

Abstract
α-Synuclein is a key pathogenic protein that aggregates in hallmark lesions in Parkinson's disease and other α-synucleinopathies. Prior in vitro studies demonstrated that it is a substrate for cross-linking by transglutaminase 2 (TG2) into higher-order species. Here we investigated whether this increased aggregation occurs in vivo and whether TG2 exacerbates α-synuclein toxicity in Mus musculus and Saccharomyces cerevisiae. Compared with α-synuclein transgenic (SynTg) mice, animals double transgenic for human α-synuclein and TG2 (TG2Tg/SynTg) manifested greater high-molecular-weight insoluble species of α-synuclein in brain lysates and developed α-synuclein aggregates in the synaptic vesicle fraction. In addition, larger proteinase K-resistant aggregates developed, along with increased thioflavin-S-positive amyloid fibrils. This correlated with an exaggerated neuroinflammatory response, as seen with more astrocytes and microglia. Further neuronal damage was suggested by greater morphological disruption of nerve fibers and a trend toward decreased c-Fos immunoreactive neurons. Finally, the performance of TG2Tg/SynTg animals on motor behavioral tasks was worse relative to SynTg mice. Greater toxicity of α-synuclein was also demonstrated in yeast cells coexpressing TG2. Our findings demonstrate that TG2 promotes the aggregation of α-synuclein in vivo and that this is associated with aggravated toxicity of α-synuclein and its downstream neuropathologic consequences.—Grosso, H., Woo, J.-M., Lee, K.-W., Im, J.-Y., Masliah, E., Junn, E., Mouradian, M. M. Transglutaminase 2 exacerbates α-synuclein toxicity in mice and yeast.
Keywords: Parkinson's disease, protein misfolding, neurodegeneration
α-Synuclein is a key protein in the pathogenesis of Parkinson's disease (PD), based on genetic, neuropathologic, and cellular/molecular lines of evidence (1). The 140-residue α-synuclein peptide is natively unfolded but forms oligomers of β-pleated sheets called protofibrils, which can proceed to form amyloid fibrils in response to diverse exogenous and endogenous factors (2). Evidence for a deleterious effect of both the protofibrillar aggregates and the mature inclusions has been generated (3). Thus, elucidating the factors that promote α-synuclein aggregation, particularly those that act early in the process, has the potential to lead to rational approaches toward neuroprotection.
Transglutaminase 2 (TG2) is one of these factors implicated in PD, as well as in other neurodegenerative diseases characterized by protein misfolding (4, 5). It is expressed in many brain regions including the cortex, hippocampus, and substantia nigra, primarily in neurons but also in glial cells (6, 7). While TG2 has several enzymatic activities and cell biological functions, its transamidation activity, which cross-links proteins between glutamine and lysine residues creating inter- or intramolecular covalent bonds, has been studied the most in relation to protein aggregation disorders (4). Among its substrates to form these isopeptide bonds that are highly resistant to proteolysis are α-synuclein, huntingtin, tau, and Aβ peptide, demonstrated in in vitro and cell-based experiments (5, 8). TG2-mediated cross-linking has been shown to alter the aggregation pathway of α-synuclein, potentially resulting in a more toxic form (9). In addition, evidence for increased TG2 activity has been found in patients with these disorders. In PD, there is increased TG2 protein and mRNA expression in the substantia nigra (10, 11) and increased TG2 protein levels in the cerebrospinal fluid (12). In addition, the isopeptide bonds formed by TG2 colocalize with α-synuclein immunoreactivity in Lewy bodies in PD- and dementia with Lewy bodies (DLB)-affected brains (8), and the 2 proteins coimmunoprecipitate in lysates of the substantia nigra from patients with PD (10). While all these provide circumstantial evidence for the role of TG2 in the pathology of α-synucleinopathies, cross-linking of α-synuclein by TG2 in vivo has not yet been demonstrated nor is there direct evidence for a toxic role of this interaction. Here, we demonstrate that increased expression of TG2 in vivo does lead to increased α-synuclein aggregation and that this function is associated with exacerbated toxicity of α-synuclein in the mouse brain as well as in yeast cells.
MATERIALS AND METHODS
Animals
TG2 transgenic (TG2Tg) mice transgenic for human TG2 expressed from the MoPrp.XhoI vector were a kind gift from Dr. Gail V. Johnson (University of Rochester, Rochester, NY, USA; ref. 13). α-Synuclein transgenic (SynTg) female mice transgenic for human wild-type (WT) α-synuclein under the control of the Thy-1 promoter (14) were cross-bred with male TG2Tg mice to create mice that were doubly transgenic for both TG2 and α-synuclein (TG2Tg/SynTg). The SynTg line was maintained by breeding SynTg females with WT males of the BDF1 background, which is mixed C57B/6-DBA2. The TG2Tg line was maintained by breeding TG2Tg mice with WT mice of the C57B/6 background. Genotypes were determined by PCR of tail DNA. Mice were housed in a 12 h light-dark cycle in a climate-controlled room, and food and water were provided ad libitum. Only male mice were used in this study. Behavior was assessed at 9 mo of age, after which animals were anesthetized and perfused with saline, and their brains were rapidly dissected. One hemibrain was immediately frozen for biochemical analyses, and the other was postfixed in 4% paraformaldehyde in PBS for immunohistochemical stains. All animal procedures were approved by the Institutional Animal Care and Use Committee of Rutgers–Robert Wood Johnson Medical School and carried out in accordance with the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Antibodies
Primary antibodies used included mouse monoclonal CUB7402 (Abcam, Cambridge, UK) against TG2, rabbit polyclonal ab73170 (Abcam) specific for human TG2, SynI (BD Transduction Laboratories, San Jose, CA, USA) against human and mouse α-synuclein, MAB 368 (Millipore, Billerica, MA, USA) against synaptophysin, anti-c-Fos (Santa Cruz Biotechnology, Dallas, TX, USA), MAB360 (Chemicon, Billerica, MA, USA) against glial fibrillary acidic protein (GFAP) for Western blotting, rabbit-anti-GFAP for immunohistochemistry (Dako, Carpinatria, CA, USA), Iba1 (Wako, Osaka, Japan) against microglia, anti-microtubule-associated protein 2 (MAP2; Santa Cruz Biotechnology) against microtubule associated protein 2, anti-yeast phosphoglycerate kinase (Invitrogen, Carlsbad, CA, USA), Ab-231 (SAB, College Park, MD, USA) against tau, and anti-β-actin (Sigma, St. Louis, MO, USA). Secondary antibodies included anti-rabbit-rhodamine (Jackson ImmunoResearch, West Grove, PA, USA), anti-mouse-FITC (Sigma), and anti-rabbit-DyLight 488 (Jackson ImmunoResearch).
Western blotting
Soluble and insoluble fractions of brain tissue lysates were prepared by first homogenizing the tissue at 4°C in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris, pH 8.0; 150 mM NaCl; 1% Nonidet P-40; 0.1% SDS; and 0.5% sodium deoxycholate) containing phosphatase inhibitors (cocktail set II; Calbiochem, Darmstadt, Germany) and protease inhibitors (cocktail set V; Calbiochem). This homogenate was centrifuged at 20,800 g in a tabletop centrifuge for 30 min, and the supernatant (soluble fraction) was saved. The pellet (insoluble fraction) was further homogenized in 1% SDS in PBS containing phosphatase and protease inhibitors.
Synaptosomal fractions were prepared as described previously (15). Briefly, tissue was first homogenized in sucrose buffer (0.32 M sucrose; 5 mM Tris, pH 7.5; and 2.5 mM phenylmethylsulfonyl fluoride) and subjected to differential centrifugation as follows. Homogenates were centrifuged at 1000 g at 4°C to obtain P1 and S1 fractions. S1 was further centrifuged at 12,000 g for 15 min to obtain P2 and S2 fractions. P2 was then resuspended in sucrose buffer, 10 vol of ice-cold deionized water was added, and the sample was homogenized. The resultant P2′ fraction was centrifuged at 25,000 g to obtain LS1 and LP1, and LS1 was centrifuged at 100,000 g for 2 h to obtain LS2 (soluble synaptosomal content) and LP2 (synaptic vesicle fraction).
Samples were electrophoresed on a NuPage 4–12% Bis-Tris Gel (Invitrogen), and separated proteins were transferred to a polyvinylidine fluoride membrane (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline and 0.1% Tween-20 before probing with antibodies. ECL Plus (Perkin-Elmer, Waltham, MA, USA) was used to develop immunoblots. Membranes were probed with CUB7402, SynI, LB509, MAB 368, yeast phosphoglycerate kinase, and β-actin. Blots were quantified using the ImageJ image analysis software (NIH, Bethesda, MD, USA).
Immunohistochemistry
Formalin-fixed hemibrains were sectioned coronally at 40 μm thickness using a vibratome. Free-floating sections were then incubated in 3% hydrogen peroxide to inhibit endogenous peroxidase activity and blocked in 5% BSA before incubation with antibodies. For proteinase K treatment, before being blocked in BSA samples were incubated in 88% formic acid for 10 min for antigen retrieval, followed by incubation in 10 μg/ml proteinase K for 10 min. For light microscopy, biotinylated HRP complex (Vector Laboratories, Burlingame, CA, USA) followed by 3,3′-diaminobenzidine was used for color development. Sections were stained with SynI, c-Fos, MAB360, and Iba1. Images of stained sections were captured using a Nikon Eclipse 55i microscope and NIS Elements D3.2 software (Nikon, Tokyo, Japan). ImageJ 1.44p (NIH) was used to threshold stained areas in a standardized manner and to automatically calculate total stained area, number of stained regions, and average size of stained particles. For all immunohistochemical analyses, results from 2–3 sections/mouse were averaged, and the means of these averages for 4 mice/group were compared. For fluorescent microscopy, images were captured using a Zeiss Axiovert 200 microscope, Zeiss AxioCam MRm camera, and AxioVision 4.7.1 software (Carl Zeiss, Oberkochen, Germany). For double immunostaining against TG2 and α-synuclein, ab73170 and SynI were used as primary antibodies. For double immunostaining against α-synuclein and GFAP, SynI and anti-rabbit-GFAP were used as primary antibodies. Fiber integrity was assessed using morphological features.
Thioflavin-S staining
Thioflavin-S staining was performed as described previously (16) with some modifications. Briefly, floating mouse brain sections were washed in PBS and mounted on Superfrost Plus slides (VWR, Radnor, PA, USA) before being processed for thioflavin-S (Thio-S) staining. Sections were incubated for 5 min in 0.25% potassium permanganate, washed in water, and incubated in 1% potassium metabisulfite and 1% oxalic acid until they appeared white. Sections were then washed in water and stained for 10 min with a solution of 0.02% thioflavin-S (Sigma). Stained sections were differentiated in 80% ethanol for 1 min twice. Finally, sections were dehydrated through an ascending alcohol series into xylene before being coverslipped with Permount (Fisher Scientific, Pittsburgh, PA, USA).
Behavioral tests
The balance beam test was adapted from a previously described report (17). Briefly, mice were habituated to a dark goal box for 3 min and then trained to walk across a narrow beam to reach that box. They were first started at 10 cm from the box. Once able to cross this distance, they were placed 25 cm from the box, then 40 cm, and finally 50 cm. On the following day, mice were again habituated to the box for 3 min, and then the time they took to cross the 50 cm length of the beam to reach the box was recorded on 3 consecutive trials and averaged for each mouse.
For the nesting behavior test, mice were housed individually for 24 h with a 5 cm square cotton nestlet (Ancare, Bellmore, NY, USA). At the end of that period, mice were removed, and a picture of the cage was taken. Pictures were then randomized and scored by an observer blinded to the mouse genotype using a scale modified from Deacon (18). Briefly, cages containing an untouched nestlet were scored as 0, and a complete nest was scored as 5. Intermediate nests were given a score between 0 and 5 on a half-point scale.
Construct preparation for yeast
WT TG2 cDNA was cloned from a human brain library and inserted into multicloning site (MCS) 2 of the galactose-inducible yeast expression vector, pESC-Leu (Stratagene, La Jolla, CA, USA), which uses the LEU2 selection marker. α-Synuclein cDNA was cloned from a human brain library and spliced into an enhanced green fluorescent protein (EGFP) expression vector. The resultant α-synuclein-EGFP fusion construct or EGFP alone was then inserted into MCS1 of the empty pESC-Leu vector, as well as this vector that already contained the TG2 cDNA in MCS2. All combinations of these vectors containing an empty MCS2 site or TG2 cDNA, and either EGFP alone or the α-synuclein-EGFP fusion construct, were then transformed into WT Saccharomyces cerevisiae BY4741 using standard methods, and the transformants were plated onto selection medium lacking leucine [SD-L medium: 20 g glucose, 6.7 g yeast nitrogen base with ammonium sulfate (MP Biomedicals, Santa Ana, CA, USA), 0.69 g Complete Supplement Mixture (CSM) minus Leu (MP Biomedicals), and 25 g agar-B (Qbiogene, Santa Ana, CA, USA) in 1 L water]. Alternatively, α-synuclein was expressed from the p426 vector with the URA3 marker [American Type Culture Collection (ATCC), Manassas, VA, USA]. pESC-Leu containing TG2 or the corresponding empty vector, and p426 containing α-synuclein or its corresponding empty vector were then cotransformed into WT S. cerevisiae BY4741, and the transformants were plated onto selection medium lacking both uracil and leucine [SD-UL: 20 g glucose, 6.7 g yeast nitrogen base with ammonium sulfate (MP Biomedicals), 0.67 g CSM minus Leu and minus Ura (MP Biomedicals), and 25 g agar-B (Qbiogene) in 1 L water].
Yeast toxicity assays
Once colonies appeared on solid selection medium, a colony of each transformant was transferred to liquid selection medium [20 g glucose, 6.7 g yeast nitrogen base with ammonium sulfate (MP Biomedicals), 0.67 g CSM minus Leu and minus Ura (MP Biomedicals), or CSM minus Leu (MP Biomedicals) in 1 L water] and allowed to grow overnight. Yeast cells were then harvested before reaching the stationary phase of growth, washed twice with water, and resuspended in liquid induction medium [SGR-L or SGR-UL: 5 g galactose, 10 g raffinose, 6.7 g yeast nitrogen base with ammonium sulfate (MP Biomedicals), 0.69 g CSM minus Leu (MP Biomedicals) or 0.67 g CSM minus Leu and minus Ura in 1 L water] at equal cell densities based on OD600 readings obtained from a SpectraMax 250 plate reader (Molecular Devices, Sunnyvale, CA, USA). At this time, samples of each condition were spotted in serial 10-fold dilutions onto solid selection medium to confirm equal cell densities at the beginning of each experiment. Periodically after induction, the OD600 of each culture was measured, and samples were spotted in serial 10-fold dilutions onto solid induction medium [5 g galactose, 10 g raffinose, 6.7 g yeast nitrogen base with ammonium sulfate (MP Biomedicals), 0.69 g CSM minus Leu (MP Biomedicals), or 0.67 g CSM minus Leu and minus Ura, 25 g agar-B (Qbiogene) in 1 L water]. At the end of the experiments, cells were harvested, and lysates were subjected to Western blot analysis to confirm TG2 and α-synuclein expression.
Statistical analysis
Data are presented as means ± se and were analyzed using a 1-way ANOVA with the Neuman-Keuls post hoc test when comparing all 4 groups. A Student's t test was used when only SynTg and SynTg/TG2Tg mice were compared, as occurred with comparisons of α-synuclein expression. Significance was defined as P < 0.05.
RESULTS
TG2 and α-synuclein colocalize in neurons in the brains of double-transgenic mice
To determine the effects of TG2 on α-synuclein in vivo, SynTg mice were cross-bred with TG2Tg mice, and the resultant 4 genotypes (SynTg, TG2Tg, TG2Tg/SynTg, and WT) were allowed to age to 9 mo. Expression and cellular localization of TG2 and α-synuclein in these transgenic lines were first confirmed by subjecting cortical, striatal, and substantia nigra sections to fluorescent immunohistochemistry for these 2 proteins. Both the mouse prion promoter driving TG2 expression (13) and the Thy1 promoter driving α-synuclein expression (14) are active in neurons. Accordingly, an antibody against human TG2 demonstrated a positive signal in the cytoplasm of cortical cells with neuronal morphology including processes in TG2Tg mouse brains (Fig. 1A). This was in agreement with Tucholski et al. (13), who originally described this line and demonstrated intraneuronal TG2 expression. Similarly, α-synuclein immunoreactivity was detected primarily in the cytoplasm of cortical neurons of SynTg mice (Fig. 1A). A strong colocalization signal was seen for both TG2 and α-synuclein in cortical neurons of TG2Tg/SynTg double-transgenic animals (Fig. 1A). A similar pattern was seen in the striatum (Fig. 1B) and substantia nigra (Fig. 1C) of these animals. These findings confirm that TG2 and α-synuclein colocalize in the same neuronal compartment, allowing for their interaction.
Figure 1.

TG2 and α-synuclein colocalize in neurons in double-transgenic mice. A) Cortical sections from each group were immunostained with SynI for α-synuclein followed by FITC-conjugated secondary antibody and with ab73170 for human TG2 followed by rhodamine-conjugated secondary antibody. Nuclei were stained with Hoechst. As expected, a strong SynI signal was detected in neurons of SynTg and TG2Tg/SynTg mouse brains. Similarly, no ab73170 signal was seen in sections from WT or SynTg mice, but a strong signal was present in the cytoplasm of TG2Tg and TG2Tg/SynTg mouse neurons. In sections from TG2Tg/SynTg mice, these signals overlapped, demonstrating colocalization of α-synuclein and TG2 in the neuronal cytoplasm (white arrows). B) Striatal sections were immunostained as above. As seen in the cortex, a strong SynI signal was detected in neurons of SynTg and double-transgenic TG2Tg/SynTg mouse brains, while an ab73170 signal was seen in TG2Tg and TG2Tg/SynTg mouse neurons. Colocalization was observed in neurons in sections from TG2Tg/SynTg mice (white arrows). C) Sections from the substantia nigra of each group were immunostained as above. As seen in the cortex and striatum, neurons of SynTg and double-transgenic TG2Tg/SynTg mouse brains had a strong SynI signal, while neurons of TG2Tg and TG2Tg/SynTg animals had a strong ab73170 signal. Again, there was colocalization of the signals in neurons of TG2Tg/SynTg mice (white arrows). Scale bars = 10 μm.
TG2 promotes accumulation of high-molecular-weight (HMW) α-synuclein in vivo
Prior in vitro and cell-based experiments have shown increased formation of HMW α-synuclein in the presence of TG2 and the necessary cofactor calcium (8). To study whether the same is true in vivo, cortical lysates from the 4 genotypes of mice were separated into detergent-soluble and -insoluble fractions and examined by Western blotting. With the use of antibody SynI, which recognizes both mouse and human α-synuclein, monomeric α-synuclein was detected in all 4 genotypes (Fig. 2A), but mice expressing the α-synuclein transgene had an increased amount of monomeric α-synuclein and a modest accumulation of HMW oligomers in the insoluble fraction. The latter was markedly increased in TG2Tg/SynTg mice compared with SynTg animals (Fig. 2A), replicating previous in vitro observations (8). The demonstrated increase of TG2 cross-linking activity by ∼70 fold in the brains of TG2Tg mice (13) explains this finding.
Figure 2.

TG2 increases the formation of higher order species of α-synuclein. A) Western blot of the insoluble fraction of cortical tissue lysates from all 4 genotypes. Vertical line marks HMW α-synuclein detected with SynI antibody. α-Synuclein monomer (arrow) is increased in mice carrying the α-synuclein transgene. TG2 (CUB7402 antibody) is detected only in TG2Tg and double-transgenic mice. β-Actin is used as loading control. B) Western blot of the vesicle fraction of the synaptosomal preparation from cortex. HMW bands of α-synuclein (vertical line) are detected only in double-transgenic SynTg/TG2Tg mice. Synaptophysin is used as loading control. C) TG2 increases HMW species of α-synuclein in the soluble synaptosomal fraction from cortex. HMW bands of α-synuclein (vertical line) are increased in double-transgenic SynTg/TG2Tg mice relative to SynTg animals. Monomeric α-synuclein is indicated with an arrow. TG2 is detected using CUB7402 antibody. D) Immunohistochemistry for proteinase K-resistant α-synuclein in striatal sections. α-Synuclein is detected only in the 2 lines carrying the α-synuclein transgene, and the size of the aggregates is greater in TG2Tg/SynTg mice relative to SynTg animals. Scale bar = 50 μm. E) Quantification of data represented in D for n = 4/group. *P < 0.05. F) Thioflavin-S staining of cortical sections. Staining is detected only in SynTg and TG2Tg/SynTg animals, with a greater signal in TG2Tg/SynTg mice. Scale bar = 100 μm.
To test the specificity of this effect on α-synuclein, brain lysates were also probed for tau, which is another substrate of TG2 (5). No increase in tau HMW species were observed in TG2Tg/SynTg mice relative to TG2Tg animals (Supplemental Fig. S1). Rather, tau HMW species were less abundant in TG2Tg/SynTg mice, likely due to competition with overexpressed α-synuclein in these double-transgenic animals. This result confirms specificity of the TG2 effect on α-synuclein in TG2Tg/SynTg mice.
Since much of the pathology seen in SynTg mice occurs in synaptic terminals (19, 20), we isolated synaptosomal fractions from the cortex of all 4 genotypes. Western blot analysis of the vesicle fraction of this synaptosomal preparation detected HMW oligomers of α-synuclein only in TG2Tg/SynTg animals and not in SynTg mice (Fig. 2B), suggesting that cross-linking of α-synuclein by TG2 induced a more stable interaction of α-synuclein with vesicular membranes in addition to increasing the amount of HMW oligomers. Monomeric α-synuclein was not detected in this subfraction in any of the genotypes, which agrees with earlier reports (15). However, in the soluble synaptosomal fraction, monomeric α-synuclein was seen, along with an increase in HMW bands of α-synuclein in TG2Tg/SynTg mice relative to SynTg animals (Fig. 2C).
We also examined proteinase K-resistant α-synuclein aggregates by immunohistochemistry in striatal sections receiving nigral nerve projections. As expected, no SynI immunoreactive signal was detected in WT or TG2Tg animals following this detergent digestion (Fig. 2D). However, specific staining of punctate aggregates was seen in SynTg mice. Notably, the size of these aggregates was significantly greater in TG2Tg/SynTg mice (Fig. 2E). In addition, these aggregates labeled with the amyloid-binding dye thioflavin-S in SynTg mice and to a greater extent in TG2Tg/SynTg animals (Fig. 2F). Based on these results, we conclude that TG2 enhances the formation of higher-order amyloid species of α-synuclein in vivo, particularly in the nerve terminals.
TG2 aggravates the neuroinflammatory effects of α-synuclein in vivo
The pathology of PD is associated with significant microglial activation, and overexpression of α-synuclein in mice is known to cause a robust neuroinflammatory reaction (21–23). To investigate whether the increased aggregation of α-synuclein associated with TG2 overexpression impacts this neuroinflammatory response in vivo, we performed immunohistochemistry for the astrocytic marker GFAP (Fig. 3A–C) and microglial marker Iba1 (Fig. 3D) on brain sections from all 4 groups. As described previously (20, 22), SynTg mice had a noticeable GFAP signal compared with no staining in the cortex and striata of WT and TG2Tg mice (Fig. 3A–C). In TG2Tg/SynTg mice, this signal was further enhanced relative to WT and TG2Tg animals (Fig. 3E). In addition, the average size of GFAP immunoreactive cells in TG2Tg/SynTg mice was significantly larger relative to that in all other genotypes, suggesting greater glial activation (24) in these double-transgenic mice (Fig. 3F). These effects do not appear to be caused by a direct action of α-synuclein within glial cells, as α-synuclein does not colocalize with astrocytes (Fig. 3H). Similarly, SynTg mice had more microglial cells than WT and TG2Tg mice, and TG2Tg/SynTg mice had even more relative to the other 3 genotypes (Fig. 3D, G). These observations indicate that TG2 exaggerates the neuroinflammatory reaction to α-synuclein.
Figure 3.

TG2 exaggerates the neuroinflammatory response to α-synuclein. A) Striatal sections from each of the 4 mouse lines stained for GFAP. B) Cortical sections from each of the 4 mouse lines stained for GFAP. C) Higher power images of cortical sections stained for GFAP. D) Striatal sections stained with Iba1 for microglia. E–G) Quantification of total GFAP-stained area for data represented in A (E), average cell size for data represented in C (F), and cell number for data represented in D (G); n = 4/group. *P < 0.05; **P < 0.01. H) Transgenic α-synuclein is not expressed in astrocytes. Cortical sections from each group were immunostained with SynI for α-synuclein followed by FITC-conjugated secondary antibody, and with anti-rabbit-GFAP followed by rhodamine-conjugated secondary antibody. Nuclei were stained with Hoechst. The anti-GFAP signal was seen in astrocytes of all mouse lines (arrowhead). SynI signal was detected in neurons of SynTg and double-transgenic TG2Tg/SynTg mouse brains (arrow) but did not colocalize with the GFAP signal, indicating no transgenic α-synuclein in astrocytes. Scale bars = 100 μm (A, B, D); 50 μm (C); 10 μm (H).
TG2 exacerbates the neuronal toxicity of α-synuclein in vivo
To investigate the effect of TG2 on the neurotoxicity of α-synuclein in vivo, MAP2 and c-Fos staining were examined next. α-Synuclein overexpression in mice is associated with impaired neuronal function and altered neuritic morphology as a manifestation of its toxicity (20, 22). These effects can be detected by depletion of MAP2, suggesting disrupted nerve fibers and decreased dendritic branching (22, 25–28), as well as reduced immunoreactivity for c-Fos, which is an immediate early gene used as a surrogate marker for neuronal activity (22, 29, 30). To determine whether the TG2-mediated exacerbation of the α-synuclein phenotype is associated with changes in the integrity of neuronal processes, brain sections from the 4 genotypes were stained for MAP2. In accordance with previous reports, MAP2 positive nerve fibers in the cortex of SynTg mice were shorter and more fragmented than those seen in WT or TG2Tg mice (Fig. 4A). These processes were disrupted even further in TG2Tg/SynTg mice. In the striatum, there was also diminished MAP2 staining in TG2Tg/SynTg mice relative to the other genotypes (Fig. 4B). In this context, the decrease in MAP2 staining is believed to represent damage to axon terminals in the striatum (26, 27).
Figure 4.

TG2 exacerbates the neuronal toxicity of α-synuclein. A) Cortical sections from all 4 genotypes were stained for MAP2. Staining intensity is decreased in SynTg mice relative to WT or TG2Tg mice. This signal is further decreased in TG2Tg/SynTg animals. n = 4 for/group. B) Striatal sections from all 4 genotypes were probed with the MAP2 antibody. As in the cortical sections, the signal in TG2Tg/SynTg animals was decreased relative to that of all other groups. C) Hippocampal sections were stained for c-Fos. There is a decrease in immunoreactive cells in SynTg mice compared with WT or TG2Tg animals, with a further decrease in TG2Tg/SynTg mice. D) Quantification of data represented in C; n = 4. Scale bars = 50 μm. *P < 0.05.
In the hippocampus, neuronal activity was assessed by counting c-Fos immunopositive cells. SynTg mice averaged 8.5 positive cells/field, which were fewer than WT (23.4 positive cells/field) and TG2Tg (37 positive cells/field) controls. TG2Tg/SynTg mice had even fewer cells than SynTg mice, with 3.6 positive cells/field, although this latter comparison did not reach statistical significance, likely due to a “floor” effect (Figs. 4C, D). Taken together, these results suggest that the neuronal damage caused by α-synuclein overexpression in these mice is exacerbated by TG2 overexpression.
TG2 exacerbates the behavioral phenotype of SynTg mice
To determine whether these biochemical and immunohistochemical changes correlate with a behavioral phenotype, we evaluated these mice for their performance on the balance beam test and for nest-building behavior. These tests were chosen because these tasks are known to be affected by nigrostriatal impairment (17, 22, 31), allowing us to better correlate the phenotype of these mice to the impairments seen in PD. On the balance beam test, as expected, SynTg mice were impaired relative to WT and TG2Tg mice, with SynTg mice averaging 12.7 s to cross the beam, while WT and TG2Tg mice took 6.6 and 4.4 s, respectively (Fig. 5A). Double-transgenic TG2Tg/SynTg mice demonstrated even greater impairment, taking an average of 23.1 s to cross the beam. A similar profile of exacerbated impairment due to coexpressing TG2 with α-synuclein was observed on nest-building ability (Fig. 5B). Nests built by SynTg mice averaged a score of 1.5 on a scale of 0–5, compared with 3.6 for WT mice and 3.1 for TG2Tg animals. Double-transgenic TG2Tg/SynTg mice performed even worse on this measure, scoring only an average of 0.5 (Fig. 5B). The related backgrounds of the SynTg line (BDF1, which is mixed C57B/6-DBA2) and the TG2Tg line (C57B/6) did not appear to affect these findings, as the motor behavior of WT animals of the BDF1 and C57B/6 backgrounds was not different (Supplemental Fig. S2).
Figure 5.
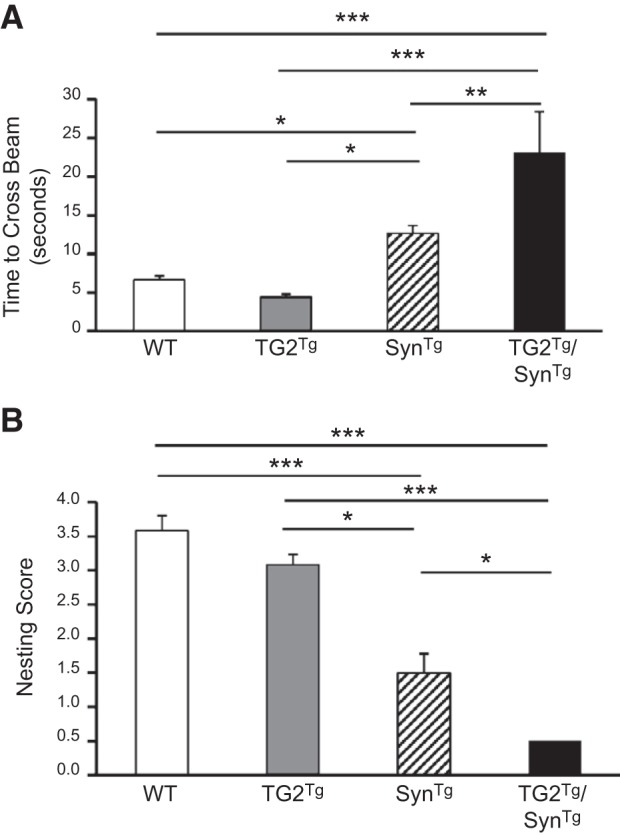
TG2 exacerbates the behavioral phenotype of SynTg mice at 9 mo of age. A) Time taken to cross the balance beam (n: WT=12, TG2Tg=6, SynTg=12, TG2Tg/SynTg=7). B) Nest building ability scored on a scale of 0 (no nest) to 5 (perfect nest) (n: WT=12, TG2Tg=6, SynTg=13, TG2Tg/SynTg=6). *P < 0.05; **P < 0.01; ***P < 0.001.
TG2 exacerbates the toxicity of α-synuclein in yeast
Yeast cells transformed with human α-synuclein are known to have growth impairment, suggesting a toxic effect of the α-synuclein protein in this model organism (32). To determine whether TG2 affects α-synuclein toxicity in yeast, cells were transformed with inducible vectors expressing either α-synuclein alone, TG2 alone, or both transgenes simultaneously. Yeast were first grown in noninducing medium and then seeded into inducing medium in equal concentrations and grown for 24 h. Expression of transgenes was confirmed by Western blots (Fig. 6A). Growth was assessed using the spotting assay and by measuring the OD600 of liquid cultures. On the spotting assay at 24 h, cells transformed with α-synuclein alone had the expected decreased growth relative to yeast containing empty vector or TG2 alone. However, the growth of cells expressing both α-synuclein and TG2 was impaired even further relative to cells expressing α-synuclein alone (Fig. 6B). Growth curves and particularly the OD600 readings at 24 h confirmed the added toxicity of α-synuclein when yeast were cotransformed with TG2 (Figs. 6C, D).
Figure 6.

TG2 exacerbates the toxicity of α-synuclein in yeast. A) Yeast cells were transformed with an inducible bidirectional vector expressing either EGFP or α-synuclein-EGFP, controlled by the GAL10 promoter, and TG2 (or no insert), controlled by the GAL1 promoter. Cell lysates were subjected to Western blotting with CUB7402 antibody to confirm expression of TG2 and with SynI to detect α-synuclein. Yeast phosphoglycerate kinase (PGK) was used as loading control. Samples were grown in liquid noninducing medium; before reaching the stationary phase, triplicates were diluted to an OD600 of 0.05 in inducing medium. B) Left panel: to confirm equal cell concentration at the start of induction, aliquots were spotted onto solid noninducing medium. B) Right panel: following 24 h of induction, samples were spotted onto solid inducing medium to determine the effects of protein expression on cell growth. α-Synuclein transformed cells demonstrate growth impairment compared with vector control or TG2 transformed cultures. Yeast transformed with both α-synuclein and TG2 show even greater growth impairment than those transformed with α-synuclein alone. C) Yeast were transformed with the p426 inducible vector that was either empty or contained α-synuclein controlled by the GALL promoter and with the inducible vector pESC-Leu that was either empty or contained TG2 controlled by the GAL1 promoter. Samples were grown in liquid noninducing medium and, before reaching the stationary phase, were diluted to an OD600 of 0.05 in inducing medium. Graph shows OD600 values of cultures at 6, 12, 24, 36, and 48 h. D) Bar graph shows OD600 value of each condition as a percentage of empty vector measured at 24 h; n = 3. All experiments were repeated ≥3 times with reproducible results. *P < 0.001.
DISCUSSION
The present investigation demonstrates that TG2 exacerbates α-synuclein aggregation into insoluble amyloid fibrils, which is associated with greater neuroinflammation and neuronal damage, as well as worse behavioral performance. The exaggeration of α-synuclein effects by TG2 is confirmed in yeast with greater growth impairment. Together, these findings provide strong evidence for TG2 playing a significant role in the toxicity of α-synuclein.
The worse phenotype of TG2Tg/SynTg mice relative to SynTg animals is accompanied by the development of HMW bands of α-synuclein associated with synaptic vesicles. Although our SynTg mouse line does not demonstrate neuronal loss in the substantia nigra, these animals exhibit considerable pathology in the nerve fibers and terminals (33). In addition, while α-synuclein is found in synaptic terminals (34) and interacts with membranes (35), monomeric α-synuclein is not detected in the synaptic vesicle fraction in any of the 4 mouse lines studied here nor has it been found in this fraction in another α-synuclein transgenic mouse line (15). Further, only the brains of TG2Tg/SynTg mice, which manifest the worst phenotype, have α-synuclein in this fraction, and only in the form of HMW species. These observations suggest that the synaptic terminals may be the site where α-synuclein exerts its toxicity and that the cross-linking of α-synuclein by TG2 into HMW aggregates leads to a more stable interaction of α-synuclein with vesicles, potentially jeopardizing their integrity (36). Interestingly, intramolecular cross-links of α-synuclein formed by TG2 differ between soluble α-synuclein compared with its membrane bound state. While multiple cross-links involving various glutamine and lysine residues can be formed in solution, the association with membranes restricts the formation of these bonds and allows only one intramolecular linkage to occur between residues glutamine-99 and lysine-58. This particular linkage can seed non-cross-linked α-synuclein and promote the growth of amyloid fibrils in vitro (37). It is conceivable that under normal conditions, monomeric α-synuclein interacts transiently with synaptic vesicle membranes (35), but on TG2 activation, these monomers become cross-linked in a way that promotes stable aggregation in association with the vesicle. Further, the propensity of the glutamine-99/lysine-58 linkage to form amyloid can explain the increase in thioflavin-S signal in TG2Tg/SynTg animals. Despite these findings, acceleration of striatal dopamine deficiency could not be demonstrated in these mice, likely because the 9-mo time point when we assessed this index was not old enough to detect an effect given that SynTg mice develop dopamine deficiency at a much older age (33).
TG2 cross-links a number of proteins that aggregate in neurodegenerative diseases including tau (5). However, we detected no evidence for TG2 causing the observed phenotype in TG2Tg/SynTg animals through cross-linking other substrates. Detailed analysis of TG2Tg mice in all outcome measures assessed in this study revealed no biochemical or behavioral deficits compared with WT animals. In addition, HMW aggregates of tau are not more abundant in TG2Tg/SynTg mice relative to TG2Tg animals. These findings suggest that the impaired phenotype of TG2Tg/SynTg mice is most likely due to the effect of TG2 on α-synuclein.
The mechanism by which TG2 exacerbates α-synuclein toxicity in the brain deserves further consideration. The cross-linking of α-synuclein by TG2 into a more toxic oligomeric form may contribute to this process (4, 5, 8). This hypothesis is supported here by the increase in HMW bands of α-synuclein by Western blotting in the brains, the accumulation of HMW bands of α-synuclein in the synaptic vesicle fraction, the increased size of proteinase K-resistant aggregates, and the increase in thioflavin-S staining in TG2Tg/SynTg animals relative to SynTg mice. However, TG2 has multiple additional functions, including activity as a GTPase, a kinase, and a protein disulfide isomerase (5, 38). One or more of these functions may contribute to its role in α-synuclein toxicity, as well. As the mechanisms by which α-synuclein exerts its toxicity unfold, we may be better able to explain how TG2 interacts with α-synuclein to affect this toxicity.
In addition to its enzymatic activities, TG2 is involved in a number of biological processes, including transcription regulation, autophagy, and inflammation (5, 38, 39), which may contribute to neuronal damage in our model. For example, TG2 participates in transcription regulation of putative neuroprotective genes, including PGC-1α and Hsp40 (40, 41), and inhibiting TG2 derepresses transcription of these genes and confers neuroprotection in models of Huntington's disease (40, 41). In addition, autophagy, which is disrupted in neurodegenerative disorders, is regulated by TG2 in complex ways that remain to be clarified (42, 43). Moreover, the role of TG2 in inflammation may be particularly important, given the large body of evidence for a role of inflammatory processes in neurodegenerative diseases (44). For example, TG2 is implicated in the activation of inflammatory mediators, such as NF-κB and TNF-α (45). In agreement with this, our data show an increase in inflammatory cells in mice transgenic for α-synuclein with a greater increase in TG2Tg/SynTg animals. Notably, neither α-synuclein nor TG2 is expressed in inflammatory cells in these mice, suggesting that this proinflammatory response in SynTg mice is initiated in neurons. Recent studies suggest that α-synuclein can be secreted from neurons (46), and through this secretion, it can interact directly with astrocytes and microglia, leading to their activation (47, 48). In our model, it is conceivable that aggregated α-synuclein secreted from neurons in the context of overexpressed or overactive TG2 might induce a greater inflammatory response than monomeric α-synuclein.
Although this study utilizes overexpression systems, these results are nevertheless relevant to human disease. Elevated levels of α-synuclein are linked to PD based on genetic evidence from families with multiplication of the SNCA gene locus, demonstrating a gene-dosage effect (49–51). In addition, polymorphic alleles at a microsatellite repeat in the promoter region of the SNCA gene leading to greater transcriptional activity are associated with sporadic disease (52, 53). Increased TG2 expression and activity are also implicated in neurodegenerative disorders. TG2 mRNA, protein, and activity have been shown to be upregulated in a number of these disorders (5) including PD (10, 11, 54). Further, TG2 is upregulated by a number of factors that are critical in these diseases. Among these factors is calcium, which is a necessary cofactor for the cross-linking activity of TG2. The loss of calcium homeostasis in neurodegenerative diseases leads to an increase in intracellular calcium to levels that may be permissive for TG2 activity (4). GTP, on the other hand, inhibits TG2 activity (55). The impaired mitochondrial respiration and the ensuing energy depletion that occurs in neurodegenerative diseases result in decreased levels of available GTP, which could disinhibit TG2 (4). Thus, the precarious biochemical environment of the brain in neurodegenerative conditions is one in which TG2 activity can be upregulated. As a result, following a primary insult or genetic predisposition that creates these conditions, TG2 may become more active, exacerbating the injury and preventing a return to homeostasis.
The results described above point to a significant contribution of TG2 to the pathogenesis of α-synucleinopathies. This exacerbated phenotype is associated with the accumulation of higher order species of α-synuclein, but other actions of TG2 may play a role in the process as well. Regardless of the mechanism, the present findings provide a rationale for investigating TG2 inhibitors as potential disease modifying therapies for PD and other α-synucleinopathies.
Supplementary Material
Acknowledgments
E.M. is supported by U.S. National Institutes of Health (NIH) grants AG-18440 and AG-022074. E.J. is supported by NIH grant NS-070898. M.M.M. is the William Dow Lovett Professor of Neurology and is supported by NIH grants NS-059869, NS-073994, and AT-006868.
The authors thank Dr. Gail V. Johnson (University of Rochester, Rochester, NY, USA) for providing TG2Tg mice, and Dr. Takeshi Iwatsubo (University of Tokyo, Tokyo, Japan) for p-α-synuclein antibody. The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CSM
- complete supplement mixture
- EGFP
- enhanced green fluorescent protein
- GFAP
- glial fibrillary acidic protein
- HMW
- high molecular weight
- MAP2
- microtubule-associated protein 2
- MCS
- multicloning site
- SynTg
- α-synuclein transgenic
- PD
- Parkinson's disease
- RIPA
- radioimmunoprecipitation assay
- TG2
- transglutaminase 2
- TG2Tg
- transglutaminase 2 transgenic
- TG2Tg/SynTg
- transglutaminase 2 transgenic and α-synuclein transgenic
- WT
- wild type
REFERENCES
- 1. Lee V. M., Trojanowski J. Q. (2006) Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52, 33–38 [DOI] [PubMed] [Google Scholar]
- 2. Goldberg M. S., Lansbury P. T., Jr. (2000) Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson's disease? Nat. Cell Biol. 2, E115–119 [DOI] [PubMed] [Google Scholar]
- 3. Cookson M. R. (2009) alpha-Synuclein and neuronal cell death. Mol. Neurodegener. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jeitner T. M., Pinto J. T., Krasnikov B. F., Horswill M., Cooper A. J. (2009) Transglutaminases and neurodegeneration. J. Neurochem. 109(Suppl. 1), 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grosso H., Mouradian M. M. (2012) Transglutaminase 2: biology, relevance to neurodegenerative diseases and therapeutic implications. Pharmacol. Ther. 133, 392–410 [DOI] [PubMed] [Google Scholar]
- 6. Campisi A., Caccamo D., Li Volti G., Curro M., Parisi G., Avola R., Vanella A., Ientile R. (2004) Glutamate-evoked redox state alterations are involved in tissue transglutaminase upregulation in primary astrocyte cultures. FEBS Lett. 578, 80–84 [DOI] [PubMed] [Google Scholar]
- 7. Filiano A. J., Bailey C. D., Tucholski J., Gundemir S., Johnson G. V. (2008) Transglutaminase 2 protects against ischemic insult, interacts with HIF1beta, and attenuates HIF1 signaling. FASEB J. 22, 2662–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Junn E., Ronchetti R. D., Quezado M. M., Kim S. Y., Mouradian M. M. (2003) Tissue transglutaminase-induced aggregation of alpha-synuclein: implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U. S. A. 100, 2047–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmid A. W., Chiappe D., Pignat V., Grimminger V., Hang I., Moniatte M., Lashuel H. A. (2009) Dissecting the mechanisms of tissue transglutaminase-induced cross-linking of alpha-synuclein: implications for the pathogenesis of Parkinson disease. J. Biol. Chem. 284, 13128–13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andringa G., Lam K. Y., Chegary M., Wang X., Chase T. N., Bennett M. C. (2004) Tissue transglutaminase catalyzes the formation of alpha-synuclein crosslinks in Parkinson's disease. FASEB J. 18, 932–934 [DOI] [PubMed] [Google Scholar]
- 11. Wilhelmus M. M., Verhaar R., Andringa G., Bol J. G., Cras P., Shan L., Hoozemans J. J., Drukarch B. (2010) Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson's disease brain. Brain Pathol. 21, 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vermes I., Steur E. N., Jirikowski G. F., Haanen C. (2004) Elevated concentration of cerebrospinal fluid tissue transglutaminase in Parkinson's disease indicating apoptosis. Mov. Disord. 19, 1252–1254 [DOI] [PubMed] [Google Scholar]
- 13. Tucholski J., Roth K. A., Johnson G. V. (2006) Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J. Neurochem. 97, 582–594 [DOI] [PubMed] [Google Scholar]
- 14. Rockenstein E., Mallory M., Hashimoto M., Song D., Shults C. W., Lang I., Masliah E. (2002) Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 68, 568–578 [DOI] [PubMed] [Google Scholar]
- 15. Kahle P. J., Neumann M., Ozmen L., Muller V., Jacobsen H., Schindzielorz A., Okochi M., Leimer U., van Der Putten H., Probst A., Kremmer E., Kretzschmar H. A., Haass C. (2000) Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 20, 6365–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun A., Nguyen X. V., Bing G. (2002) Comparative analysis of an improved thioflavin-s stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J. Histochem. Cytochem. 50, 463–472 [DOI] [PubMed] [Google Scholar]
- 17. Quinn L. P., Crook B., Hows M. E., Vidgeon-Hart M., Chapman H., Upton N., Medhurst A. D., Virley D. J. (2008) The PPARgamma agonist pioglitazone is effective in the MPTP mouse model of Parkinson's disease through inhibition of monoamine oxidase B. Br. J. Pharmacol. 154, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deacon R. M. (2006) Assessing nest building in mice. Nat. Protoc. 1, 1117–1119 [DOI] [PubMed] [Google Scholar]
- 19. Hashimoto M., Rockenstein E., Mante M., Mallory M., Masliah E. (2001) beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223 [DOI] [PubMed] [Google Scholar]
- 20. Masliah E., Rockenstein E., Adame A., Alford M., Crews L., Hashimoto M., Seubert P., Lee M., Goldstein J., Chilcote T., Games D., Schenk D. (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron 46, 857–868 [DOI] [PubMed] [Google Scholar]
- 21. Su X., Maguire-Zeiss K. A., Giuliano R., Prifti L., Venkatesh K., Federoff H. J. (2008) Synuclein activates microglia in a model of Parkinson's disease. Neurobiol. Aging 29, 1690–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee K. W., Chen W., Junn E., Im J. Y., Grosso H., Sonsalla P. K., Feng X., Ray N., Fernandez J. R., Chao Y., Masliah E., Voronkov M., Braithwaite S. P., Stock J. B., Mouradian M. M. (2011) Enhanced phosphatase activity attenuates alpha-Synucleinopathy in a mouse model. J. Neurosci. 31, 6963–6971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kurz A., May C., Schmidt O., Muller T., Stephan C., Meyer H. E., Gispert S., Auburger G., Marcus K. (2011) A53T-alpha-synuclein-overexpression in the mouse nigrostriatal pathway leads to early increase of 14-3-3 epsilon and late increase of GFAP. J. Neural Transm. 119, 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kajihara H., Tsutsumi E., Kinoshita A., Nakano J., Takagi K., Takeo S. (2001) Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: immunohistochemical and electron microscopic studies. Brain Res. 909, 92–101 [DOI] [PubMed] [Google Scholar]
- 25. Harada A., Teng J., Takei Y., Oguchi K., Hirokawa N. (2002) MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol. 158, 541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwata M., Muneoka K. T., Shirayama Y., Yamamoto A., Kawahara R. (2005) A study of a dendritic marker, microtubule-associated protein 2 (MAP-2), in rats neonatally treated neurosteroids, pregnenolone and dehydroepiandrosterone (DHEA). Neurosci. Lett. 386, 145–149 [DOI] [PubMed] [Google Scholar]
- 27. Zhang X., Dong F., Mayer G. E., Bruch D. C., Ren J., Culver B. (2007) Selective inhibition of cyclooxygenase-2 exacerbates methamphetamine-induced dopamine depletion in the striatum in rats. Neuroscience 150, 950–958 [DOI] [PubMed] [Google Scholar]
- 28. Koob A. O., Ubhi K., Paulsson J. F., Kelly J., Rockenstein E., Mante M., Adame A., Masliah E. (2010) Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp. Neurol. 221, 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kano T., Suzuki Y., Shibuya M., Kiuchi K., Hagiwara M. (1995) Cocaine-induced CREB phosphorylation and c-Fos expression are suppressed in Parkinsonism model mice. Neuroreport 6, 2197–2200 [DOI] [PubMed] [Google Scholar]
- 30. Palop J. J., Jones B., Kekonius L., Chin J., Yu G. Q., Raber J., Masliah E., Mucke L. (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc. Natl. Acad. Sci. U. S. A. 100, 9572–9577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sager T. N., Kirchhoff J., Mork A., Van Beek J., Thirstrup K., Didriksen M., Lauridsen J. B. (2010) Nest building performance following MPTP toxicity in mice. Behav. Brain Res. 208, 444–449 [DOI] [PubMed] [Google Scholar]
- 32. Outeiro T. F., Lindquist S. (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lam H. A., Wu N., Cely I., Kelly R. L., Hean S., Richter F., Magen I., Cepeda C., Ackerson L. C., Walwyn W., Masliah E., Chesselet M. F., Levine M. S., Maidment N. T. (2011) Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human alpha-synuclein. J. Neurosci. Res. 89, 1091–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clayton D. F., George J. M. (1999) Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 58, 120–129 [PubMed] [Google Scholar]
- 35. Auluck P. K., Caraveo G., Lindquist S. (2010) alpha-Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu. Rev. Cell Dev. Biol. 26, 211–233 [DOI] [PubMed] [Google Scholar]
- 36. Butterfield S. M., Lashuel H. A. (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 49, 5628–5654 [DOI] [PubMed] [Google Scholar]
- 37. Nemes Z., Petrovski G., Aerts M., Sergeant K., Devreese B., Fesus L. (2009) Transglutaminase-mediated intramolecular cross-linking of membrane-bound alpha-synuclein promotes amyloid formation in Lewy bodies. J. Biol. Chem. 284, 27252–27264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Siegel M., Khosla C. (2007) Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol. Ther. 115, 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gundemir S., Colak G., Tucholski J., Johnson G. V. (2012) Transglutaminase 2: A molecular Swiss army knife. Biochim. Biophys. Acta 1823, 406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karpuj M. V., Becher M. W., Springer J. E., Chabas D., Youssef S., Pedotti R., Mitchell D., Steinman L. (2002) Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 8, 143–149 [DOI] [PubMed] [Google Scholar]
- 41. McConoughey S. J., Basso M., Niatsetskaya Z. V., Sleiman S. F., Smirnova N. A., Langley B. C., Mahishi L., Cooper A. J., Antonyak M. A., Cerione R. A., Li B., Starkov A., Chaturvedi R. K., Beal M. F., Coppola G., Geschwind D. H., Ryu H., Xia L., Iismaa S. E., Pallos J., Pasternack R., Hils M., Fan J., Raymond L. A., Marsh J. L., Thompson L. M., Ratan R. R. (2010) Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol. Med. 2, 349–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mastroberardino P. G., Iannicola C., Nardacci R., Bernassola F., De Laurenzi V., Melino G., Moreno S., Pavone F., Oliverio S., Fesus L., Piacentini M. (2002) ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ. 9, 873–880 [DOI] [PubMed] [Google Scholar]
- 43. D'Eletto M., Farrace M. G., Falasca L., Reali V., Oliverio S., Melino G., Griffin M., Fimia G. M., Piacentini M. (2009) Transglutaminase 2 is involved in autophagosome maturation. Autophagy 5, 1145–1154 [DOI] [PubMed] [Google Scholar]
- 44. Przedborski S. (2010) Inflammation and Parkinson's disease pathogenesis. Mov. Disord. 25(Suppl. 1), S55–S57 [DOI] [PubMed] [Google Scholar]
- 45. Iismaa S. E., Mearns B. M., Lorand L., Graham R. M. (2009) Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 89, 991–1023 [DOI] [PubMed] [Google Scholar]
- 46. Desplats P., Lee H. J., Bae E. J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S. J. (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U. S. A. 106, 13010–13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee H. J., Suk J. E., Patrick C., Bae E. J., Cho J. H., Rho S., Hwang D., Masliah E., Lee S. J. (2010) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim C., Ho D. H., Suk J. E., You S., Michael S., Kang J., Joong Lee S., Masliah E., Hwang D., Lee H. J., Lee S. J. (2013) Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) alpha-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- 50. Farrer M., Kachergus J., Forno L., Lincoln S., Wang D. S., Hulihan M., Maraganore D., Gwinn-Hardy K., Wszolek Z., Dickson D., Langston J. W. (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 55, 174–179 [DOI] [PubMed] [Google Scholar]
- 51. Chartier-Harlin M. C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M., Waucquier N., Defebvre L., Amouyel P., Farrer M., Destee A. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1167–1169 [DOI] [PubMed] [Google Scholar]
- 52. Farrer M., Maraganore D. M., Lockhart P., Singleton A., Lesnick T. G., de Andrade M., West A., de Silva R., Hardy J., Hernandez D. (2001) alpha-Synuclein gene haplotypes are associated with Parkinson's disease. Hum. Mol. Genet. 10, 1847–1851 [DOI] [PubMed] [Google Scholar]
- 53. Chiba-Falek O., Nussbaum R. L. (2001) Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 10, 3101–3109 [DOI] [PubMed] [Google Scholar]
- 54. Citron B. A., Suo Z., SantaCruz K., Davies P. J., Qin F., Festoff B. W. (2002) Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem. Int. 40, 69–78 [DOI] [PubMed] [Google Scholar]
- 55. Smethurst P. A., Griffin M. (1996) Measurement of tissue transglutaminase activity in a permeabilized cell system: its regulation by Ca2+ and nucleotides. Biochem. J. 313, 803–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.