

Abstract
Abstract
Attempted oxidation of a TADDOL-derived bisthioacetate resulted in a rather unexpected and remarkable cyclization and deprotection reaction, giving a thiolane-1,1-dioxide as the main product. Systematic in situ ESI-HRMS studies revealed a bicyclic, highly acid labile key intermediate of this reaction. Supported by force field calculations, the high sensitivity of this intermediate was judged to be due to the formation of a highly strained trans-configured bicyclo[3.3.0]skeleton.
Graphical abstract
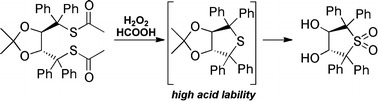
Keywords: Oxidation, Molecular modeling, Cyclization, Mass spectrometry, Tartaric acid, Organocatalysis
Introduction
Amongst the various ways of creating enantiomerically enriched products, catalytic methods are nowadays considered to be the most appealing because the use of stoichiometric amounts of valuable chiral reagents can be avoided. Besides enzymatic and metal-catalyzed asymmetric transformations, the use of sub-stoichiometric amounts of organic molecules (organocatalysts) has proven to possess an enormous potential [1–3].
Recently List et al. [4] described the application of chiral binol-based disulfonimides as chiral counteranions in Mukaiyama aldol reactions. As this elegant approach and the suggested mechanism provide a very useful methodology for the general activation of carbonyl groups towards a variety of nucleophiles, the synthesis of analogous sulfonimides based on different chiral sources is an opportunity to broaden the scope of this strategy.
Among the easily available natural chiral sources, tartaric acid (1) has obtained a prominent position not only for historical reasons, but especially due to the fact that both enantiomers are readily available from natural sources. Thus, 1 has become a valuable and cheap source of primary chiral information for asymmetric catalysis. Tartaric acid derived tetraaryl-2,2-dimethyl-1,3-dioxolan-4,5-dimethanol (TADDOL, 2) and analogous compounds are easily synthesized and extraordinarily versatile chiral auxiliaries which have found numerous and widespread applications as chiral ligands in asymmetric metal-catalyzed transformations (for a leading review about TADDOLs see [5]; for a recent review see [6]). Surprisingly, their use as chiral organocatalysts (avoiding the use of precious or potentially toxic metals) has so far been limited to a few applications only [7–10].
As part of a program aimed at the synthesis of novel tartaric acid derived organocatalysts, we therefore targeted the synthesis of the chiral disulfonimide 3 starting from the readily available chiral source 1 (Fig. 1). The synthesis of this compound and its use as a chiral organocatalyst would not only broaden the field of tartaric acid derived catalysis but also the field of chiral counterion directed applications.
Fig. 1.
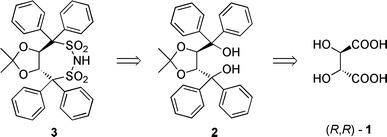
Retrosynthetic analysis for the disulfonimide 3
Results and discussion
The syntheses of TADDOL derivatives were thoroughly investigated over the last few decades [5, 6]. Variations of the aryl substituents or in the ketal group have to be introduced early in the synthesis of TADDOLs from 1, whereas functionalizations of the hydroxyl groups are most commonly achieved via conversion of 2 to the dichloro compound 4 which can then be treated with different nucleophiles (so far mainly nitrogen-, sulfur-, and oxygen-containing nucleophiles have been reported in literature [5, 11–13]).
For the synthesis of the disulfonimide 3 a direct approach via synthesis of the dithiol 5 and subsequent oxidation followed by imide formation appears to be the most straightforward one. However, investigations by Seebach et al. [5, 11] showed that synthesis of 5 is rather difficult and, furthermore, this dithiol is prone to the formation of a disulfide bond under oxidative conditions. Therefore we decided to introduce the sulfur functionality via formation of a bisthioacetate (giving compound 6). As thioacetates are known to be easily oxidized to sulfonic acids upon treatment with a variety of oxidants (e.g. peracids or dioxirane) [14–16] this approach should give access to the disulfonic acid 7, one step away from the target 3 (Fig. 2).
Fig. 2.
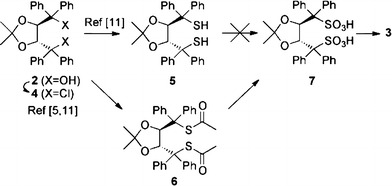
Intended synthesis of 3 via the bisthioacetate 6
Synthesis of 6 could be achieved by treatment of 4 (synthesized according to Ref. [11]) with an excess of thioacetic acid (HSAc) at 50 °C in moderate yield (36%, not optimized). It is worth noting that neither treatment of 4 with stoichiometric amounts of thioacetic acid nor with KSAc or Hg(SAc)2 gave any conversion.
Oxidation of 6 was first attempted using Oxone® (2KHSO5·KHSO4·K2SO4) under a variety of conditions but no conversion was obtained. Treatment with an excess of performic acid (generated in situ by treatment of HCOOH with H2O2) at room temperature resulted in full conversion of 6. However, detailed NMR and HRMS analysis of the obtained product showed that no desired disulfonic acid 7 was formed. Instead, the reaction led to the formation of a mixture of the cyclic sulfoxide 8 and the corresponding sulfone 9 (ratio ~1:4) exclusively after 12 h reaction time. After 20 h reaction time, no sulfoxide was detected anymore and 9 was isolated in 55% yield after column chromatography (Fig. 3). Also the use of peracetic acid did not give any disulfonic acid 7 but several decomposition products and small amounts of 8 and 9, whereas peroxide-mediated oxidation under slightly basic conditions did not result in any conversion at all.
Fig. 3.
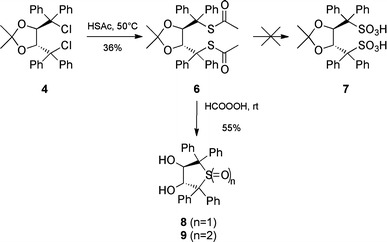
Attempted synthesis of 3 giving the thiolanes 8 and 9 instead
This remarkable formation of the thiolane products 8 and 9 was unexpected as, to the best of our knowledge, no comparable single-step transformation has been reported so far. Besides the unexpected cyclization forming the thiolane ring, the cleavage of the acetonide under these reaction conditions is also remarkable. Normally the TADDOL acetonide group is known to be rather stable under acidic conditions, surviving acidic work-up without any problem [5]. In addition, the ketal proved to be rather stable even under the acidic conditions during the synthesis of 6 at 50 °C (some unidentified by-products have been formed, so a partial cleavage can not be excluded), whereas during the oxidation reaction full hydrolysis occurred at much lower temperature. Accordingly, there must be a significant decrease in stability of the dioxolane ring during this sequence, making it more sensitive towards acid-mediated cleavage.
To get a clearer picture of this reaction, in situ mass spectrometry studies and force field calculations were undertaken. Several possible intermediates for the conversion of 6 to 9 might be possible. Out of a variety of possible reaction mechanisms, two pathways seemed to be the most probable: ketal cleavage first (due to the acidic reaction conditions) followed by cyclization and subsequent oxidation (path A, Fig. 4), or cyclization first followed by ketal cleavage and oxidation (path B, Fig. 4).
Fig. 4.
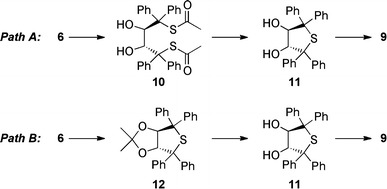
Proposed pathways for the conversion of 6 to 9
We therefore carried out the reaction and monitored its progress by electrospray ionization high resolution mass spectrometry (ESI-HRMS). After 15–30 min at 0 °C we were able to clearly identify the bicyclic intermediate 12 as its Na and K adducts in the positive ion mode with accuracies of 1.29 and 3.12 ppm. Furthermore, at this point of the reaction, small amounts of the sulfoxide 8 were already detected (besides unconverted starting material 6). Warming up the reaction mixture to room temperature, after further 30 min significant amounts of sulfoxide 8 accompanied by traces of sulfone 9 were detected. During all these measurements no ketal cleavage product 10 could be detected. In addition, the bicyclic intermediate 12 was hardly detectable at ambient temperature, indicating the high instability of this compound under the acidic reaction conditions.
These studies unambiguously proved that the reaction proceeds via pathway B (Fig. 4), with the bicyclic compound 12 as a key intermediate (12 is a known compound synthesized by Seebach et al. [13]).
The remarkable cyclization step most probably proceeds via oxidation of one thioacetate group first (giving the corresponding mono-sulfonic acid), followed by a desulfonation, intramolecular SN1 cyclization, and deacetylation sequence resulting in 12. The high tendency of 6 to undergo this cyclization might be due to the easy formation of a highly reactive tertiary carbocation under acidic conditions, thus facilitating the desulfonation and favoring the intramolecular cyclization. However, none of the possible highly reactive intermediates in this step could be detected during our HRMS investigations, making an exact determination of the mechanism and a proof of this hypothesis impossible.
The increased sensitivity of the dioxolane ring may be due to the highly strained trans-configured bicyclo[3.3.0]skeleton, which destabilizes the ketal moiety significantly as compared to monocyclic TADDOL derivatives. Molecular mechanics calculations support this hypothesis. Therefore, the conformations of 2, 6, and the intermediate 12 were optimized by using the MMFF94 force field (MM3 and MMX yielded similar results). From these minimum energy conformations, the relative stabilities of the dioxolane rings of the three compounds were calculated without minimization after replacing the substituents for H, just leaving the comparable (4S,5S)-2,2,4,5-tetramethyl-1,3-dioxolane conformations A (derived from the minimum energy conformation of 2), B (derived from the minimum energy conformation of 6), and C (derived from the minimum energy conformation of 12), for comparison of their relative steric energies. It was found that the dioxolane rings A and B were significantly more stable than C (A: 142, B: 172, and C: 188 kJ/mol). This result can be rationalized by considering the contribution of the individual steric energy components, especially bond stretching, angle bending, and torsion. It was shown that the bond lengths in the dioxolane rings are approximately the same in 2 and 6, whereas in the case of 12 the C–C bond between the bridgehead atoms is remarkably shorter (1.50 Å) in comparison to the monocyclic 2 (1.54 Å) and 6 (1.55 Å). It is worth noting that the calculated value for 2 is in accordance with published X-ray structures of 2 [17, 18]. Comparing the bond (α) and dihedral (φ) angles of the TADDOL-derived conformations A–C with the values of the force field minimized most stable conformation of (4S,5S)-2,2,4,5-tetramethyl-1,3-dioxolane (D), significant differences were observed. The dihedral angles (φ) of the O–C–C–O bond of 2 (A, φ = 38°) and 6 (B, φ = 37°) were slightly smaller compared to D (φ = 40°) while the bicyclo-derived structure C showed the highest torsion (φ = 43°). Comparing the bond angles at the two stereogenic centers even more striking differences were observed. TADDOL (2) (=A) showed only a small deformation compared to the optimized conformation D, mainly due to the steric repulsion of the phenyl groups. However, the bisthioacetate (6)-derived B already showed a higher angle distortion, explaining the higher steric energy of B compared to A. Finally, the trans-configured bicyclo[3.3.0]skeleton of 12 leads to a significant angle deformation at the bridgehead carbons (Fig. 5).
Fig. 5.
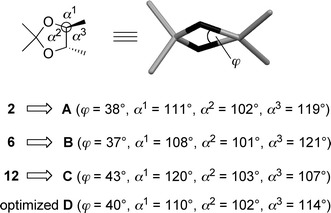
Force field calculations revealed the conformational differences between the monocyclic 2 and 6 and the bicyclic 12
Accordingly, the enhanced acid-sensitivity of the trans-configured bicyclic compound 12 is due to a high bond angle deformation and bond compression resulting in a significantly higher ring strain in comparison to the monocyclic 2 and 6. The driving force of the hydrolysis is therefore derived from the considerable relief of strain energy.
Conclusion
The attempted synthesis of the disulfonic acid 7 by oxidation of the bisthioacetate 6 failed completely, giving the unexpected sulfone 9 as the major product instead. In situ ESI-HRMS investigations proved that this unique reaction proceeds via the bicyclic intermediate 12. No further intermediates of this surprising cyclization could be detected. As a result of its highly strained trans-configured bicyclic skeleton, intermediate 12 was found to be extraordinarily acid-sensitive, resulting in full hydrolysis of the acetonide group under mild conditions. This instability was also corroborated by force field calculations which showed that the bicyclo[3.3.0]skeleton of 12 is highly strained and therefore the dioxolane ring is significantly more labile to ring-opening reactions than usually is the case for TADDOLs. Thus, as a result of their reduced stability, bicyclo[3.3.0]-based TADDOL derivatives seem to be less suited for further applications in chiral catalysis (the comparably high acid sensitivity of a pyrrolidine-containing TADDOL-derived bicyclo[3.3.0]skeleton was recently reported [19]).
Experimental
Melting points were measured on a Kofler melting point microscope (Reichert, Vienna). 1H and 13C NMR spectra were recorded on a Bruker Avance DRX 500 MHz spectrometer using a TXI cryoprobe with z-gradient coil. Typical resolutions and chemical shift precisions were ±1 Hz for 1H and ±0.8 Hz for 13C. All NMR spectra were referenced to the solvent peak. High resolution mass spectra were obtained by using an Agilent 6520 Q-TOF mass spectrometer with an ESI source and an Agilent G1607A coaxial sprayer. All analyses were made in the positive ionization mode. Purine (exact mass for [M + H]+ = 121.050873) and 1,2,3,4,5,6-hexakis(2,2,3,3-tetrafluoropropoxy)-1,3,5,2,4,6-triazatriphosphinane (exact mass for [M + H]+ = 922.009798) were used for internal mass calibration. [α]20D values were recorded on a Perkin Elmer 241 MC Polarimeter. Force field calculations were done with Pcmodel 9.0 (Serena Software). All chemicals were purchased from commercial suppliers and used without further purification. (R,R)-TADDOL dichloride (4) was synthesized starting from l-tartaric acid according to Ref. [11].
S,S’-((4R,5R)-2,2-Dimethyl-1,3-dioxolane-4,5-diyl)bis(diphenylmethylene) diethanethioate (6, C35H34O4S2)
A mixture of 390 mg (R,R)-4 (0.775 mmol) and 2.25 cm3 AcSH was stirred at 50 °C for 15 h, cooled, and extracted with aqueous NaHCO3 (sat.) and EtOAc. The organic layer was washed with brine, dried over Na2SO4, and evaporated to dryness. Purification by column chromatography (silica gel, hexanes/EtOAc = 5:1) yielded 162 mg (36%) 6 as a white solid. M.p.: 192–196 °C; 1H NMR (500 MHz, CDCl3): δ = 7.58 (d, J = 7.4 Hz, 4H, Ar–H), 7.42–7.12 (m, 16H, Ar–H), 5.95 (s, 2H, –CH–), 2.17 (s, 6H, –COCH3), 0.35 (s, 6H, –CH3) ppm; 13C NMR (125 MHz, CDCl3): δ = 194.0 (–CO–), 144.8 (Ar–C), 140.3 (Ar–C), 131.8 (Ar–C), 131.3 (Ar–C), 127.9 (Ar–C), 127.5 (Ar–C), 126.5 (Ar–C), 126.4 (Ar–C), 110.8 (C(CH3)2), 79.9 (–CH–), 65.4 (CPh2), 31.1 (–COCH3), 28.0 (–CH3) ppm; HRMS (ESI+): m/z calcd for C35H34O4S2 + Na: 605.1791 [M + Na]+, found: 605.1785; [α]20D = −282° cm2 g−1 (c = 3.4, CHCl3).
(3R,4R)-2,2,5,5-Tetraphenyltetrahydrothiophene-3,4-diol-1,1-dioxide (9, C28H24O4S)
A mixture of 8 cm3 HCOOH and 0.8 cm3 H2O2 (30%) was stirred at room temperature for 1 h, cooled to 0 °C, and a solution of 72 mg 6 (0.124 mmol) in 4 cm3 CH2Cl2 was added dropwise (15 min). The mixture was slowly warmed up to room temperature (1 h) and stirred for an additional 19 h. The mixture was neutralized with aqueous Na2CO3 (sat.) and extracted with CH2Cl2 twice. The combined organic layers were washed with brine, dried over Na2SO4, and evaporated to dryness. Purification by column chromatography (silica gel, hexanes/EtOAc = 3:1) yielded 31 mg (55%) 9 as an oily residue. 1H NMR (500 MHz, CDCl3): δ = 7.63–7.48 (m, 8H, Ar–H), 7.37–7.10 (m, 12H, Ar–H), 5.18 (s, 2H, –CH–) ppm; 13C NMR (125 MHz, CDCl3): δ = 136.4 (Ar–C), 134.9 (Ar–C), 132.0 (Ar–C), 130.0 (Ar–C), 129.1 (Ar–C), 128.8 (Ar–C), 128.5 (Ar–C), 128.0 (Ar–C), 80.7 (CPh2), 74.1 (–CH–) ppm; HRMS (ESI+): m/z calcd for C28H24O4S + NH4: 474.1734 [M + NH4]+, found: 474.1736; [α]20D = −199.5° cm2 g−1 (c = 0.6, CHCl3).
Acknowledgments
This work was supported by the FWF (Austrian Science Funds) Project No. P22508-N17. We are grateful to Dr. Christoph Etzlstorfer for his support with the force field calculations, and Prof. Dr. Norbert Müller and Prof. Dr. Heinz Falk for inspiring discussions.
References
- 1.Berkessel A, Gröger H. Asymmetric organocatalysis: from biomimetic concepts to applications in asymmetric synthesis. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- 2.Dalko I, editor. Enantioselective organocatalysis. Weinheim: Wiley-VCH; 2007. [Google Scholar]
- 3.Seayad J, List B. Org Biomol Chem. 2005;3:719. doi: 10.1039/b415217b. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Garcia P, Lay F, Garcia-Garcia P, Rabalako C, List B. Angew Chem Int Ed. 2009;48:4363. doi: 10.1002/anie.200901768. [DOI] [PubMed] [Google Scholar]
- 5.Seebach D, Beck AK, Heckel A. Angew Chem Int Ed. 2001;40:92. doi: 10.1002/1521-3773(20010105)40:1<92::AID-ANIE92>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 6.Pellissier H. Tetrahedron. 2008;64:10279. doi: 10.1016/j.tet.2008.08.029. [DOI] [Google Scholar]
- 7.Akiyama T, Saitoh Y, Morita H, Fuchibe K. Adv Synth Catal. 2008;347:1523. doi: 10.1002/adsc.200505167. [DOI] [Google Scholar]
- 8.Belokon YN, Kochetkov KA, Churkina TD, Ikonnikov NS, Chesnokov AA, Larionov OV, Singh I, Parmar VS, Vyskocil S, Kagan HB. J Org Chem. 2000;65:7041. doi: 10.1021/jo000719p. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Unni AK, Thadani AN, Rawal VH. Nature. 2003;424:146. doi: 10.1038/424146a. [DOI] [PubMed] [Google Scholar]
- 10.McGilvra JD, Unii AK, Modi K, Rawal VH. Angew Chem Int Ed. 2006;45:6130. doi: 10.1002/anie.200601638. [DOI] [PubMed] [Google Scholar]
- 11.Seebach D, Beck AK, Hayakawa M, Jaeschke G, Kühnle FNM, Nägeli I, Pinkerton AB, Rheiner PB, Duthaler RO, Rothe PM, Weigand W, Wünsch R, Dick S, Nesper R, Wörle M, Gramlich V. Bull Soc Chim Fr. 1997;134:315. [Google Scholar]
- 12.Gerbino DC, Mandolesi SD, Koll LC, Podesta JC. Synthesis. 2005;15:2491. [Google Scholar]
- 13.Seebach D, Hayakawa M, Sakaki J, Schweizer B. Tetrahedron. 1993;49:1711. doi: 10.1016/S0040-4020(01)80529-9. [DOI] [Google Scholar]
- 14.Koenig NH, Swern D. J Am Chem Soc. 1957;79:4235. doi: 10.1021/ja01572a067. [DOI] [Google Scholar]
- 15.Belecki K, Berliner M, Bibart RT, Meltz C, Ng K, Phillips J, Ripin DHB, Vetelino M. Org Process Res Dev. 2007;11:754. doi: 10.1021/op7000386. [DOI] [Google Scholar]
- 16.Knapp S, Darout E, Amorelli B. J Org Chem. 2006;71:1380. doi: 10.1021/jo0520386. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg I, Stein Z, Weber E, Dörpinghaus N, Franken S. J Chem Soc Perkin Trans. 1990;2:953. [Google Scholar]
- 18.Beck AK, Bastani B, Plattner DA, Petter W, Seebach D, Braunschweiger H, Gysi P, Vecchia LL. Chimia. 1991;45:238. [Google Scholar]
- 19.Weibel D (2003) PhD thesis (Nr. 15290), ETH Zürich