, p65 exhibited nuclear localization (solid arrowheads), while in the cortical peri-MCA region, p65 diffused in the cytoplasm (open arrowheads). Scale bars = 20 μm. B) Schematic diagram shows the relative location of the putative NFκB binding sites on the human MIF promoter, and the corresponding sequence of the oligonucleotides. C, D) GSAs were performed using a wt-NFκB consensus oligonucleotide labeled with IRDye 700 as the probe. Lane 1 is labeled probe alone without nuclear extract. Incubation of the wt-NFκB probe with nuclear extract resulted in a shifted band composed of DNA-protein complex (lane 2). Addition of anti-NFκB p65 monoclonal antibody in the incubation mixture as in lane 2 resulted in a supershifted band (C, lane 9). Competition assays were performed by adding 5- or 50-fold molar excess of unlabeled wt-NFκB oligonucleotide (lanes 3, 4), mu-NFκB oligonucleotide (lanes 5, 6), MIF-NFκB (C, lanes 7, 8), MIF-NFκB-A (D, lanes 7, 8), MIF-NFκB-B (D, lanes 9, 10), MIF-NFκB-C (D, lanes 11, 12), and 50-fold molar excess of wt-NFκB (C, lane 10). E, F) GSAs and supershift assays were performed using MIF-NFκB-A oligonucleotide (E) or MIF-NFκB-C oligonucleotide (F) labeled with IRDye 700 as probes. Lane 1 is labeled probe alone without nuclear extract. Incubation of IRDye 700-MIF-NFκB-A or -C with nuclear extracts resulted in a shifted band (lane 2). Addition of anti-NFκB p65 monoclonal antibody resulted in a supershifted band (lane 5). Competition assays were achieved by adding 100-fold molar excess of unlabeled wt-NFκB (lanes 3, 6) or the same amount of mu-NFκB (lane 4). G) ChIP assay; details are described in Materials and Methods. Monoclonal rabbit-anti-p65 antibody was used to precipitate DNA fragments that could bind to NFκB p65. Normal rabbit serum was used for sham IP control. Target DNA fragments containing sequences corresponding to MIF-NFκB-A, -B, and -C were amplified by PCR. PCR products were resolved on 1.5% agarose gel. β-Actin was amplified as control.
To determine which binding sites acts as the cis-acting element for NFκB, GSAs were performed with individual putative MIF-NFκB binding oligonucleotides using wt-NFκB-IRDye 700 as the probe. MIF-NFκB-A, located at −2536 to −1517 bp, and MIF-NFκB-C, at −513 to −504 bp, reduced the binding intensity of the probe significantly (Fig. 2D, lanes 7, 8, 11, 12), whereas MIF-NFκB-B, at −1027 to −1018 bp, did not compete with the probe (Fig. 2C, lanes 9, 10). This result indicated that MIF-NFκB-A and MIF-NFκB-C contained the NFκB cis-acting element that could bind with transcription factor NFκB p65 in vitro. To further confirm the binding, MIF-NFκB-A and MIF-NFκB-C were labeled with IRDye 700 and used as probes. After incubating MIF-NFκB-A probe with NFκB p65-enriched HEK nuclear extract, a shifted band was observed (Fig. 2E, lane 2), which was completely abolished by adding 100-fold molar excess of unlabeled wt-NFκB as the competitor (Fig. 2E, lane 3) but not by the same amount of mu-NFκB (Fig. 2E, lane 4). Addition of anti-NFκB p65 monoclonal antibody further retarded the shift of the DNA-protein complex (Fig. 2E, lane 5), and the supershifted band was abolished by adding 100-fold molar excess of the unlabeled wt-NFκB (Fig. 2E, lane 6). The same results were observed for MIF-NFκB-C (Fig. 2F). These results clearly demonstrate that the MIF promoter contains 2 cis-acting elements for NFκB binding.
Next, a ChIP assay was performed to examine whether NFκB could recognize the consensus sequences on the MIF promoter under physiological conditions. NFκB p65 monoclonal antibody was used to precipitate the DNA fragments that could bind to NFκB p65. DNA fragments from the immunoprecipitates pulled down by NFκB p65 antibody were amplified by PCR using 3 pairs of MIF-NFκB putative consensus binding site-specific primers. The results confirmed that sequences corresponding to MIF-NFκB-A and -C but not MIF-NFκB-B were amplified (Fig. 2G). β-Actin, as the control for antibody specificity, could not be amplified after immunoprecipitation (Fig. 2G). Sheared gDNA without immunoprecipitation was amplified by all the pairs of primers, indicating that the target DNA sequences on the MIF promoter were intact due to their binding to NFκB p65, while after sham immunoprecipitation, none of these pairs of primers amplified the targeting sequences, indicating that none of our tested DNA sequences could be precipitated nonspecifically (Fig. 2G). Taken together, these results confirm that the two cis-acting elements MIF-NFκB-A and -C on the human MIF promoter were specifically recognized by transcription factor NFκB under physiological conditions.
NFκB down-regulates MIF gene transcription under hypoxia
To investigate whether NFκB signaling regulates transcriptional activation of MIF gene expression via the cis-acting elements identified in the MIF promoter, a series of MIF gene promoter deletion plasmids were constructed and assayed. Previously, a 2.6 kb human MIF promoter from −2634 to +35 bp was cloned into a luciferase reporter plasmid to generate phMIF-luc, and this promoter activity was up-regulated under hypoxia (21). In the present study, MIF promoter constructs were cotransfected with an NFκB p65 expression plasmid or its backbone as the control into cells, and the promoter activities were measured by luciferase assay. Overexpression of NFκB p65 significantly increased the MIF promoter activity to 1.99 ± 0.09-fold of the control (P<0.05; Fig. 3A), further confirmed by Western blot showing that the luciferase protein level was increased to 1.33 ± 0.12-fold of the control (P<0.05; Supplemental Fig. S1A, B). To further investigate the effect of NFκB signaling on the MIF promoter, plasmid phMIF-5kluc, containing −5415 to +35 bp of the MIF promoter, was cotransfected with an NFκB p65 expression plasmid. The promoter activity of phMIF-5kluc was elevated by 2.33 ± 0.19-fold compared to control (P<0.05), similar to the effect on phMIF-luc (Fig. 3A). To confirm the cis-acting elements' effect, the MIF-NFκB oligonucleotides containing the binding sites (Fig. 2A) were cloned into a modified luciferase reporter vector, pGL-pl (41), to generate the plasmid pMIF-NFκBluc. This plasmid was created to exclude the interference of other elements with the interaction between NFκB and the MIF promoter. NFκB p65 overexpression markedly increased the promoter activity of pMIF-NFκBluc by 33.3 ± 1.9-fold (P<0.001; Fig. 3A). To determine the contribution of the individual binding sites to the promoter activity on NFκB activation, plasmids containing MIF promoter fragments with different NFκB binding sites and/or pseudo-binding sites were generated (Fig. 3B). Notably, overexpression of NFκB did not affect the promoter activity of phMIF-Dluc that lacked NFκB binding sites. In contrast, the promoter activities of phMIF-luc, phMIF-Bluc, and phMIF-Cluc were significantly increased on overexpression of NFκB p65 by 1.53 ± 0.17-, 1.33 ± 0.11-, and 1.38 ± 0.06-fold (P<0.05; Fig. 3B). The results clearly demonstrate that activation of NFκB signaling enhances MIF promoter activity.
Figure 3.

Regulation of human MIF gene expression by NFκB signaling. A) HEK293 cells were cotransfected with MIF promoter plasmids and NFκB p65 expression plasmid or its backbone. Cells were harvested with passive lysis buffer and measured for luciferase activity. Firefly luciferase activity was normalized to Renilla luciferase activity for transfection efficiency control, and expressed as RLU to reflect the promoter activity. Values are expressed as means ± sd, n = 6. *P < 0.001 vs. controls; 2-way ANOVA with Bonferroni posttests. B) Schematic diagram of deletion plasmids containing different human MIF promoter fragments in front of the firefly luciferase reporter gene. Vertical arrows indicate starting location of the putative NFκB binding sites. Deletion plasmids were confirmed by sequencing and restriction enzyme digestion checking, and digested samples were analyzed on a 0.7% agarose gel. Vector size is 4.7 kb, and the MIF promoter fragment inserts range from ∼230 bp to 2.7 kb. Luciferase assay was performed as in A. Values are expressed as means ± sd, n = 6. *P < 0.001 vs. controls; 2-way ANOVA with Bonferroni posttests. C) N2A cells were transfected with NFκB expression plasmid. Total RNA was extracted from cells by Tri reagent, and semiquantitative RT-PCR was used to measure mouse MIF mRNA in N2A cells with mouse MIF gene-specific primers. GAPDH mRNA was used as the internal control. D) Quantification of C. Level of MIF mRNA is expressed as relative ratio to GAPDH. Values are expressed as means ± sd, n = 3. *P < 0.01 vs. controls; Student's t test. E) N2A cells were transfected with NFκB expression plasmid. Protein levels of MIF in N2A cells were assessed by Western blot on a 12% Tris-tricine SDS-PAGE gel. MIF was detected by anti-MIF antibody; β-actin was used as internal control. F) Quantification of E. Level of MIF protein is expressed as relative ratio to β-actin. Values are expressed as means ± sd, n = 3. *P < 0.01 vs. controls; Student's t test. G) HEK293 cells were transfected with MIF promoter plasmids and subjected to hypoxia treatment or cotransfected with NFκB p65 expression plasmid. Promoter activities were evaluated as in A. Values are expressed as means ± sd, n = 6. *P < 0.01 vs. controls; 2-way ANOVA with Bonferroni posttest. H) HEK293 cells were cotransfected with NFκB p65 expression plasmid with additional hypoxia treatment, and promoter activities were evaluated as in A. Values are expressed as means ± sd, n = 6. *P < 0.01 vs. controls; 2-way ANOVA with Bonferroni posttests. I) HEK293 cells were transfected with NFκB expression plasmid and subjected to hypoxia (2% of oxygen balanced by nitrogen). Total RNA was extracted from cells by Tri reagent, and semiquantitative RT-PCR was used to measure MIF mRNA with MIF gene specific primers. GAPDH mRNA was used as the internal control. J) Quantification of I. Values are expressed as means ± sd, n = 3. *P < 0.001 vs. controls; Student's t test. K) HEK293 cells were transfected with NFκB expression plasmid and subjected to hypoxia (2% of oxygen balanced by nitrogen). Protein levels of MIF were assessed by Western blot on a 12% Tris-tricine SDS-PAGE gel. L) Quantification of K. Values are expressed as means ± sd, n = 3. *P < 0.01 vs. controls; Student's t test.
To examine the effect of NFκB signaling on endogenous MIF gene expression, an NFκB p65 expression plasmid was transfected into N2A and HEK293 cells. At 24 h after transfection, cells were harvested for mRNA and protein level analysis. NFκB p65 expression significantly increased MIF expression at both the mRNA (Fig. 3C) and protein (Fig. 3E) levels to ∼1.68 ± 0.11-fold (P<0.05; Fig. 3D) and 2.42 ± 0.36-fold of the controls (P<0.05; Fig. 3F) in N2A cells, respectively. The endogenous levels of MIF mRNA (Supplemental Fig. S1C) and protein (Supplemental Fig. S1E) further were significantly increased in HEK293 cells after transfection with NFκB p65 expression plasmid. The level of MIF mRNA was increased to 1.29 ± 0.08-fold of the control (P<0.05; Supplemental Fig. S1D), and the protein level was increased to 1.70 ± 0.25-fold of the control (P<0.05; Supplemental Fig. S1F).
Microglia cells are the major immune cell type in the brain responding to stroke. Therefore, we investigated the effect of NFκB signaling activation on MIF expression in BV-2 cells, a mouse microglia cell line. NFκB p65 expression plasmid was transfected into BV-2 cells. At 24 h after transfection, cells were harvested for mRNA and protein level analysis. MIF mRNA expression in BV-2 cells was significantly up-regulated by 1.42 ± 0.16-fold of the control (P<0.05; Supplemental Fig. S1G, H); and the protein level of MIF also was elevated by 1.93 ± 0.12-fold of the control (P<0.05; Supplemental Fig. S1I, J).
Taken together, these results clearly demonstrate that MIF gene expression is up-regulated by activation of NFκB signaling. We have shown that both hypoxia (21) and activation of NFκB signaling alone result in up-regulation of MIF gene expression. It remained intriguing as to why MIF expression was down-regulated in the MCA territory at 6 h after the onset of stroke, when massive tissue decomposition has yet to occur. A previous report showed that, although the CRE on the MIF promoter was responsive to CRF-mediated MIF gene up-regulation (27), it also is involved in MIF gene down-regulation under hypoxic conditions (28). Given that the HREs on the MIF promoter were 33 and 43 bp upstream of MIF-NFκB-A and -C, respectively (Supplemental Fig. S1K), we examined the effect of NFκB signaling on MIF promoter activity under hypoxia, which better mimics the condition of NFκB activation following ischemia. The promoter activity of phMIF-luc was elevated by either hypoxia (2.66±0.20-fold of the control, P<0.05) or transfection of NFκB p65 alone (5.66±0.17-fold of the control, P<0.05), whereas HREs-deficient plasmid phMIFδH-luc responded to NFκB p65 overexpression with a 10.43 ± 0.20-fold increase in activity (P<0.05), but did not respond to hypoxia (Fig. 3G). Successful induction of signaling pathways were confirmed by enhanced promoter activities of positive control plasmids pEpoE-luc by hypoxia and pNFκB-luc by NFκB (data not shown). To assess the effect of hypoxia on NFκB signaling-mediated MIF promoter activity, the NFκB p65 expression plasmid, cotransfected cells were subjected to hypoxia treatment. Dual activation of hypoxia and NFκB signaling resulted in significant reduction of the luciferase activity of phMIF-luc to 0.86 ± 0.03 of the control (P<0.05), while the elevation of the promoter activity of phMIFδH-luc by NFκB signaling was not affected by additional activation of the hypoxia signaling pathway (Fig. 3H). These data indicate that induction of HIF-1α by hypoxia does not directly affect the ability of the cis-acting elements in the MIF gene promoter to bind NFκB and the reduction of promoter activity under dual signals most likely was due to the interaction between the HRE and NFκB binding motif on the MIF promoter.
We further examined the effect of NFκB signaling on endogenous MIF expression under hypoxic conditions. Dual activation of hypoxia and NFκB signaling was achieved by overexpressing NFκB p65 expression plasmid under hypoxic condition. MIF mRNA (Fig. 3I) and protein (Fig. 3K) expression was reduced significantly to 0.80 ± 0.05-fold (P<0.05; Fig. 3J) and 0.16 ± 0.01-fold (P<0.05; Fig. 3L), respectively. Taken together, these results indicate that activation NFκB signaling under hypoxic condition contributes to down-regulation of endogenous MIF gene expression.
MIF protects neurons from oxidative stress- and in vitro ischemia/reperfusion-induced apoptosis by attenuating caspase-3 activation
Our results suggested that activation of NFκB signaling under hypoxic conditions contributes to down-regulation of MIF expression in the MCA territory during the acute phase of stroke. To further determine the role of MIF in stroke, we first assessed the function of MIF on cultured primary cortical neurons under strokelike conditions. Strokelike conditions were induced in cultured mouse cortical neurons by an in vitro model of ischemia/reperfusion. The primary neuronal culture was challenged in glucose-free medium under hypoxic condition for 2 h (ischemic period), the culture medium then replaced by normal medium, followed by 16 h of incubation under normoxic conditions (reperfusion). In vitro ischemia/reperfusion resulted in significant loss of neurons, 88.08 ± 2.93% viability (P<0.01), measured by a MTS assay (Fig. 4A). The effect of MIF was assessed by adding human purified MIF protein (Supplemental Fig. S2A). Treatment of cortical neurons with purified human MIF at a concentration of 200 ng/ml during ischemia and reperfusion periods completely rescued the detrimental effect induced by in vitro ischemia/reperfusion with cell viability of 100.04%±3.34% (P<0.001; Fig. 4A). These results suggest a protective role for MIF on neurons under in vitro ischemia-reperfusion-like conditions.
Figure 4.
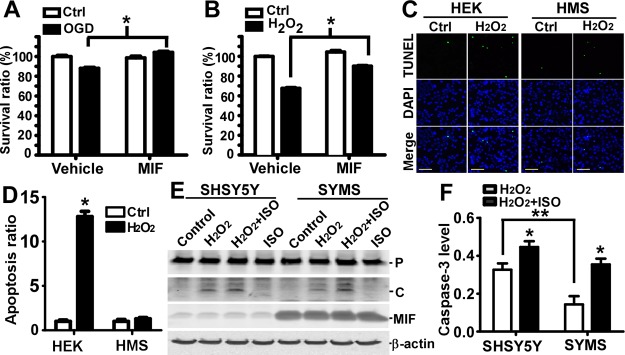
MIF protected neurons by inhibiting caspase-3 activation. Cortical neurons cultured from E14 mouse embryos were subjected to an in vitro model of ischemia/reperfusion or oxidative stress. A) In vitro ischemia/reperfusion was achieved by challenging the primary neuronal culture in glucose-free medium under hypoxic conditions for 2 h (ischemia period), followed by 16 h incubation under normoxic conditions after medium exchange to normal medium (reperfusion period). Purified human his-tagged MIF (200 ng/ml) or vehicle was added in the medium during the entire experiment. Cell viability was assessed by using MTS assays. Survival ratio represents the average absorbance to control absorbance. Values represent means ± sd, n = 10. *P < 0.01; Student's t test. B) Oxidative stress was induced by 50 μM of H2O2 for 16 h in normal medium. Purified human his-tagged MIF (200 ng/ml) or vehicle was added in the medium during the entire experiment. Cell viability was assessed by using MTS assays. Survival ratio represents the average absorbance to control absorbance. Values represent means ± sd, n = 6, *P < 0.001; Student's t test. C) HEK293 and MIF stable HMS cells were treated with 50 μM H2O2 for 24 h in medium lacking sodium pyruvate, followed by TUNEL assay (green channel). DAPI (blue channel) was used to stain nuclei. Scale bars = 100 μm. D) Quantification of cell numbers identified by DAPI staining and apoptotic cells identified by TUNEL labeling by ImageJ. Signals were averaged from 5 randomly selected views. Values represent means ± sd, n = 3. *P < 0.01 vs. cells without H2O2 treatment; 2-way ANOVA with Bonferroni posttests. E) SHSY5Y and MIF stable SYMS cells were treated with 200 μM H2O2 for 16 h. MIF inhibitor ISO-1 (50 μM) was added to block the effect of MIF. Cells were lysed by Chap cell extract buffer. Lysate was resolved on a 12% Tris-tricine SDS-PAGE gel, and protein levels were analyzed by Western blot. An anti-caspase-3 antibody recognizing both pro- and cleaved caspase-3 was used. F) Quantification of E. Ratio of the cleaved form of caspase-3 to β-actin level was analyzed. Values represent means ± sd, n = 3. *P < 0.01; 2-way ANOVA with Bonferroni posttests.
Oxidative stress contributes to cell death induced by ischemia, particularly following reperfusion (50). To determine the effect of MIF on oxidative stress-induced neuronal death, primary cultures of cortical neurons isolated from mouse embryos were treated with H2O2. H2O2 treatment of cortical neurons resulted in a significant decrease in neuronal viability to 67.52 ± 1.18% vs. control (P<0.01), as assessed by the MTS assay (Fig. 4B). When cortical neurons were incubated with purified MIF at a concentration of 200 ng/ml, H2O2-induced cell death was significantly prevented, and cell viability was increased to 90.06 ± 1.03% relative to control (P<0.01; Fig. 4B). These results clearly demonstrate that MIF rescues neuronal loss following strokelike conditions in vitro and may be particularly protective against reperfusion induced oxidative stress.
To study the effect of MIF on neuronal apoptosis, HEK293 and SHSY5Y were transfected with a human MIF expression plasmid to generate HMS (Supplemental Fig. S2B) and SYMS stable cell lines (Supplemental Fig. S2D). HMS and SYMS robustly express human MIF protein 2.46 ± 0.14-fold (Supplemental Fig. S2C) and 7.02 ± 0.40-fold (Supplemental Fig. S2E) compared to control in cell lysates detected by Western blot. MIF levels in culture medium of these cell lines, as detected by ELISA, were also significantly increased from 18.5 ± 1.11 to 54.30 ± 3.29 ng/ml in SYMS (P<0.01) and 32.00 ± 2.36 to 384.39 ± 20.05 ng/ml in HMS (P<0.01) stable cells, respectively (Supplemental Fig. S2F). To further examine the protective role of MIF, TUNEL assay was performed. H2O2 treatment at a concentration of 50 μM significantly induced cell death in HEK293 cells (Fig. 4C), resulting in 12.81 ± 1.01-fold increase of TUNEL-positive cells (P<0.01; Fig. 4D). In contrast, overexpression of MIF in HMS cells (Fig. 4C) blocked the H2O2-induced cell death (Fig. 4D). H2O2 treatment at a concentration of 100 μM resulted in complete detachment of HEK cells 24 h after treatment, whereas 33.44 ± 2.52% HMS cells survived after treatment, and 7.82 ± 1.70% of the surviving cells were TUNEL positive (data not shown). Our results clearly demonstrate that MIF is essential in combating oxidative stress-induced cell death and promoting cell survival under such conditions.
Since MIF exhibited strong rescue effects on neurons subjected to H2O2 treatment, and H2O2-induced oxidative stress triggers activation of the caspase cascade, we sought to investigate whether MIF's protective effect was mediated through the caspase signaling pathway. SYMS and SHSY5Y cells were treated with H2O2 for 16 h, and caspase-3 cleavage was analyzed by Western blot. In the control groups, no cleaved form could be detected in both cell lines (Fig. 4E). H2O2 treatment markedly induced the cleaved active form of caspase-3 in both cell lines (Fig. 4E). However, there was 38.40 ± 6.45% (P<0.01) lower levels of the cleaved caspase-3 form in SYMS than in SH-SY5Y control cells. Furthermore, addition of the MIF inhibitor ISO-1 significantly increased the level of the cleaved form of caspase-3 by 1.36 ± 0.05-fold in SHSY5Y and 2.47 ± 0.12-fold in SYMS (P<0.001; Fig. 4F).
Disruption of MIF gene expression accelerates neuronal death within the MCA territory
To examine the effect of MIF on neuronal apoptosis during the acute phase of stroke in vivo, TUNEL assays were performed on MIF-knockout (Mif−/−) and wild-type (Mif+/+) mice subjected to experimental stroke. The results showed that Mif−/− mice developed TUNEL-positive cells in the MCA territory as early as 6 h after the onset of ischemia, and nuclei in this area appeared to be condensed (Fig. 5B). In contrast, nuclei in the MCA territory displayed healthy appearance, and no TUNEL-positive cells appeared in Mif+/+ mice (Fig. 5A). At 8 h after ischemia onset, the number and area of TUNEL-positive cells were significantly increased in Mif−/− mice, while scarce TUNEL-positive cells developed in Mif+/+ mice (data not shown). Consistent with the TTC staining results, the difference between Mif−/− and Mif+/+ mice is less evident at 24 h after stroke, 54.65 ± 9.17% in Mif+/+ vs. 65.58 ± 6.75% in Mif−/− mice (P>0.05; Fig. 5B).
Figure 5.
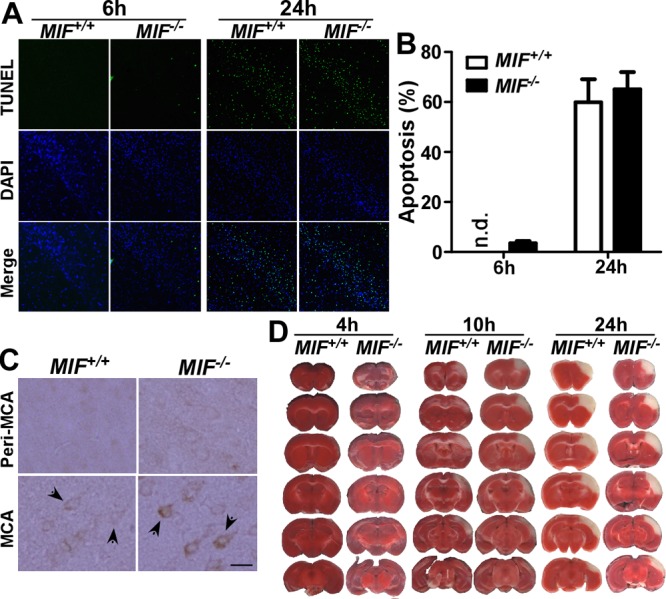
MIF reduced cell death and infarct development during stroke. A) Wild-type (Mif+/+) and MIF-knockout (Mif −/−) mice were subjected to 2 h tMCAl, followed by 4 or 22 h reperfusion, and the brain slices were analyzed for cell apoptosis by TUNEL assay (green channel). Nuclei were stained with DAPI (blue channel). Representative images were taken from the same location in the MCA territory. B) Average counts of the nuclei and TUNEL-positive cells from 5 selected views were quantified by ImageJ; bars represent percentage of TUNEL signal. Values represent means ± sd, n = 3. P > 0.05; Student's t test. C) Cleaved caspase-3 was determined histologically in wild-type and MIF-knockout mice 4 h after 2 h tMCAl. Sample images were taken from the cortical peri-MCA region and MCA territory. Activation of caspase-3 is indicated by positive staining of the cleaved form of caspase-3. Arrows indicate caspase-3-positive cells. Scale bar = 20 μm. D) Mif+/+ and Mif−/− mice were subjected to 2 h tMCAl on the right hemisphere, followed by 2, 8 or 22 h reperfusion. The brain was freshly cut into 1 mm slices and subjected to TTC staining.
We have shown that MIF protects neurons from oxidative stress induced apoptosis by inhibiting activation of caspase-3 in vitro. To examine whether MIF could exert the same effect in vivo following experimental stroke, Mif−/− and Mif+/+ mice were subjected to tMCAl, and the cleaved form of caspase-3 was determined histologically 2 h after reperfusion. To avoid batch to batch variation, immunostaining was performed side by side for Mif+/+ and Mif−/− mice. Sample images were taken from the peri-MCA region and MCA territory for evaluation of caspase-3 activation (Fig. 5C). In the peri-MCA region, both wild-type and knockout mice displayed weak background caspase-3 signal (Fig. 5C, top panels). Cleaved caspase-3 was detected in the MCA territory in Mif−/− and Mif+/+ mice, and Mif−/− mice displayed stronger staining signals for the cleaved form of caspase-3 in the MCA territory compared to Mif+/+ wild-type mice (Fig. 5C, bottom panels). Taken together, these results demonstrate that MIF negatively regulates caspase-3 and, in turn, protects against neuronal death during the acute phase of stroke, and disruption of MIF gene expression accelerates neuronal death and infarct development within the MCA territory.
Focal ischemia-induced infarction was routinely examined to assess stroke outcome. Since MIF promoted neuronal survival, we assessed whether it affected infarct formation in vivo. Wild-type and MIF-knockout mice were subjected to tMCAl. At 4, 10 and 24 h after the onset of ischemia, TTC staining was performed to assess infarct development during the acute phase of stroke (Fig. 5D). Loss of TTC staining was not significant at 4 h after stroke onset in both wild-type and MIF-knockout mice. Impaired cell activity/incomplete infarction in the MCA-supplied region, indicated by partial loss of TTC staining, was observed at 10 h after stroke onset, and Mif−/− mice exhibited a significantly greater loss of TTC staining, including the development of a complete infarct, compared to the wild-type mice (Fig. 5D). The infarct volume was 6.51 ± 1.48% for the wild-type mice and 9.07 ± 1.32% for MIF-knockout mice (n=4, P<0.05). However, both Mif+/+ and Mif−/− mice exhibited complete loss of TTC staining 24 h after stroke onset, and no difference was found in terms of the infarct volume (Fig. 5D). The infarct volume was 9.91 ± 0.69% and 9.13 ± 0.71% for wild-type and knockout mice, respectively (n=3, P>0.05). These results indicate that MIF deficiency results in loss of cell function relatively early following stroke and accelerates development of an infarct. Interestingly, the protective effect of MIF in the MCA territory in MIF+/+ mice diminished during later phases of infarct development.
DISCUSSION
MIF is expressed in many organs, including the brain, and as a pleiotropic protein is involved in a variety of biological processes. It plays fundamental roles in inducing inflammatory responses and regulating cell death and survival (11, 13–15). A recent review systematically summarized the most up-to-date studies on the roles of MIF on cardiomyocytes in vitro and in vivo, and its relevance to clinical outcomes after heart ischemia, and suggested multiple mechanisms underlying the protective effect of MIF (51). Stroke is detrimental to neurons in a similar way as a myocardial infarct is to cardiomyocytes. However, the effect of MIF on neurons experiencing ischemia remains elusive. A recent study showed that Mif−/− mice had smaller infarcts compared to wild-type mice following focal ischemia (23). We previously demonstrated altered MIF expression following stroke with initial up-regulation and eventual down-regulation (21), which suggests the expression pattern of MIF could affect its function. In the present study, we examined the temporospatial expression of MIF and its role during stroke. We adapted a stroke model that produced infarction in the frontal and parietal cortex, which is supplied mainly by the MCA. Infarction induced by this stroke model is highly reproducible. Compared to other focal stroke models, such as proximal MCA occlusion (MCAo), in which the developed infarct spreads beyond the cerebral cortex, this model produces smaller infarcts restricted to the MCA-supplied cerebral cortex, providing us an opportunity to assess the expression and function of MIF in a relatively homogenous cell group, especially for neurons. Our results demonstrate that MIF expression is selectively down-regulated in the infarcted MCA territory, while MIF expression is not affected in the posterior cerebral artery/anterior cerebral artery-supplied peri-MCA area, despite its direct connection to the infarcted MCA territory. Our observations are consistent with a previous study showing that MIF is expressed in the peri-infarct area up to 72 h after stroke, and diminishes in the ischemic core 3 h after ischemia (23).
Both hypoxia and NFκB signaling pathways are activated following stroke in the infarct area (35, 49, 52). In previous studies, activation of NFκB signaling within the area that later became infarcted was shown by EMSA using brain lysates, and nuclear localization was observed histologically (35, 49). Stephenson et al. (49) further attributed early activation of NFκB signaling to neuronal rather than glial cells. In this study, we confirmed nuclear localization of NFκB in the MCA territory, and it was mainly diffuse in the adjacent peri-MCA area. Hypoxia activates HIF-1α, which mediates up-regulation of MIF gene expression via interaction with HREs on the MIF promoter (21, 28, 29). The present study identifies two functional cis-acting NFκB binding elements in the MIF promoter through which activation of NFκB signaling regulates MIF gene expression by transcriptional activation of the MIF promoter. We found that activation of either hypoxia or the NFκB pathway alone drives up-regulation of MIF gene transcription. However, NFκB signaling was activated under hypoxic conditions, leading to an inhibition of MIF gene expression. Since deletion of HREs in the MIF promoter eliminated the direct effect of HIF-1α on promoter activity and preserved the ability of NFκB to up-regulate gene expression during hypoxia, it was likely that an interaction could have occurred between HRE and NFκB binding elements on the MIF promoter under hypoxic conditions. Previous studies demonstrated that HIF-1α is significantly induced in the infarcted area 1 h after stroke onset, peaks at 12 h, and gradually declines thereafter (52), and a significant up-regulation of NFκB signaling was also observed in a similar time window (49). Furthermore, it was reported that, under hypoxia, the induction of a HIF-1α mediated HRE reporter gene occurred before NFκB signaling mediated the NFκB responsive element (NRE) reporter gene (53), indicating that activation of HIF-1α precedes that of NFκB signaling. Taken together, it is likely that, in the infarcted area, rapid activation of HIF-1α contributes to the initial up-regulation of MIF and that subsequent activation of NFκB signaling suppresses MIF expression thereafter. Activation of NFκB signaling after stroke is evident despite the controversial conclusions as to whether it is protective or deleterious to stroke outcome (36). It is plausible that MIF may serve as a downstream effector responsible for synchronizing the upstream regulatory signals, such as NFκB signaling, and, in turn, determining cell fate. Our results suggest that MIF expression is tightly regulated by NFκB and hypoxia and could serve as an effector that directly regulates cell survival during the acute phase of stroke.
Anti-inflammation and inhibition of apoptosis are two key therapeutic approaches to reduce stroke-related neurological dysfunction. Interestingly, the function of MIF has been implicated in both of these processes, being antiapoptotic but proinflammatory. MIF exhibits strong antiapoptotic properties by suppressing p53 activity by transcription inhibition and negatively regulating JNK signaling (13–15). Under ischemic conditions, MIF is protective by stimulating AMP-activated protein kinase during ischemia-reperfusion induced energy deprivation in the heart (17). However, the main effector of this pathway, GLUT4, only has been shown to function in the cerebellum (54). Recently, a similar study in heart ischemia observed increased infarction in Mif−/− mice and suggested a protective effect of MIF via reduction of oxidative stress-induced reactive oxygen species (ROS) accumulation and cytochrome c release (55). Oxidative stress induced by massive ROS generation also occurs after stroke and is one of the major insults leading to neuronal apoptosis (50). Neurons undergoing apoptosis during stroke are often the target for therapeutic interventions. Cell apoptosis under stroke conditions may be activated through caspase-dependent pathways involving the dysfunction of mitochondria (2). It was reported that postischemic reperfusion resulted in the overproduction of ROS in mitochondria (56), in turn affecting the release of cytochrome c and leading to caspase-3 activation. The role of MIF in reducing ROS levels has been observed during heart ischemia (55), and it was suggested that MIF's CXXC motif (Cys57-Ala-Leu-Cys60) plays a role in redox balance under stressed conditions (19, 20). In our study, we clearly demonstrate that stably expressing MIF in a human neuronal cell line reduced casapase-3 cleavage induced by H2O2 treatment, and this effect was diminished after addition of ISO-1, a MIF inhibitor (57). The effect of MIF expression on inhibition of caspase-3 activation under oxidative stress and reduction of TUNEL staining confirmed the protective role of MIF. We also observed these characteristics of MIF in vivo. In MIF knockout mice, caspase-3 activation in the infarct area was markedly increased when compared to wild-type mice.
An antiapoptotic role for MIF in suppressing JNK activation by direct interaction with its coactivator Jab1 also has been suggested (14). It also has been reported that knockout of JNK3, which among the 3 JNKs (JNK1, JNK2, and JNK3) is expressed primarily in the brain, contributes to neuronal resistance to excitotoxicity (58, 59). Also of note, we observed a protective effect of MIF in reducing NMDA induced excitotoxicity in primary neuronal cultures (data not shown). Furthermore, we showed that MIF exhibited a strong effect in combating oxidative stress induced by H2O2. In addition, we observed faster neuronal apoptosis in Mif−/− mice at 6 and 8 h after stroke onset, and deletion of the MIF gene accelerated infarct development during the acute phase of stroke. Taken together, these data clearly support a neuroprotective role for MIF during stroke.
The area of the infarct gradually extends to adjacent areas, lasting for several days following the acute phase of stroke. This late-phase process is slower when compared to the infarction that evolves in the area directly supplied by the blocked artery during the acute phase of stroke. This circumstance suggests that different mechanisms may be involved between acute and late phase infarct development. Interestingly, a recent study assessed the infarct volume at d 7 following transient MCAo and found that Mif+/+ mice developed slightly larger infarcts when compared to Mif−/− mice (23). Although no differences in cytokine production were found between the Mif+/+ and Mif−/− mice during these 7 d (22), the results indeed demonstrated that MIF mediated the response of macrophages/microglia to ischemia (23). The role of MIF in initiating and sustaining inflammatory responses has been well characterized outside of the central nervous system. It has been shown that constitutive expression of MIF is essential to combat common infections, and knockout of the Mif gene can result in higher mortality to certain infections (60) but protection from excessive host inflammation (61). A protective effect of MIF has been attributed to its unique ability to override the anti-inflammatory effects of glucocorticoids (61), thereby promoting the production of proinflammatory cytokines (60, 61). In addition, constitutive expression of MIF maintains the surface expression of TLR4, which, in turn, is responsible for sensing infection and initiating a host defense response (11). These characteristics of MIF suggest that MIF both has an antistress role and is necessary to activate the first line of host defense. In contrast, under chronic inflammatory conditions, such as rheumatoid arthritis, atherosclerosis, and Alzheimer's disease, MIF is persistently up-regulated (62, 63). Recruitment of macrophages is involved in the pathology of rheumatoid arthritis and atherosclerosis and was shown to be mediated by the interaction between MIF expressed at the lesion site and the chemokine receptors on inflammatory cells (64). Although the mechanism for sustained MIF expression is unknown, in comparison to the effect of MIF during acute inflammation, MIF appears to have a predominant deleterious effects in chronic inflammation. Similar mechanisms may explain the late-phase infarct development. Up-regulation of MIF expression in the peri-infarct area in response to acute stress induces a macrophage/microglia response, which, in turn, contributes to a larger infarct volume by prolonging infarct development. MIF could play dual roles following ischemia depending on the time window. It worth noting that a recent study on heart ischemia took into consideration the severity of the ischemic insult and assessed infarct volume during both early and late phases of infarct development. It reported that mice lacking MIF expression developed larger infarcts following mild ischemic insult (15 min occlusion) during the early phase (4 h after the ischemic insult) of infarct development, while this protective effect of MIF was not observed at 24 h following an ischemic insult induced for 30 min (55). This observation is in concordance with our results and is likely due to the loss of MIF expression.
To date, inhibition of MIF has been suggested in many therapeutic strategies, especially in inflammation-induced diseases. Our results show that down-regulation of MIF by NFκB signaling accelerates neuronal apoptosis during stroke, which suggests that use of a MIF inhibitor may reduce the already narrow therapeutic windows following stroke and confer a higher risk of ischemic damage. Our study indicates that MIF protects neurons against ischemia and a strategy to control MIF expression at a therapeutic level could have beneficial effects for patients with stroke.
Supplementary Material
Acknowledgments
The authors thank Dr. Cheryl Wellington [University of British Columbia (UBC)] for providing the BV-2 cell line.
This work was supported by Canadian Institutes of Health Research (CIHR) operating grant MOP-97825, the Jack Brown and Family Alzheimer's Research Foundation (to W.S.), and U.S. National Institutes of Health grant AI042310 (to R.B.). W.S. is the holder of the Tier 1 Canada Research Chair in Alzheimer's Disease. Si Z. is the recipient of a National Sciences and Engineering Research Council of Canada–Canada Graduate Scholarships Doctoral (NSERC-CGSD) scholarship and a UBC 4YF Scholarship. M.Z. is supported by UBC 4YF Scholarship. Sh.Z. was supported by the Chinese Scholarship Council award.
The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ChIP
- chromatin immunoprecipitation
- CRE
- cyclic-AMP response element
- E
- embryonic day
- EMSA
- electrophoretic mobility shift assay
- GSA
- gel shift assay
- HIF-1α
- hypoxia-inducible factor 1α
- HEK293
- human embryonic kidney 293
- HRE
- hypoxia responsive element
- IP
- immunoprecipitation
- MCA
- middle cerebral artery
- MCAl
- middle cerebral artery ligation
- MCAo
- middle cerebral artery occlusion
- MIF
- macrophage migration inhibitory factor
- mu-NFκB
- mutant nuclear factor κB
- NFκB
- nuclear factor κB
- OGD
- oxygen-glucose deprivation
- PFA
- paraformaldehyde
- rCCA
- right common carotid artery
- RLU
- relative luciferase unit
- rMCA
- right middle cerebral artery
- ROS
- reactive oxygen species
- tMCAl
- transient distal middle cerebral artery ligation
- TTC
- 2,3,5-triphenyl-tetrazolium chloride
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
- wt-NFκB
- wild-type nuclear factor κB
REFERENCES
- 1. Donnan G. A., Fisher M., Macleod M., Davis S. M. (2008) Stroke Lancet 371, 1612–1623 [DOI] [PubMed] [Google Scholar]
- 2. Broughton B. R., Reutens D. C., Sobey C. G. (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40, e331–339 [DOI] [PubMed] [Google Scholar]
- 3. Iadecola C., Anrather J. (2011) The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lipton P. (1999) Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568 [DOI] [PubMed] [Google Scholar]
- 5. Liu Y., Wong T. P., Aarts M., Rooyakkers A., Liu L., Lai T. W., Wu D. C., Lu J., Tymianski M., Craig A. M., Wang Y. T. (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amor S., Puentes F., Baker D., van der Valk P. (2010) Inflammation in neurodegenerative diseases. Immunology 129, 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiong X., Barreto G. E., Xu L., Ouyang Y. B., Xie X., Giffard R. G. (2011) Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42, 2026–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ziegler G., Freyer D., Harhausen D., Khojasteh U., Nietfeld W., Trendelenburg G. (2011) Blocking TLR2 in vivo protects against accumulation of inflammatory cells and neuronal injury in experimental stroke. J. Cereb. Blood Flow Metab. 31, 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang L., Zhang Z. G., Zhang R. L., Lu M., Krams M., Chopp M. (2003) Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 34, 1790–1795 [DOI] [PubMed] [Google Scholar]
- 10. Lazovic J., Basu A., Lin H. W., Rothstein R. P., Krady J. K., Smith M. B., Levison S. W. (2005) Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin-1 type 1 receptor-deficient mice conferring neuroprotection. Stroke 36, 2226–2231 [DOI] [PubMed] [Google Scholar]
- 11. Calandra T., Roger T. (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 3, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernhagen J., Calandra T., Mitchell R. A., Martin S. B., Tracey K. J., Voelter W., Manogue K. R., Cerami A., Bucala R. (1993) MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365, 756–759 [DOI] [PubMed] [Google Scholar]
- 13. Hudson J. D., Shoaibi M. A., Maestro R., Carnero A., Hannon G. J., Beach D. H. (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 190, 1375–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleemann R., Hausser A., Geiger G., Mischke R., Burger-Kentischer A., Flieger O., Johannes F. J., Roger T., Calandra T., Kapurniotu A., Grell M., Finkelmeier D., Brunner H., Bernhagen J. (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408, 211–216 [DOI] [PubMed] [Google Scholar]
- 15. Mitchell R. A., Liao H., Chesney J., Fingerle-Rowson G., Baugh J., David J., Bucala R. (2002) Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc. Natl. Acad. Sci. U. S. A. 99, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amaral F. A., Fagundes C. T., Guabiraba R., Vieira A. T., Souza A. L., Russo R. C., Soares M. P., Teixeira M. M., Souza D. G. (2007) The role of macrophage migration inhibitory factor in the cascade of events leading to reperfusion-induced inflammatory injury and lethality. Am. J. Pathol. 171, 1887–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller E. J., Li J., Leng L., McDonald C., Atsumi T., Bucala R., Young L. H. (2008) Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451, 578–582 [DOI] [PubMed] [Google Scholar]
- 18. Koga K., Kenessey A., Powell S., Sison C. P., Miller E. J., Ojamaa K. (2011) Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid. Redox Signal. 14, 1191–1202 [DOI] [PubMed] [Google Scholar]
- 19. Nguyen M. T., Beck J., Lue H., Funfzig H., Kleemann R., Koolwijk P., Kapurniotu A., Bernhagen J. (2003) A 16-residue peptide fragment of macrophage migration inhibitory factor, MIF-(50–65), exhibits redox activity and has MIF-like biological functions. J. Biol. Chem. 278, 33654–33671 [DOI] [PubMed] [Google Scholar]
- 20. Nguyen M. T., Lue H., Kleemann R., Thiele M., Tolle G., Finkelmeier D., Wagner E., Braun A., Bernhagen J. (2003) The cytokine macrophage migration inhibitory factor reduces pro-oxidative stress-induced apoptosis. J. Immunol. 170, 3337–3347 [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Zis O., Ma G., Shan Z., Zhang X., Wang S., Dai C., Zhao J., Lin Q., Lin S., Song W. (2009) Upregulation of macrophage migration inhibitory factor gene expression in stroke. Stroke 40, 973–976 [DOI] [PubMed] [Google Scholar]
- 22. Inacio A. R., Bucala R., Deierborg T. (2011) Lack of macrophage migration inhibitory factor in mice does not affect hallmarks of the inflammatory/immune response during the first week after stroke. J. Neuroinflammation 8, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inacio A. R., Ruscher K., Leng L., Bucala R., Deierborg T. (2011) Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J. Cereb. Blood Flow Metab. 31, 1093–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paralkar V., Wistow G. (1994) Cloning the human gene for macrophage migration inhibitory factor (MIF). Genomics 19, 48–51 [DOI] [PubMed] [Google Scholar]
- 25. Bacher M., Meinhardt A., Lan H. Y., Dhabhar F. S., Mu W., Metz C. N., Chesney J. A., Gemsa D., Donnelly T., Atkins R. C., Bucala R. (1998) MIF expression in the rat brain: implications for neuronal function. Mol. Med. 4, 217–230 [PMC free article] [PubMed] [Google Scholar]
- 26. Calandra T., Bernhagen J., Mitchell R. A., Bucala R. (1994) The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 179, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waeber G., Thompson N., Chautard T., Steinmann M., Nicod P., Pralong F. P., Calandra T., Gaillard R. C. (1998) Transcriptional activation of the macrophage migration-inhibitory factor gene by the corticotropin-releasing factor is mediated by the cyclic adenosine 3′,5′- monophosphate responsive element-binding protein CREB in pituitary cells. Mol. Endocrinol. 12, 698–705 [DOI] [PubMed] [Google Scholar]
- 28. Baugh J. A., Gantier M., Li L., Byrne A., Buckley A., Donnelly S. C. (2006) Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochem. Biophys. Res. Commun. 347, 895–903 [DOI] [PubMed] [Google Scholar]
- 29. Welford S. M., Bedogni B., Gradin K., Poellinger L., Broome Powell M., Giaccia A. J. (2006) HIF1alpha delays premature senescence through the activation of MIF. Genes Dev. 20, 3366–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schreck R., Albermann K., Baeuerle P. A. (1992) Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 17, 221–237 [DOI] [PubMed] [Google Scholar]
- 31. Baeuerle P. A., Henkel T. (1994) Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 12, 141–179 [DOI] [PubMed] [Google Scholar]
- 32. Stockley J. H., O'Neill C. (2007) The proteins BACE1 and BACE2 and beta-secretase activity in normal and Alzheimer's disease brain. Biochem. Soc. Trans. 35, 574–576 [DOI] [PubMed] [Google Scholar]
- 33. Chen C. H., Zhou W., Liu S., Deng Y., Cai F., Tone M., Tone Y., Tong Y., Song W. (2012) Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int. J. Neuropsychopharmacol. 15, 77–90 [DOI] [PubMed] [Google Scholar]
- 34. Tong Y., Zhou W., Fung V., Christensen M. A., Qing H., Sun X., Song W. (2005) Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural. Transm. 112, 455–469 [DOI] [PubMed] [Google Scholar]
- 35. Schneider A., Martin-Villalba A., Weih F., Vogel J., Wirth T., Schwaninger M. (1999) NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5, 554–559 [DOI] [PubMed] [Google Scholar]
- 36. Ridder D. A., Schwaninger M. (2009) NF-kappaB signaling in cerebral ischemia. Neuroscience 158, 995–1006 [DOI] [PubMed] [Google Scholar]
- 37. Mattson M. P., Culmsee C., Yu Z. F. (2000) Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 301, 173–187 [DOI] [PubMed] [Google Scholar]
- 38. Baldwin A. S., Jr. (1996) The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14, 649–683 [DOI] [PubMed] [Google Scholar]
- 39. Miyamoto S., Verma I. M. (1995) Rel/NF-kappa B/I kappa B story. Adv. Cancer Res. 66, 255–292 [PubMed] [Google Scholar]
- 40. Huang F. P., Wang Z. Q., Wu D. C., Schielke G. P., Sun Y., Yang G. Y. (2003) Early NFkappaB activation is inhibited during focal cerebral ischemia in interleukin-1beta-converting enzyme deficient mice. J. Neurosci. Res. 73, 698–707 [DOI] [PubMed] [Google Scholar]
- 41. Cai F., Chen B., Zhou W., Zis O., Liu S., Holt R. A., Honer W. G., Song W. (2008) SP1 regulates a human SNAP-25 gene expression. J. Neurochem. 105, 512–523 [DOI] [PubMed] [Google Scholar]
- 42. Zhou W., Song W. (2006) Leaky scanning and reinitiation regulate BACE1 gene expression. Mol. Cell. Biol. 26, 3353–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ly P. T., Wu Y., Zou H., Wang R., Zhou W., Kinoshita A., Zhang M., Yang Y., Cai F., Woodgett J., Song W. (2013) Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123, 224–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen C. H., Zhou W., Liu S., Deng Y., Cai F., Tone M., Tone Y., Tong Y., Song W. (2011) Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int. J. Neuropsychopharmacol. 18, 1–14 [DOI] [PubMed] [Google Scholar]
- 45. Xu Q., Guo H., Zhang X., Tang B., Cai F., Zhou W., Song W. (2012) Hypoxia regulation of ATP13A2 (PARK9) gene transcription. J. Neurochem. 122, 251–259 [DOI] [PubMed] [Google Scholar]
- 46. Mizue Y., Ghani S., Leng L., McDonald C., Kong P., Baugh J., Lane S. J., Craft J., Nishihira J., Donnelly S. C., Zhu Z., Bucala R. (2005) Role for macrophage migration inhibitory factor in asthma. Proc. Natl. Acad. Sci. U. S. A. 102, 14410–14415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen S. T., Hsu C. Y., Hogan E. L., Maricq H., Balentine J. D. (1986) A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke 17, 738–743 [DOI] [PubMed] [Google Scholar]
- 48. Sun X., He G., Song W. (2006) BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer's disease in Down syndrome. FASEB J. 20, 1369–1376 [DOI] [PubMed] [Google Scholar]
- 49. Stephenson D., Yin T., Smalstig E. B., Hsu M. A., Panetta J., Little S., Clemens J. (2000) Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 20, 592–603 [DOI] [PubMed] [Google Scholar]
- 50. Niizuma K., Endo H., Chan P. H. (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 109(Suppl. 1), 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rassaf T., Weber C., Bernhagen J. (2014) Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 102, 321–328 [DOI] [PubMed] [Google Scholar]
- 52. Zhang X., Deguchi K., Yamashita T., Ohta Y., Shang J., Tian F., Liu N., Panin V. L., Ikeda Y., Matsuura T., Abe K. (2010) Temporal and spatial differences of multiple protein expression in the ischemic penumbra after transient MCAO in rats. Brain Res. 1343, 143–152 [DOI] [PubMed] [Google Scholar]
- 53. Nakayama K. (2013) cAMP-response element-binding protein (CREB) and NF-kappaB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase MMP1. J. Biol. Chem. 288, 22584–22595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bakirtzi K., Belfort G., Lopez-Coviella I., Kuruppu D., Cao L., Abel E. D., Brownell A. L., Kandror K. V. (2009) Cerebellar neurons possess a vesicular compartment structurally and functionally similar to Glut4-storage vesicles from peripheral insulin-sensitive tissues. J. Neurosci. 29, 5193–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koga K., Kenessey A., Powell S. R., Sison C. P., Miller E. J., Ojamaa K. (2011) Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid. Redox Signal. 14, 1191–1202 [DOI] [PubMed] [Google Scholar]
- 56. Szeto H. H. (2006) Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 8, E521–E531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Al-Abed Y., Dabideen D., Aljabari B., Valster A., Messmer D., Ochani M., Tanovic M., Ochani K., Bacher M., Nicoletti F., Metz C., Pavlov V. A., Miller E. J., Tracey K. J. (2005) ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J. Biol. Chem. 280, 36541–36544 [DOI] [PubMed] [Google Scholar]
- 58. Leppa S., Bohmann D. (1999) Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 18, 6158–6162 [DOI] [PubMed] [Google Scholar]
- 59. Wagner E. F., Nebreda A. R. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]
- 60. Koebernick H., Grode L., David J. R., Rohde W., Rolph M. S., Mittrucker H. W., Kaufmann S. H. (2002) Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 99, 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Calandra T., Bernhagen J., Metz C. N., Spiegel L. A., Bacher M., Donnelly T., Cerami A., Bucala R. (1995) MIF as a glucocorticoid-induced modulator of cytokine production. Nature 377, 68–71 [DOI] [PubMed] [Google Scholar]
- 62. Bacher M., Deuster O., Aljabari B., Egensperger R., Neff F., Jessen F., Popp J., Noelker C., Reese J. P., Al-Abed Y., Dodel R. The role of macrophage migration inhibitory factor in Alzheimer's disease. Mol. Med. 16, 116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morand E. F., Leech M., Bernhagen J. (2006) MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 5, 399–410 [DOI] [PubMed] [Google Scholar]
- 64. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., Bucala R., Hickey M. J., Weber C. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.