

Abstract
A practical experimental strategy is proposed that could potentially enable greater control of the tip apex in non-contact atomic force microscopy experiments. It is based on a preparation of a structure of interest alongside a reference surface reconstruction on the same sample. Our proposed strategy is as follows. Spectroscopy measurements are first performed on the reference surface to identify the tip apex structure using a previously collected database of responses of different tips to this surface. Next, immediately following the tip identification protocol, the surface of interest is studied (imaging, manipulation and/or spectroscopy). The prototype system we choose is the mixed Si(111)-7×7 and 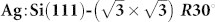 surface which can be prepared on the same sample with a controlled ratio of reactive and passivated regions. Using an “in silico” approach based on ab initio density functional calculations and a set of tips with varying chemical reactivities, we show how one can perform tip fingerprinting using the Si(111)-7×7 reference surface. Then it is found by examining the imaging of a naphthalene tetracarboxylic diimide (NTCDI) molecule adsorbed on
surface which can be prepared on the same sample with a controlled ratio of reactive and passivated regions. Using an “in silico” approach based on ab initio density functional calculations and a set of tips with varying chemical reactivities, we show how one can perform tip fingerprinting using the Si(111)-7×7 reference surface. Then it is found by examining the imaging of a naphthalene tetracarboxylic diimide (NTCDI) molecule adsorbed on 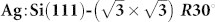 surface that negatively charged tips produce the best intramolecular contrast attributed to the enhancement of repulsive interactions.
surface that negatively charged tips produce the best intramolecular contrast attributed to the enhancement of repulsive interactions.
Scanning probe microscopy (SPM) has become the tool of choice for providing detailed information on real-space atomic and molecular structure at surface interfaces. The high sensitivity of SPM to tip-sample interactions makes it a particularly powerful method of studying single molecules with exceptionally high resolution. In scanning tunneling microscopy (STM), for instance, it has been shown that submolecular resolution is achievable by imaging molecular orbital density1,2. This is possible because the image contrast is sensitive to the density of states of the molecular orbitals within an energy window around the Fermi level. However, whilst impressive resolution can be achieved, relating the observed image to the atomic structure is challanging and generally requires detailed theoretical consideration of the electronic structure. Non-contact atomic force microscopy3 (NC-AFM) operated in the frequency modulation mode4 can avoid this problem as the contrast in AFM relies on the variation of the short-range interaction between tip and surface. Moreover, NC-AFM offers the possibility to quantitatively measure tip-sample forces allowing, for instance, the chemical identification of a surface5, measurement of molecular pair potentials6,7 and measurement of the lateral forces during atomic manipulation8. By using an “on-off” approach9,10 the short-range interaction forces and potential maps can be obtained directly experimentally and a quantitative comparison of these with density function theory (DFT) calculations is possible. Most importantly for molecular study, by functionalising the scanning tip NC-AFM is capable of revealing the internal structure of planar molecules with unprecedented resolution. Since individual bonds were first resolved by Gross et al11, NC-AFM has been shown to reveal the bond order within an organic molecule12, molecular charge distribution13, conformation14 and the stages of a chemical reaction15. More recently intermolecular features have also been resolved for hydrogen-bonded molecules16,17 although the interpretation of the contrast is far from trivial and requires very careful analysis of tip-sample interactions17,45.
Although in a simplistic view NC-AFM can be considered a probe of the total surface electron density18, the exact imaging mechanism of NC-AFM is complex due to the interplay of Pauli repulsion, covalent and Coulomb interactions, and van der Waals attraction. Thus far the mechanism of sub-molecular contrast was assigned solely to Pauli repulsion, with the long-range attraction being considered to only contribute to a site-independent background11. To attain intramolecular resolution, the tip first needs to be functionalized by picking up individual atoms or molecules19 to passivate the reactive tip apex. If the tip is not passivated the attractive tip-sample interaction becomes too high and the molecule is manipulated before submolecular resolution can be achieved11. Only in exceptional cases, such as for large graphene sheets20 or molecules bound on semiconductor surfaces21, can reactive tips acheive similar resolution. This presents a challenge for non-metallic systems such as semiconductor surfaces where controlled tip termination is significantly more difficult. Consequently, in semiconductor systems sophisticated theoretical modeling involving a large number of conceivable tip models22,23,24 is essential in order to correctly interpret the experimental images. However, this is impossible to do reliably without knowing the actual tip termination.
To overcome these difficulties, several tip identification protocols have been proposed, such as determining the tip apex chemical identity by performing spectroscopy measurements on ionic KBr25, imaging of the Cu:O surface26, TiO227, MgO28, Si(100)29,30 and the hydrogen-passivated Si(111) surface24. Fuiju and Fijihira31 proposed a new sample holder where two surfaces can be mounted side by side: the CaF2(111) surface to be used for tip characterisation, while the other contained a mixed assembly of thiolates on the Au(111) surface. A similar method was proposed by Naydenov et al.32 in the context of STM where the tip characterisation was performed on the Pt(111) surface before moving onto studying a single molecule adsorbed on the Si(001) surface, both samples mounted in the same sample holder. One method, the so-called CO front atom identification (COFI) protocol33, exploits the small spatial extent of a CO molecule adsorbed on a Cu(111) surface to reverse image the tip state prior to using it for force spectroscopy measurements. This method was later used to distinguish atomic species at the tip apex and its orientation34. Interestingly, it has been shown that a tip characterised by the COFI method can in principle be transferred to another surface33.
Results
Proposed tip identification protocol
Motivated by previous studies31,33, we propose a practical two-step experimental strategy which has the potential to enable not only identification, but accurate control of the apex of a NC-AFM tip. Our proposal, shown schematically in Fig. 1, is based on characterising AFM probes by “reverse imaging”7,35 the tip state using a reference system (step one) followed by performing a measurement on a system of interest with the now characterised tip (step two) during the same experimental run, thereby ensuring minimal tip change.
Figure 1.

(a) Schematic of the two-step tip control strategy we propose. The tip is first characterised on the Si(111)-7×7 surface (left) after which it is moved to the actual sample of interest. In our case this is the Ag-terminated Si(111) reconstruction (right). By comparing the experimentally measured tip response above specific surface sites on the Si(111)-7×7 surface with the corresponding data in our tip database, the tip termination can be characterised prior to the second step. Since the macroscopic part of the tip remains unknown, the “on minus off” method is used both in experiment and simulations to minimise the effect of the long-ranged background forces. (b) STM image of a mixed sample containing regions of the Si(111) -7×7 and 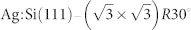 surfaces. (c) Higher resolution image of the region highlighted in (b).
surfaces. (c) Higher resolution image of the region highlighted in (b).
The example system we choose is the mixed Si(111)-7×7 and 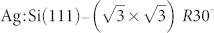 surface which can be prepared on the same sample with a controlled coverage ratio of chemically reactive and passivated regions (see Fig. 1B and C). Although force spectroscopy measurements can be performed on various lattice sites of the Si(111)-7×7 surface, the response depends crucially on the (unknown) tip structure. To identify the tip apex structure, we propose that an extended database of theoretically calculated spectroscopy responses for different tip terminations be generated which can then be compared with the experimental observations. This procedure would allow the composition, structure, and relative orientation of the tip apex to be characterised on the reference surface (in the case described below, the Si(111)-7×7 reconstruction) before moving to the surface and/or adsorbates of interest. If during imaging or manipulation of the surface of interest the tip structure has changed (which can be ascertained via changes in contrast or the spectroscopic response), the first step can be repeated to recharacterise the tip. The tip preparation protocol (e.g. controlled tip crashes and STM bias pulsing) can be repeated until a desirable tip is identified by comparing force spectroscopy measurements to the entries in the theoretical database. The key advantage of this strategy is that specific tip states can be generated and, in principle, recovered in the event that an inadvertent tip change occurs.
surface which can be prepared on the same sample with a controlled coverage ratio of chemically reactive and passivated regions (see Fig. 1B and C). Although force spectroscopy measurements can be performed on various lattice sites of the Si(111)-7×7 surface, the response depends crucially on the (unknown) tip structure. To identify the tip apex structure, we propose that an extended database of theoretically calculated spectroscopy responses for different tip terminations be generated which can then be compared with the experimental observations. This procedure would allow the composition, structure, and relative orientation of the tip apex to be characterised on the reference surface (in the case described below, the Si(111)-7×7 reconstruction) before moving to the surface and/or adsorbates of interest. If during imaging or manipulation of the surface of interest the tip structure has changed (which can be ascertained via changes in contrast or the spectroscopic response), the first step can be repeated to recharacterise the tip. The tip preparation protocol (e.g. controlled tip crashes and STM bias pulsing) can be repeated until a desirable tip is identified by comparing force spectroscopy measurements to the entries in the theoretical database. The key advantage of this strategy is that specific tip states can be generated and, in principle, recovered in the event that an inadvertent tip change occurs.
Following the tip characterisation process one can image the surface or adsorbate of interest. In this work we chose a single naphthalene tetracarboxylic diimide (NTCDI) molecule adsorbed on the 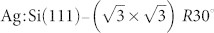 surface as our prototype. The imaging performances of different tips have been evaluated to understand which tip terminations may be suitable to resolve the molecular skeleton, i.e. are capable of intramolecular resolution.
surface as our prototype. The imaging performances of different tips have been evaluated to understand which tip terminations may be suitable to resolve the molecular skeleton, i.e. are capable of intramolecular resolution.
Si(111)-7×7 was chosen as the reference to build the database for characterising tips because it contains a number of well-established and easy-to-identify sites (adatoms, rest atoms, corner hole, etc.) on which to perform the spectroscopy measurements. We note that this surface has been previously suggested33 as a good candidate for tip characterisation.
Fig. 2 shows the top and side views of the dimer-adatom-stacking fault (DAS) model of the Si(111)-7×7 surface with all essential atomic sites indicated. Unlike the bare Si(111) surface, which contains unsaturated dangling bond orbitals, the 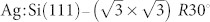 surface is an ideal prototype of a chemically inert substrate for studies of deposited (e.g. physisorbed) molecules. Previous work has shown that NTCDI forms hydrogen-bonded 2D networks on the
surface is an ideal prototype of a chemically inert substrate for studies of deposited (e.g. physisorbed) molecules. Previous work has shown that NTCDI forms hydrogen-bonded 2D networks on the 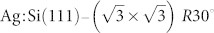 surface36,37 which show both intra- and intermolecular contrast when imaged with NC-AFM17.
surface36,37 which show both intra- and intermolecular contrast when imaged with NC-AFM17.
Figure 2. Top (top image) and side (bottom image) views of the DAS model of the Si(111)-7×7 surface.

The bottom image also shows a 2D map of the surface electrostatic potential (with red color indicating the lowest, and the blue the highest electrostatic potential). The solid black lines in the bottom image highlight the areas of the lowest electrostatic potential above rest and corner-hole atoms. The pink line corresponds to an electron density of 0.001 a.u.
Experimentally, the 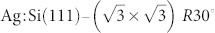 surface can be produced by first preparing a pristine Si(111)-7×7 sample by standard flashing/annealing methods. The sample is then annealed at ~480°C during controlled deposition of silver until the desired coverage is reached and annealed at ~520°C for a further 10 minutes. If the deposition time is controlled carefully, it is possible to prepare sub-monolayer coverages of silver leading to a relatively defect free mixed surface containing regions of both Si(111)-7×7 and
surface can be produced by first preparing a pristine Si(111)-7×7 sample by standard flashing/annealing methods. The sample is then annealed at ~480°C during controlled deposition of silver until the desired coverage is reached and annealed at ~520°C for a further 10 minutes. If the deposition time is controlled carefully, it is possible to prepare sub-monolayer coverages of silver leading to a relatively defect free mixed surface containing regions of both Si(111)-7×7 and 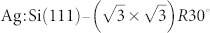 as shown in Figure 1(b,c). A detailed description of the
as shown in Figure 1(b,c). A detailed description of the 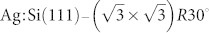 surface is given elsewhere38.
surface is given elsewhere38.
In this paper we use sophisticated ab initio simulations to calculate the response of multiple NC-AFM tip terminations to an NTCDI molecule adsorbed on the 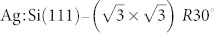 surface. Through an examination of the calculated F(z) curves beyond the force turnaround we identify tip structures capable of providing intramolecular resolution of planar molecules. Based on our findings we propose a two-stage strategy to: (i) identify the experimental NC-AFM tip structure against a database of theoretically modelled tip apices, then (ii) determine whether the experimental tip is capable of providing intramolecular resolution of planar molecules. In the real experiment (which we mimic here with purely numerical methods) step (i) can be achieved by comparing measured spectra taken above certain lattice sites of the reference surface to a database of theoretical F(z) spectroscopy curves for a large number of possible tip structures. Knowledge of the tip apex structure elucidates the imaging mechanism. Classes of possible tips can then be selected which are capable of providing intramolecular resolution of the molecules.
surface. Through an examination of the calculated F(z) curves beyond the force turnaround we identify tip structures capable of providing intramolecular resolution of planar molecules. Based on our findings we propose a two-stage strategy to: (i) identify the experimental NC-AFM tip structure against a database of theoretically modelled tip apices, then (ii) determine whether the experimental tip is capable of providing intramolecular resolution of planar molecules. In the real experiment (which we mimic here with purely numerical methods) step (i) can be achieved by comparing measured spectra taken above certain lattice sites of the reference surface to a database of theoretical F(z) spectroscopy curves for a large number of possible tip structures. Knowledge of the tip apex structure elucidates the imaging mechanism. Classes of possible tips can then be selected which are capable of providing intramolecular resolution of the molecules.
In our simulations, a number of different tip models were considered, as explained below. It was assumed that these nanotips are responsible for the short-range interaction between the tip and surface. Since we are only interested here in the tip force relative to a certain surface site, the macroscopic contribution to the tip force due to the unknown macroscopic part of the tip was not taken into account, so that we only considered the interaction of the nanotip (to be called simply “tip” in what follows) with either of the two surfaces.
Seven atomic tip models have been considered which vary in geometry, chemical structure, electronic properties, and their relative orientation with respect to the surface. The tip candidates are shown in Fig. 3 in which the electrostatic potential maps are also plotted. The Si-based tips were modelled by two different Si clusters built upon either (111) (H3 configuration) or (100) surface (dimer configuration) orientations, and terminated with either Si, H, or Ag atoms to simulate a range of possible types of apex following tip crashes. It is also possible that the tip is terminated by the molecule used in the imaging, so NTCDI molecules in two orientations, O-down and H-down, were also considered. Since a CO-terminated tip is known to yield intramolecular resolution for a number of organic molecules on metal surfaces19, this tip was also included in our analysis for comparison. The adsorbed CO is most stable when back-bonded via its C atom39,40. In total, including both orientations of the NTCDI tip, eight tip models were considered. Since the tips we consider here are assumed to be covalently bound to a macroscopic part of the tip, upper atoms of the tips were fixed in all our simulations as indicated in Fig. 3.
Figure 3.

Schematic view of all seven tip models with their electrostatic potential maps calculated for an electron density isosurface of 0.001 a.u.: (a) Si based H3 tip; (b) Si dimer tip; (c) H passivated H3 tip; (d) passivated dimer tip; (e) Ag passivated H3 tip; (f) NTCDI and (g) CO tip. The tips (a) – (e) are Si-based clusters built up using different surface orientations and some terminated at their apices with either H or Ag. Two NTCDI tips were considered differing in their orientation, O-down and H down. Red indicates the lowest, and blue the highest electrostatic potential of the tip molecule.
To identify the tips we defined defined the following criteria and categories. (i) The bare Si tips in H3 or dimer configurations are classified as reactive because of the existence of dangling bonds directed towards the surface on the apices of these tips, while the passivated Si tips, NTCDI tip and CO tip are chemically inert. (ii) The Ag-terminated Si tip and H-down NTCDI tip are classified as positively charged considering their electrostatic potentials around the apex. The electrostatic potential surface (EPS) for different tips calculated using DFT electron density is shown in Fig. 3, in which a surface of constant electron density of 0.001 a.u. is coloured according to the electrostatic potential to guide the comparison between different tips.
Results on Si(111)-7×7: identification of tips
Force mapping and imaging of Si(111)-7×7
In order to characterise each tip model, we calculated F(z) above preselected lattice sites for all eight tip structures. The following lattice sites on the Si(111)-7×7 surface were used: corner adatoms (CoA), rest atoms (R), atoms at the hexagon centre (Hex) and centre (or central) adatoms (CeA), all in either of the two halves, as well as the centre (dimer) site (Ce) and the corner hole (CoH), i.e. ten sites in total, see Fig. 2. The sites were chosen on the basis of differences in their chemical reactivity41. For instance, the fully occupied dangling-bond site localised on the rest atoms should produce significantly different behaviour to that of the partially occupied adatom sites42. This is also evidenced by the calculated surface electrostatic potential shown in the bottom panel of Fig. 2. In addition, it has been shown24 that Si(111)-7×7 can be controllably dosed with single hydrogen atoms producing a chemically unreactive site in NC-AFM measurements. Therefore, to complement complement the chemically distinct sites already available within the Si(111)-7×7 reconstruction we also modelled a hydrogen-passivated centre adatom located on the unfaulted half of the unit cell (H-CeA-U). Our reference surface thus consists of eleven distinct sites with at least three significantly different reactivities (adatom, restatom and passivated adatom). The calculated spectra provide a comprehensive description of the response of each tip to the surface which can then be used for tip fingerprinting.
In STM measurements of the Si (111)-7×7 surface, negative sample bias images enable the faulted and unfaulted halves of the unit cell to be easily distinguished. For NC-AFM studies, however, despite expected differences in the reactivity for each half of the unit cell, the variation is much less noticeable and has only been observed when operating in the very weakly attractive regime7,43. The fact that the tip forces are very similar for the same types of sites on faulted and unfaulted halves of the surface unit cell is generally confirmed by our simulations. We also note that the corner adatom in the faulted half (CoA-F) is found to be slightly more attractive than the corner adatom at unfaulted half (CoA-U) at higher distance and less attractive at close approach except for the O-down NTCDI tip. The calculated raw forces for all ten sites including the corner hole can be found in the Supporting information (SI). For clarity, we will focus here on force spectra calculated for the unfaulted half only (5 sites). In addition to these, the central and the passivated sites will also be used as shown in Figure 4. In these calculations, to mimic experimental data we used the “on minus off” method, such that only the short-range parts of the forces were used obtained by subtracting the force curve above the corner hole.
Figure 4. Force versus distance curves of tip candidates approaching selected sites on Si(111)-7×7.

The values of the maximum attractive force above various lattice sites serve as the simplest comparison one can make between force spectra. As such, we use this to start our discussion on tip fingerprinting. The magnitude of attraction depends on the reactivity of the tip but differs from site to site and with height z. Using bare Si tips in H3 or dimer configurations, the maximum attractive forces above Si adatoms (CoA-U and CeA-U sites) reaches 2.0 nN and 1.7 nN, respectively, while the maximum attractive forces above Si rest atoms (R-U) are 2.8 nN and 2.2 nN, respectively. However, in the case of the H3 tip the maximum force above the adatom sites happens at 0.9 Å larger height than above the rest atom, while in the case of the dimer tip the difference in heights is much larger, 2.1 Å. The maximum force for the O-down NTCDI tip is 2.1 nN above the adatom sites, similar to the Si tips, however, it has almost no attraction above the rest atom site until the very close approach of the tip-sample distance of 1.9 Å. In addition, and qualitatively the opposite of the Si tips responses, the force reaches its maximum value above the adatom sites at 1.2 Å closer to the surface than for the rest atom. Figure 4(h) shows the force plotted on two scales where a sharp increase in force is calculated over the adatoms at 3Å. This corresponds to a snap-to-contact between the silicon adatom and the oxygen atom on the NTCDI as a bond is formed. This is likely due to the lone pair electrons of the O in NTCDI which allow the formation of a strong bond with the half-filled adatom of the surface. The data reported above should enable telling these three tips apart quite easily.
The maximum attractive forces above the adatoms and rest atom sites observed for chemically inert tips (both passivated Si tips, as well as for H-down NTCDI and CO tips) are between three to ten times smaller, around 0.2–0.7 nN, i.e. these tips clearly form a second family of tips which is quite different from the first set (reactive tips) discussed above. By examining the force spectroscopy curves in detail, it is possible to differentiate between them. For both passivated Si tips and the CO tip the maximum forces at the rest atom site are at least two times smaller than above the adatom sites, while for the H-down NTCDI tip it is the opposite: the maximum force above the rest atom (0.27 nN) is much larger than above the adatom sites (0.04–0.05 nN). Additionally, the H:Si dimer tip differs from the H:Si H3 tip in that the maximum force above the adatom sites is more than three times larger, however, as will be discussed below, this is also related to instabilities of the adsorbed hydrogen on the dimer tip. The CO tip has a similar pattern of maximum forces above the adatom and rest atoms sites to those for the H:Si H3 tip, however, the maximum force above the rest atom site happens at 0.3 Å closer to the surface than for the adatom sites for the CO tip, while this is the other way round for the H:Si H3 tip. The Ag-terminated Si tip has the maximum forces above the adatom and rest atom sites very close to each other and lying somewhere in between the values for the two families of tips discussed above. The maximum in the force above the rest atom site happens 0.8 Å closer to the surface than for the adatom sites.
Based on these observations a general distinction between reactive and inert tip structures can be made. Reactive tips are identified as those that undergo a strong interaction (>2 nN) with both the adatom sites (and possibly with the restatom site) of the Si(111)-7×7. Chemically inert tips are instead calculated to interact weakly with these two sites, producing a maximum attractive force of no more than 0.3 nN (see later discussion regarding stability of the H:Si dimer tip). Finally, there could be tips of intermediate reactivity with the maximum forces lying between these two characteristic values. Not only can the family of chemically inert tips be clearly distinguished from the reactive tips, but it is also possible to tell them apart within the family by looking at the particular pattern of maximum forces above the rest atom and adatom sites. However, identification of tips demonstrating intermediate reactivity may require applying additional criteria.
Note that for all tips discussed here no large difference has been found in the force spectroscopy curves between the centre and corner adatom sites. It is possible however that the results would be different for other tips, not considered here.
To enable a complete characterisation of the tip whereby all of our structures (including the O-down NTCDI termination) can be distinguished we also considered a hydrogen-passivated adatom as a third chemically distinct site. The maximum attractive force above the H passivated Si adatoms varies much less from tip to tip. Indeed, for reactive H3 and dimer Si tips the force is 0.6 nN and 0.25 nN, respectively, for the Ag-terminated tip it is 0.35 nN, while for all other tips the interaction is very weak (attractive forces are less than 0.1 nN). These trends follow the general observation that chemically inert tips - which we later show to be essential for intramolecular resolution - always show weaker interaction than more reactive structures. Importantly, the O-down NTCDI tip also behaves as a non-reactive tip above this lattice site. Therefore it is clear that the fully occupied restatom and passivated hydrogen sites on Si(111)-7×7 provide the best test to identify chemically inert tips (which we later show most commonly provide intramolecular resolution), and must be considered in addition to the half-filled adatom site to provide a fingerprint for the tip structure. We stress again, all of the forces forces discussed here have been processed using the “on minus off” method to remove the long-range component.
It is interesting now to discuss the stability of our of our tip models. If a tip undergoes a modification during the spectroscopy experiment, e.g. acquires an atom from the surface or loses one to the surface, it may not be appropriate for further studies and may be discarded. These kind of events, seen for some of our tips, manifest themselves in the force spectroscopy curves by abrupt changes of the force often with a dissipation signal (not studied here). Indeed, it is seen from Fig. 4(a) that the Si H3 tip becomes unstable at around 4.2 Å above the passivated surface site. We find that this is because it picks up the H atom from the surface, effectively turning itself turning itself into the H passivated Si H3 tip. This was confirmed by our retraction calculations which are consistent with previous observations on both Si(100)29 and Si(111)-7×724. Therefore, if this tip is to be used in further studies at stage two as a reactive tip, passivated sites are to be avoided during stage one as otherwise the tip would change its reactivity to inert. The dimer tip, however, does not pick up the H atom from the passivated adatom site which is explained by comparing the H atom removal energies for the two tips and the adatom site. We have previously shown that the adsorption energy for hydrogen on the Si H3 tip is approximately 0.8 eV larger than for the dimer tip structure29. Additional calculations (using the SIESTA code44 as in the previous work29) show that the adsorption energy for H on a Si(111)-7×7 adatom (which adopts a T4 configuration) is also larger than the dimer tip structure, but now only by 0.45 eV, thus explaining the stability of the H:Si H3 tip.
At the same time, the dimer tip, see Fig. 4(b), experiences an abrupt increase of the force at 4.5 Å above the adatom sites due to formation of a covalent bond with the dangling bond of the Si adatom. This triggers a reversible (as was shown by as was shown by our calculations of the force-distance curve for retraction of the tip) reconfiguration of the Si dimer at the tip apex. We have previously shown that this sudden distortion of the tip is associated with energy dissipation and hence may manifest itself in the dissipation signal, providing an additional distinct feature for the dimer tip30.
The passivated Si H3 tip remains stable for all investigated lattice sites. The passivated dimer tip, Fig. 4(d), on the other hand, loses its H Fig. 4(d), loses its H atom when approaching an adatom site, in agreement with both the suggested ordering of adsorption energy outlined above and the observation that the reactive tip does not pick up reactive dimer tip does not pick up an H atom from the passivated site on the surface. Upon retraction, retraction the tip becomes reactive suggesting that the adatom sites may sometimes be used to depassivate certain tips if necessary.
As mentioned above, when the O-down, when the O-down NTCDI tip approaches the Si adatoms, the formation of a covalent bond between the O atom of the tip and the Si adatom is seen. Upon retraction, the tip remains intact meaning that this instability is reversible. Similarly, the Ag-terminated Si tip, Fig. 4(e), shows unstable behaviour above adatom sites as the Ag atom jumps to the surface. It does not, however, remain there remain there upon retraction, i.e. this instability is also reversible.
Finally, the CO-terminated and the H-down NTCDI NTCDI tips were found to be stable above all of the investigated investigated lattice sites of the Si(111) surface. Note that NTCDI and CO tips are prone to bending or tilting upon close approach17,45. Moreover, this flexibility, especially for the CO tip, may result in an apparent enhancement of contrast12,45.
Image contrast
Thus far our consideration has been limited to analysing F(z) spectroscopy for the different tip structures. To provide more immediate insight into the tip characterisation it may be instructive to discuss what kind of images one might expect from these tips. We shall consider the relative differences in contrast that would be expected expect from the different lattice sites. The image contrast will be discussed assuming that the oscillation amplitude is small and the AFM is operated in the constant height mode. In this case the image contrast corresponds to the frequency shift of the cantilever (which, we find, follows a similar profile to the force). For convenience, we shall adopt the following convention frequently used in frequently used in NC-AFM experiments: dark features: dark features correspond to more negative frequency shifts (more attractive regions) and bright features correspond to more positive frequency shifts (repulsive, or less attractive regions). We shall also assume that a series of images are taken in which the height is each time reduced moving the oscillating tip closer to the surface. Our discussion will remain qualitative and will be based entirely on the force versus distance curves calculated for ten lattice sites and the H-passivated site as explained above. Also recall that the forces we consider are only short-range forces as in each case the contribution from the corner-hole site has been subtracted.
Consider first the two reactive bare Si tips. At large tip-sample distances, the long-range attractive interaction is nearly site-independent and leads to faint contrast approaching the noise level of the experimental equipment. At closer approach, the Si adatoms appear more attractive and hence will appear as dark features, the restatoms and H passivated sites with intermediate intensity and the central and hexagon sites as less attractive features, with a similar brightness to the image background. Appoaching even closer, at around 3 Å and below, the adatoms and H passivated sites are expected to appear as bright features following turn-arounds in force whilst the rest atoms remain dark in appearance. For the chemically inert tips, however, the situation is different46. For the passivated tip structures the attractive interaction is suppressed and the relatively high geometric position of the H atom adsorbed on the silicon surface is much more noticeable. In this case, images taken at high tip-sample separations (>4.5 Å) would reveal the H passivated sites as the most notable features appearing as dark features (depressions), while other sites would appear with a similar brightness to the image background. At closer approach, passivated sites will turnaround in force appearing much brighter than any other feature, followed by hexagon and rest atoms sites, with adatom sites being the darkest. Recall, however, that the passivated dimer tip becomes unstable above the adatom sites in this distances range. Finally, at even closer approach, the rest atom sites will also become distinctively bright.
Imaging with the CO tip would at large heights first reveal hexagonal and rest atom sites as brightest, with the H-passivated sites being the darkest. At closer approach, the H-passivated site becomes the brightest, while the adatom sites the darkest. Finally, at even closer approach adatom sites become brighter than the rest atom sites, although the passivated sites still appear as brightest.
Let us finally consider the two NTCDI based tips. In both cases the passivated site appears the brightest starting from some tip height. It is followed by the adatom sites for the H-down tip and the rest atom site for the O-down tip. The darkest features in this distance range are the rest atoms and adatom sites for the H-down and O-down tips, respectively.
Finally, for most tips the rest atoms would not manifest themselves in the image until a very close tip approach where the repulsive interaction becomes too high to maintain a stable tip above adatom sites. Therefore for a full characterisation of the tip beyond the general classification discussed here, quantitative F(z) spectroscopy is essential. Taking spectroscopy curves above specific lattice sites might also technically be more preferable than taking a set of images as discussed above.
A short discussion of the interaction mechanisms in relation to the image contrast is contained in the Supporting Information.
Results for an example test system: NTCDI adsorbed on the 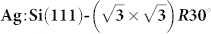 surface
surface
Force mapping and imaging of NTCDI
To understand in detail the mechanism responsible for the possible intramolecular contrasts to be observed in experiment with different tips, an example system was built up in which a single NTCDI molecule was adsorbed on the 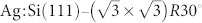 surface as shown in Fig. 5. NTCDI is adsorbed on the surface via relatively weak van der Waals forces with an adsorption energy of 1.454 eV (physisorption). Description of the van der Waals interaction, see Section Methods, is important in our DFT simulations as failure to do so may result in a significant underestimation of the molecular interactions. For instance, the calculated adsorption energy of NTCDI on the
surface as shown in Fig. 5. NTCDI is adsorbed on the surface via relatively weak van der Waals forces with an adsorption energy of 1.454 eV (physisorption). Description of the van der Waals interaction, see Section Methods, is important in our DFT simulations as failure to do so may result in a significant underestimation of the molecular interactions. For instance, the calculated adsorption energy of NTCDI on the 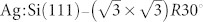 surface is found to be only 0.142 eV without accounting for the dispersion interactions.
surface is found to be only 0.142 eV without accounting for the dispersion interactions.
Figure 5.

The DFT-relaxed structure of a single NTCDI molecule adsorbed on the Ag:Si surface: (a) top and (b) side views. Only the NTCDI molecule and top layer Ag atoms of the surface are shown in (a). The black arrows labeled with A and B indicate the C-C bond at the centre of the molecule and the centre of a C5N hexagon, respectively, chosen to represent two sites that to be distinguished during imaging if submolecular resolution to be established.
As our goal here is to assess the ability of the tips to provide intramolecular resolution, two sites were considered and are marked as A and B in the figure: the C-C bond in the centre of the molecule (site A) and the centre of a hexagon (site B). If there is a clear contrast between sites A and B, then one may expect that the C-C bond which is surrounded by two hexagon sites will be visible in the image. It is assumed that the contrast in all four hexagon sites of the molecule will be very similar.
In experiment, In experiment17, clear inter- and intramolecular contrast appears only when the repulsive contribution to the tip-sample interaction causes the force to turn around beyond the minimum in the force curve. When the AFM is operated before the turnaround point, the molecule will appear as a dark diffuse feature. As the tip is approached and intramolecular features become visible, the attractive well will appear as a dark halo12,47 and the molecular skeleton as bright features. Therefore, if we are to see the C-C bond as a bright protrusion, the interaction above the bond (site A) must be less attractive than in the middle of the hexagon (site B). If, however, the force above the C-C bond is more attractive, then the C-C bond may still be visible, but as a dark feature. The larger the difference between the forces, the larger the frequency shift above the two sites, and hence the more clearly the skeleton of the molecule is expected to be visualized in AFM. The frequency shifts were simulated assuming that these are small compared with the fundamental frequency of the sensor and can be found in the Supplementary information.
The DFT-calculated force distance curves for the two sites A and B and all eight tips are given in Fig. 6. Let us first discuss the bare Si H3 and dimer tips. From the experimental point of view, strong bonding between the tip and the adsorbed molecule is undesirable as it might result in a manipulation of the molecule or reconfiguration of the tip. We find in our simulations for a single molecule adsorbed on the surface that, at very close approach, bare Si and the Ag-terminated tips strongly covalently bind to the molecule and may even pick it up upon retraction. Hence they may not be ideal for imaging and achieving intramolecular contrast when scanning a single molecule. The reactive tips need to be passivated by an atom or a molecule to avoid this happening. The situation will be different however when molecules are locked in an island (see, e.g.17) or strongly bound to a surface21. In this case it's possible that adsorbed molecules may still be scanned stably by these rather reactive tips. Although we have not carried out simulations of the force spectroscopy curves for a molecule in an island, for some of the tips considered here (CO, NTCDI, as well as bare, hydrogenated and Ag-terminated Si H3 tips) previously reported calculations17 show that qualitatively calculated forces for a single molecule may be indicative of the expected contrast for a molecule in an island if the region of very close approach is disregarded.
Figure 6. Calculated force versus distance curves for different tips approaching the C-C bond at the centre of the molecule (site A, red) and the centre of a C5N hexagon (site B, green).

Looking at Fig. 6(a), we see that the contrast of the C-C bond with respect to the hexagon centre with the bare Si H3 tip would change several times: if at large distances the C-C bonds image as darker features, at intermediate distances the contrast changes to a brighter feature, which changes again at much closer approach. In the case of the bare dimer tip, Fig. 6(b), such a change happens only once: if at large and intermediate distances the C-C bond imaged as a dark feature, at closer approach it is imaged as a brighter feature. The situation for the dimer tip is qualitatively similar to that for the Ag-terminated Si tip, Fig. 6(e), although the maximum attractive force in this case is observed at much closer approach. (Note, however, that however, that this distance cannot be measured experimentally). In all three cases the contrast changes once in the turn-around region, with the maximum attractive force being in the region of 0.2-0.35 nN. Importantly, for both bare Si tips the difference in the forces above the two sites is rather small, less than 0.03 nN, while it is only slightly larger for the Ag-terminated tip (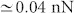 ). Therefore, the contrast in these three cases is expected to be weak. The maximum attractive forces between H passivated Si tips in H3 or dimer configurations, Fig. 6(c,d), are about 0.15 nN. The interaction above site A is initially more attractive, but changes to more repulsive than above site B in the turn-around region. The difference in forces in the latter region reaches 0.05 nN for the H passivated Si H3 tip, while it is only 0.02 nN for the passivated dimer tip, so that the expected contrast must be weak, as in the previous case. The H-down NTCDI tip, Fig. 6(f), behaves in a similar manner. In the turn-around region the turn-around region this tip images the C-C bond initially dark, but then bright at closer approach, with the contrast reaching around 0.05 nN.
). Therefore, the contrast in these three cases is expected to be weak. The maximum attractive forces between H passivated Si tips in H3 or dimer configurations, Fig. 6(c,d), are about 0.15 nN. The interaction above site A is initially more attractive, but changes to more repulsive than above site B in the turn-around region. The difference in forces in the latter region reaches 0.05 nN for the H passivated Si H3 tip, while it is only 0.02 nN for the passivated dimer tip, so that the expected contrast must be weak, as in the previous case. The H-down NTCDI tip, Fig. 6(f), behaves in a similar manner. In the turn-around region the turn-around region this tip images the C-C bond initially dark, but then bright at closer approach, with the contrast reaching around 0.05 nN.
Consider now the O-down NTCDI and CO tips. Both show qualitatively similar behaviour. Indeed, the maximum attractive forces in both cases are around 0.2 nN. Both O-down tips in the turn-around region show stronger repulsion on the C-C bond making it appear brighter than the hexagon centres. An important observation, however, concerns the magnitude of the difference in forces for these two sites. In this case, one would expect time one would expect a strong contrast as this difference reaches about 0.1 nN in both cases, i.e. a significantly larger value than for e.g. passivated tips discussed above. This means that O-down tips can produce much better intramolecular contrast of the NTCDI molecule than the other tips.
Understanding the interaction mechanism and image contrast
In order to rationalize the image contrast in AFM operated in the region where repulsive forces begin to make a strong contribution let us consider the main factors determining the interaction between the tip and the adsorbed molecule. Electron-rich areas are located above atoms or chemical bonds, and the total electron density (TED) distribution associated with the molecule would most likely represent its skeleton in the image, as these will be the regions where the Pauli repulsion is the largest. By probing the repulsive force, the TED and the skeleton of the molecule could be visualized in experiment. As we have shown in our previous study of NTCDI networks17 examination of the electron density difference (EDD) reveals that only a small proportion of the TED contributes to the repulsive interactions that underpin submolecular contrast in NC-AFM. This raises an interesting question regarding a particular tip's ability to enhance or suppress repulsive interactions.
Our results show that some tips are more efficient than others at probing the TED across a molecule. For instance, if the tip produces a positive electrostatic potential, such as the H-down NTCDI tip or Ag-terminated Si tip, there will be a strong Coulomb attractive force between the tip and the molecule adsorbed on the surface which will offset the repulsive force leading to reduced (or missing) contrast. On the other hand, using tips with negative electrostatic potential, such as e.g. the O-down tips, the repulsive forces due to Pauli repulsion are enhanced by the Coulomb repulsion in electron-rich areas leading to improved contrast. In our previous study we proposed a mechanism of spontaneous tip-termination17 with an NTCDI molecule which enabled the reported intramolecular resolution in NC-AFM. It is perhaps worth noting that despite the large number of previous studies of molecular systems in NC-AFM, our report was the first observation of “spontaneous” intramolecular resolution of organic molecules (i.e. without the need to directly functionalise the tip. The requirement of electronegative and inert tip terminations, however, offers a reasonable explanation as to why such a tip is rare, i.e. the molecule under study must contain electronegative groups otherwise the spontaneous tip termination would not be capable of intramolecular resolution. In the instances where the molecules under study possess electronegative end groups, we expect that submolecular resolution in NC-AFM through spontaneous/deliberate tip pick-up should be possible.
Discussion
In conclusion, we have investigated the efficacy of various tip structures in producing intramolecular resolution in NC-AFM experiments on planar molecules. To identify tip structures of this type we have proposed a strategy for performing in situ tip identification using the Si(111)-7×7 reconstruction as a reference surface. The essential point of our strategy is based on the availability, prior to experiment, of a theoretical database containing detailed responses of various tips of different structure and chemical composition to the reference system (revealed via spectroscopy above selected lattice sites and images at different scan heights). The database may also contain some dynamical information on tip responses, e.g. energy barriers for picking up (depositing) atoms to (from) the tips which can be used for assessing the tips stabilities at various temperatures. Based on this method we propose that spectroscopy and imaging measurements can be performed as a first stage on the reference surface and the results compared with the entries in the theoretical database. By making this comparison, detailed information regarding the structure and chemical composition of the tip apex can be inferred allowing one to “fingerprint” each tip structure. With our implementation of the “on minus off” method, measured forces on the reference system can be directly compared with the entries in the database which were also calculated using the same method and are better comparable with experiment as they represent relative forces. We propose that this tip identification process can be done on the fly immediately prior to further experiments. Therefore, as a second stage the now-characterised tip can be used to perform measurements on the system of interest, which in our example is the submolecular study of planar NTCDI molecules. The second surface is suggested to be prepared on the same sample next to the reference one, seperated only by a few nanometres to preserve the AFM tip and easily recharacterise its structure. We demonstrate that such a sample can be routinely prepared containing adjacent regions of both Si(111)-7×7 and 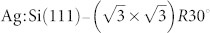 surfaces. Moreover, there is exciting potential for coupling the tip fingerprinting strategy we describe here with the automated probe optimisation protocols introduced by Woolley et al.48.
surfaces. Moreover, there is exciting potential for coupling the tip fingerprinting strategy we describe here with the automated probe optimisation protocols introduced by Woolley et al.48.
To demonstrate how this strategy may work, we have performed an “in silico” study, in which both stages of the proposed procedure are performed in a way that would mimic the actual experiment. Here the Si(111)-7×7 surface is considered as a reference system for tip fingerprinting, while the 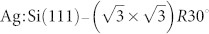 surface with a single NTCDI molecule adsorbed on it is considered as an example of the system of interest. Eight tip models have been considered ranging from reactive to nonreactive tips, and for each of them force spectroscopy calculations have been performed above eleven lattice sites on the Si(111)-7×7 surface including a H-passivated adatom site. The calculated spectroscopy curves form example entries to the database; with time many other tips may be simulated and added to it eventually enabling one to achieve precise characterization of the tip in the actual experiment. The chosen spectra positions on the Si(111)-7×7 surface represent chemically distinct sites allowing full characterisation of the tip. In particular, the force curves taken over the half-occupied adatom site, the fully occupied rest atom site and the passivated hydrogen terminated adatom reveal a significant amount of information regarding tips chemical structure. The chemical reactivity of the tip could be identified by checking the maximum attractive forces. We find that any value larger than 1 nN above the adatoms indicates the formation of a covalent bond between tip and surface. In the case that rest atom spectra also exceeds 1 nN the tip can be identified as reactive and unsuitable for intramolecular resolution in NC-AFM. In contrast, chemically inert tips are calculated to interact more weakly with these two sites, producing a maximum attractive force of no more than 0.3 nN. Finally, in the case of tips that show both strong attraction over adatom sites but weak attraction over rest atom sites, the suitability for submolecular NC-AFM experiments can be identified via a weak interaction above a hydrogen passivated site (i.e. <0.1 nN).
surface with a single NTCDI molecule adsorbed on it is considered as an example of the system of interest. Eight tip models have been considered ranging from reactive to nonreactive tips, and for each of them force spectroscopy calculations have been performed above eleven lattice sites on the Si(111)-7×7 surface including a H-passivated adatom site. The calculated spectroscopy curves form example entries to the database; with time many other tips may be simulated and added to it eventually enabling one to achieve precise characterization of the tip in the actual experiment. The chosen spectra positions on the Si(111)-7×7 surface represent chemically distinct sites allowing full characterisation of the tip. In particular, the force curves taken over the half-occupied adatom site, the fully occupied rest atom site and the passivated hydrogen terminated adatom reveal a significant amount of information regarding tips chemical structure. The chemical reactivity of the tip could be identified by checking the maximum attractive forces. We find that any value larger than 1 nN above the adatoms indicates the formation of a covalent bond between tip and surface. In the case that rest atom spectra also exceeds 1 nN the tip can be identified as reactive and unsuitable for intramolecular resolution in NC-AFM. In contrast, chemically inert tips are calculated to interact more weakly with these two sites, producing a maximum attractive force of no more than 0.3 nN. Finally, in the case of tips that show both strong attraction over adatom sites but weak attraction over rest atom sites, the suitability for submolecular NC-AFM experiments can be identified via a weak interaction above a hydrogen passivated site (i.e. <0.1 nN).
Moving now to the question of the ability of different tip apices to provide intramolecular resolution to provide intramolecular resolution, it is shown that negatively charged tips produce the clearest intramolecular contrast in NC-AFM when operated in the Pauli repulsive regime, followed by hydrogen-terminated silicon clusters. At the same time, positively charged tips or the Ag-terminated Si tip considered in our study are much less capable of imaging the skeleton of the molecule. We propose that AFM tip terminations with negative electrostatic potentials, such as e.g. the O-down NTCDI and CO tips, enhance the repulsive forces due to Coulomb repulsion in electron-rich areas leading to slightly improved contrast.
The tip identification and control (TIC) protocol we have described has the potential to address arguably the key challenge facing NC-AFM microscopists: characterisation, generation, and recovery of a specific, well-defined probe apex. The easily prepared Si(111)-7×7 surface contains a number of well recognisable sites and can serve as one possible example of a reference surface that has the potential to distinguish many classes of different tips. But it is not the only one: one may try other surfaces containing well distinguished lattice sites as well. It is however essential that it is possible to prepare the reference surface in proximity to the surface of interest on the same sample. The contiguous chemically inert Ag:Si surface used in this study is an ideal substrate for studying molecular systems in a physisorbed environment, which enables the quantitative study of weak intermolecular interactions.
We hope that this study will stimulate additional calculations in order to extend and create databases for the fingerprinting of NC-AFM tip apices.
Methods
We used the CP2K Quickstep package49,50 employing a hybrid Gaussian and plane-wave method. A double-zeta Gaussian molecularly optimized basis set plus polarization (DZVP-MOLOPT)51 was used. The Goedecker-Teter-Hutter (GTH) pseudopotentials and the Perde-Burke-Ernzerhof (PBE) exchange-correlation density functional, together with a 300 Ry plane-wave energy cutoff, were used. And the van der Waals forces were taken into account within the Grimme D3 method52. The force convergence criterion used for geometry relaxations was 0.005 eV/Å.
Although we did use a rather extended localised basis set in our calculations, the counterpoise correction53 was used for a number of systems to test the importance of the basis set superposition error (BSSE). As discussed in the Supporting Information, these calculations indicated that this correction is in all cases relatively small and hence neglecting it cannot change qualitative conclusions of our study.
The Si(111)-7×7 and 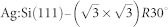 unit cells were constructed using three bilayers plus the surface adatoms, with a vacuum gap (the distance between the adlayer and the bottom surface of the next slab) of 25 Å. The bottom bilayer, whose dangling bonds were terminated with hydrogen atoms, was frozen during geometry optimizations in both cases.
unit cells were constructed using three bilayers plus the surface adatoms, with a vacuum gap (the distance between the adlayer and the bottom surface of the next slab) of 25 Å. The bottom bilayer, whose dangling bonds were terminated with hydrogen atoms, was frozen during geometry optimizations in both cases.
To calculate the force-distance curves for a given tip, we started with the tip and surface at a certain distance from each other, and then the tip approached the surface system in a quasi-static manner with equal steps of 0.1 Å. The tip displacement in the simulations is measured with respect to the fixed layers in the tip and surface, and thus is consistent with the piezodisplacement in AFM. The calculated total energies were then fitted by a sum of inverse powers up to 6-th order. Subsequently, the tip force F(z) between the nanometer-sized tip at the end of the cantilever and the sample were obtained by differentiating the energy U(z) curve.
To avoid the problem of considering the (unknown) long-range interaction due to a macroscopic part of the tip, the corresponding attractive background force was subtracted in all cases using an “on minus off” method commonly used in experiments7,9. We used the “off” spectra taken over the corner holes of the Si(111)-7×7 surface, which correspond to the weakest short range force. The calculated spectrum is subtracted from the spectra calculated over other lattice sites so that reliable short range force curves may be obtained in the case of each tip. The force curves, F(z), obtained in this way are better suited for characterising the response of every tip apex without the uncontrolled contribution from the (unknown) macroscopic part of the tip.
Two imaging modes in FM-AFM, topographic and constant height, are available. Since it is known11 that intramolecular contrast can be obtained in the constant height mode when the force gradient becomes negative, i.e. when the repulsive contribution to the tip-sample interaction causes the force to turn around beyond the minimum in the force curve, the constant height mode is employed in our analysis.
Drawings of the electronic density and electrostatic potential were produced using VESTA54.
Author Contributions
P.M., J.W., Y.W. and L.K. conceived the project. H.S., S.J. and Z.Z. performed the DFT calculations. P.S. provided the experimental images. H.S., S.J., P.M., J.W., Y.W. and L.K. analyzed the results of the DFT calculations and prepared the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
H.S., Z.Z., J.W. and Y.W. acknowledge the financial support from the 973 Program (2011CB933300), the National Natural Science Foundation of China (51271134, 11328403 and J1210061), the Fundamental Research Funds for the Central Universities, and the CERS-1-26 (CERS-China Equipment and Education Resources System). Via our membership of the UK's HPC Materials Chemistry Consortium, this work made use of the facilities of ARCHER, the UK's national highperformance computing service, which is funded by the Office of Science and Technology through EPSRC's High End Computing Programme, grant number EP/L000202. S.P.J. thanks the Engineering and Physical Sciences Research Council (EPSRC) for Grant No. EP/J500483/1. We are also grateful for access to the University of Nottingham High Performance Computing Facility. P.M. thanks EPSRC for the award of a Leadership Fellowship (EP/G007837/1). We also acknowledge funding from the European Comission's ICT-FET programme via the Atomic Scale and Single Molecule Logic gate Technologies (AtMol) project, Contract No. 270028.
References
- Repp J., Meyer G., Stojković S. M., Gourdon A. & Joachim C. Molecules on Insulating Films: Scanning-Tunneling Microscopy Imaging of Individual Molecular Orbitals. Phys. Rev. Lett. 94, 026803 (2005). [DOI] [PubMed] [Google Scholar]
- Liljeroth P., Repp J. & Meyer G. Current-Induced Hydrogen Tautomerization and Conductance Switching of Naphthalocyanine Molecules. Science 317, 1203–1206 (2007). [DOI] [PubMed] [Google Scholar]
- Morita S., Wiesendanger R. & Meyer E. Noncontact Atomic Force Microscopy, Vol. 1. (Springer, 2002). [Google Scholar]
- Albrecht T. R. et al. Frequency modulation detection using high Q cantilevers for enhanced force microscope sensitivity. J. Appl. Phys. 69, 668–673 (1991). [Google Scholar]
- Sugimoto Y. et al. Chemical identification of individual surface atoms by atomic force microscopy. Nature 446, 64–67 (2007). [DOI] [PubMed] [Google Scholar]
- Sun Z., Boneschanscher M. P., Swart I., Vanmaekelbergh D. & Liljeroth P. Quantitative Atomic Force Microscopy with Carbon Monoxide Terminated Tips. Phys. Rev. Lett. 106, 046104 (2011). [DOI] [PubMed] [Google Scholar]
- Chiutu C. et al. Precise Orientation of a Single C60 Molecule on the Tip of a Scanning Probe Microscope. Phys. Rev. Lett. 108, 268302 (2012). [DOI] [PubMed] [Google Scholar]
- Ternes M., Lutz C. P., Hirjibehedin C. F., Giessibl F. J. & Heinrich A. J. The force needed to move an atom on a surface. Science 319, 1066–1069 (2008). [DOI] [PubMed] [Google Scholar]
- Lantz M. A. et al. Quantitative Measurement of Short-Range Chemical Bonding Forces. Science 291, 2580–2583 (2001). [DOI] [PubMed] [Google Scholar]
- Ternes M. et al. Interplay of Conductance, Force, and Structural Change in Metallic Point Contacts. Phys. Rev. Lett. 106, 016802 (2011). [DOI] [PubMed] [Google Scholar]
- Gross L., Mohn F., Moll N., Liljeroth P. & Meyer G. The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009). [DOI] [PubMed] [Google Scholar]
- Gross L. L. et al. Bond-order discrimination by atomic force microscopy. Science 337, 1326–1329 (2012). [DOI] [PubMed] [Google Scholar]
- Mohn F., Gross L., Moll N. & Meyer G. Imaging the charge distribution within a single molecule. Nat. Nanotechnol. 7, 227–31 (2012). [DOI] [PubMed] [Google Scholar]
- Pavliček N. et al. Atomic Force Microscopy Reveals Bistable Configurations of Dibenzo[a,h]thianthrene and their Interconversion Pathway. Phys. Rev. Lett. 108, 086101 (2012). [DOI] [PubMed] [Google Scholar]
- de Oteyza D. G. et al. Direct Imaging of Covalent Bond Structure in Single-Molecule Chemical Reactions. Science 340, 1434–1437 (2013). [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. Real-space identification of intermolecular bonding with atomic force microscopy. Science 342, 611–614 (2013). [DOI] [PubMed] [Google Scholar]
- Sweetman A. M. et al. Mapping the force-field of a hydrogen-bonded assembly. Nat. Commun. 5, 3931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty P. Atom-technology and beyond: manipulating matter using scanning probes. Nanoscience 1, 116–144 (2013). [Google Scholar]
- Mohn F., Schuler B., Gross L. & Meyer G. Different tips for high-resolution atomic force microscopy and scanning tunneling microscopy of single molecules. Appl. Phys. Lett. 102, 073109 (2013). [Google Scholar]
- Boneschanscher M. P. et al. Quantitative atomic resolution force imaging on epitaxial graphene with reactive and nonreactive AFM probes. ACS Nano 6, 10216–10221 (2012). [DOI] [PubMed] [Google Scholar]
- Sweetman A. M. et al. Intramolecular bonds resolved on a semiconductor surface. Phys. Rev. B (accepted). [Google Scholar]
- Martsinovich N. & Kantorovich L. Theoretical modelling of tip effects in the pushing manipulation of C 60 on the Si(001) surface. Nanotechnology 19, 235702 (2008). [DOI] [PubMed] [Google Scholar]
- Pou P. et al. Structure and stability of semiconductor tip apexes for atomic force microscopy. Nanotechnology 20, 264015 (2009). [DOI] [PubMed] [Google Scholar]
- Yurtsever A. et al. Force mapping on a partially H-covered Si(111)-(7×7) surface: Influence of tip and surface reactivity. Phys. Rev. B 87, 155403 (2013). [Google Scholar]
- Hoffmann R., Kantorovich L. N., Baratoff A., Hug H. J. & Guntherodt H. J. Sublattice identification in scanning force microscopy on alkali halide surfaces. Phys. Rev. Lett. 92, 146103 (2004). [DOI] [PubMed] [Google Scholar]
- Bamidele J. et al. Chemical tip fingerprinting in scanning probe microscopy of an oxidized Cu (110) surface. Phys. Rev. B 86, 155422 (2012). [Google Scholar]
- Bechstein R. et al. ‘All-inclusive' imaging of the rutile TiO2(110) surface using NC-AFM. Nanotechnology 20, 505703 (2009). [DOI] [PubMed] [Google Scholar]
- Trevethan T., Watkins M. & Shluger A. L. Models of the interaction of metal tips with insulating surfaces. Beilstein J. Nanotechnol. 3, 329–335 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis S., Sweetman A., Bamidele J., Kantorovich L. & Moriarty P. Role of orbital overlap in atomic manipulation. Phys. Rev. B 85, 235305 (2012). [Google Scholar]
- Jarvis S. P., Kantorovich L. & Moriarty P. Structural development and energy dissipation in simulated silicon apices. Beilstein J. Nanotechnol. 4, 941–948 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S. & Fujihira M. Differentiation of molecules in a mixed self-assembled monolayer of h- and cl-terminated bicyclo[2.2.2]octane derivatives. Nanotechnology 17, S112 (2006). [DOI] [PubMed] [Google Scholar]
- Naydenov B., Ryan P., Teague L. C. & Boland J. J. Measuring the force of interaction between a metallic probe and a single molecule. Phys. Rev. Lett. 97, 098304 (2006). [DOI] [PubMed] [Google Scholar]
- Welker J., Weymouth A. J. & Giessibl F. J. The influence of chemical bonding configuration on atomic identification by force spectroscopy. ACS Nano 7, 7377–7382 (2013). [DOI] [PubMed] [Google Scholar]
- Hofmann T., Pielmeier F. & Giessibl F. J. Chemical and crystallographic characterization of the tip apex in scanning probe microscopy. Phys. Rev. Lett. 112, 066101 (2014). [DOI] [PubMed] [Google Scholar]
- Giessibl F. J., Hembacher S., Bielefeldt H. & Mannhart J. Subatomic Features on the Silicon (111)-(7×7) Surface Observed by Atomic Force Microscopy. Science 289, 422–425 (2000). [DOI] [PubMed] [Google Scholar]
- Keeling D. et al. Assembly and Processing of Hydrogen Bond Induced Supramolecular Nanostructures. Nano Lett. 3, 9–12 (2003). [Google Scholar]
- Perdigão L. et al. Coadsorbed NTCDI-melamine mixed phases on Ag-Si(111). Phys. Rev. B 76, 245402 (2007). [Google Scholar]
-
Sweetman A.
et al.
Simultaneous noncontact AFM and STM of
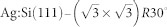 . Phys. Rev. B
87, 075310 (2013). [Google Scholar]
. Phys. Rev. B
87, 075310 (2013). [Google Scholar] - Bacalzo F. T., Musaev D. G. & Lin M. C. Theoretical Studies of CO Adsorption on Si(100)-2×1 Surface. J. Phys. Chem. B 102, 2221–2225 (1998). [Google Scholar]
- Gajdo M., Eichler A. & Hafner J. CO adsorption on close-packed transition and noble metal surfaces: trends from ab initiocalculations. J. Phys.-Condes. Matter 16, 1141–1164 (2004). [Google Scholar]
- Tao F. & Xu G. Q. Attachment Chemistry of Organic Molecules on Si(111)-7×7. Accounts Chem. Res. 37, 882–893 (2004). [DOI] [PubMed] [Google Scholar]
- Northrup J. E. Origin of Surface States on Si(111)(7×7). Phys. Rev. Lett. 57, 154 (1986). [DOI] [PubMed] [Google Scholar]
- Lantz M. A. et al. Short-range electrostatic interactions in atomic-resolution scanning force microscopy on the Si(111)-7×7 surface. Phys. Rev. B 68, 035324 (2003). [Google Scholar]
- Soler J. M. et al. The SIESTA method for ab initio order-N materials simulation. J. Phys.-Condes. Matter 14, 2745–2779 (2002). [Google Scholar]
- Hapala P. Kichin G. Wagner C. Tautz F. S. Temirov R. & Jelinek P. Mechanism of high-resolution STMAFM imaging with functionalized tips. Phys. Rev. B 90, 085421 (2014). [DOI] [PubMed] [Google Scholar]
- Sweetman A., Rahe P. & Moriarty P. Unique determination of “subatomic” contrast by imaging covalent backbonding. Nano Letter (2014). [DOI] [PubMed] [Google Scholar]
- Guo C.-S., Van Hove M. A., Zhang R.-Q. & Minot C. Prospects for Resolving Chemical Structure by Atomic Force Microscopy: A First-Principles Study. Langmuir 26, 16271–16277 (2010). [DOI] [PubMed] [Google Scholar]
- Woolley R. A. J., Stirling J., Radocea A., Ktasnogor N. & P M. Automated probe microscopy via evolutionary optimization at the atomic scale. Appl. Phys. Lett. 98, 253104 (2011). [Google Scholar]
- Hutter J., Iannuzzi M., Schiffmann F. & VandeVondele J. CP2K:atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 4, 15–25 (2013). [Google Scholar]
- VandeVondele J. et al. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 167, 103–128 (2004). [Google Scholar]
- VandeVondele J. & Hutter J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007). [DOI] [PubMed] [Google Scholar]
- Grimme S., Antony J., Ehrlich S. & Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- Boys S. F. & Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566 (1970). [Google Scholar]
- Momma K. & Izumi F. VESTA for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information