

Abstract
Fibrodysplasia ossificans progressiva formerly known as Myositis ossificans progressiva is a rare hereditary mesodermal disorder. It is characterized by congenital skeletal anomalies and progressive ectopic bone formation in connective tissue, resulting in mature ossification within soft tissues and bridging between osseous structures. It is extremely rare and has an incidence of one in two million people. Usually, it has typical clinical and radiographic features. Here, we present a case of a young patient diagnosed to have an advanced fibrodysplasia ossificans progressiva. Plain radiographs provide characteristic findings, and radiologists may play a major role in diagnosing and preventing invasive procedures or further traumatic insults to the affected patient. Though rare, diagnosis of fibrodysplasia ossificans progressiva should be considered whenever characteristic radiographic features of multifocal heterotopic bone formation is seen along with the valgus deformities of the big toes. Being a rare condition, treatment guidelines are not clear and this condition needs further research.
Keywords
Myositis ossificans progressiva; Fibrodysplasia ossificans progressiva; Heterotopic ossification; Myositis ossificans.
Introduction
Myositis ossificans progressiva or fibrodysplasia ossificans progressiva (FOP) is a rare connective tissue disease characterized by widespread, progressive, ectopic ossification of soft tissues (striated muscles, tendons, fasciae, ligaments and subcutaneous tissues).1 It is an autosomal dominant inherited disease. However, most of the cases are sporadic with one person affected in a family.2 Characteristic feature of the disease during childhood is the deformity of the big toes and thumbs.2 Classically, the disease is also characterized by heterotopic ossification of soft tissues, which is usually complicated by restriction of movements at the affected sites. It usually starts and progresses in a cranio-caudal dorsal-ventral and proximo-distal manner.3 However, flares of the disease might occur at sites of trauma or injury.2 The tongue, smooth muscles, diaphragmatic muscles and cardiac muscles are fortunately spared.4 It is estimated that the incidence of the disease is 1 per 2 million people and the prevalence is 2500 cases worldwide.3 Plain radiographs provide characteristic findings and radiologists may play a major role in diagnosing the disease and avoiding further invasive tests.
We report a case of a young male who was diagnosed during childhood and experienced the classical features of the disease.
Case Report
The patient was a 21-year-old male who was referred to radiology department with progressive inability to open his jaw since 2 months. On further questioning, he was noted by his parents to have deformities and shortening of the great toes since childhood. He was operated at the age of 10 for excision of a bony outgrowth in the left second toe. The patient's past medical history revealed multiple episodes of spontaneous swellings at multiple sites, including chin, thyroid region, back and lower limbs. At the age of 20, right femoral extension osteotomy and supracondylar osteotomy were done in a different hospital. He was able to mobilize with a single elbow crutch. He improved temporarily after the surgery. However, there was progressive restriction of movements of the hips, neck, left upper limb and lumbar spine with noticeable deformities. There was no familial relationship between the parents and none of his family members had a similar problem. On examination, the patient was noted to have multiple non-tender firm subcutaneous nodules in the chin, neck, and back with scoliosis of the lumbar spine. Both ankles showed equines deformity and the great toes were short with valgus deformity (Fig. 1a). The movements at the following areas: the mandible, neck, lumbar spine, left arm and both hips were restricted.
Figure 1.

Clinical photograph (1a) and radiograph of both feet AP views (1b) shows bilateral hallux valgus deformity with short great toes. Deformed short proximal phalanx of great toe with absent distal phalanx is seen on radiograph.
CT of the maxillofacial bones and a skeletal survey were performed. Maxillofacial CT revealed normal temporomandibular joints. However, posterior elements of the cervical vertebrae were expanded and the facet joints of C2-C7 vertebrae were fused (Fig. 2b). Lateral view radiograph of the cervical spine (Fig. 2a) showed complete fusion of the posterior column of cervical vertebrae, as well as calcification of the anterior and posterior vertebral ligaments with straightening of the cervical spine.
Figure 2.

Lateral cervical spine radiograph and 3D CT of cervical spine shows expansion of posterior elements with ossification and bony fusion.
Lumbosacral spine radiographs showed severe scoliosis with heterotopic ossified pars along the soft tissues dorsally (Fig. 3a). Radiograph of the pelvis showed ossified bridges around the hip joints, extending from the ischium to the greater and lesser trochanters and to the proximal shaft of femur (Fig. 3b). An ossified bridge was also seen in the left humerus (Fig. 3c). Exostoses around the right knee joint and right elbow were also seen. Radiographs of the feet showed the characteristic finding of this disease; shortening of the first metatarsal and absence of a phalanx with valgus deformity of the great toes (Fig. 1b)
Figure 3.
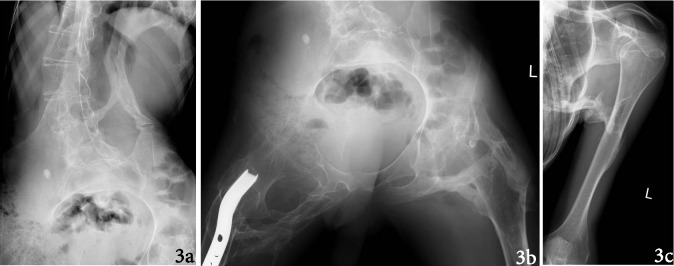
AP view of lumbar spine, pelvis and left shoulder shows mature multifocal heterotopic ossification and bridging between osseous structures between ribs and humerus as well as between lumbar spine and pelvis.
Discussion
Myositis ossificans progressiva is a rare autosomal dominant disease, inherited as a trait with variable expression and complete penetrance. It is clinically characterized by two main features, anomalies of the great toes and thumbs and progressive ectopic ossification of soft tissues causing limitation of movements. A study for identifying the gene responsible for the disease concluded that there is a specific linkage to the 2q23-24 region of chromosome 2 for the classical type of the disease.5 However, different studies suggested different genes responsible for the disease. Examples of these genes include 4q27-31 and 17q21-22.2,6 The heterotopic ossification in soft tissues was examined and found to be of normal bone by all means.7 The pathogenesis of ectopic bone formation is unknown, but overproduction on bone morphogenetic protein-4 in lesion cells and lymphocytic cells is reported as one of the factors.6
Although the average onset age of the disease is about 5 years,6 the affected child might be noticed after birth to have short (macrodactylia with hypoplasia or synostosis of the phalanges) and deformed great toes (Valgus deformity, Fig. 1) which occurs in most of the cases of myositis ossificans progressiva. This was noticed by the parents of the patient during childhood. From birth until adolescence, the patient experiences spontaneous hard soft tissue swellings, which are usually painful.2 These symptoms are usually the initial presentation of the patient and there might be flare-ups after trauma. Some cases present intensely with acute torticollis and painful mass in the sternocleidomastoid.6 In most cases, it starts at the neck progressing downwards to affect the thoracic and lumbar regions, and then the limbs.2 Sternocleidomastoid is often the initial site of involvement, progresses to shoulder girdle, upper arms, spine and pelvis. End result is bridging between extremities and torso, between ribs and between thorax and pelvis with severe restriction of motion.6 Eventually, the affected patients develop bridging between extremities and torso, between ribs and between thorax and pelvis with severe restriction of motion.3 As it was the case in our patient, scoliosis is the end result of this heterotopic ossification that occurs more on one side.2 Even though, the lungs are not usually affected directly by the disease, recurrent infections and atelectasis usually occur due to restriction of chest expansion secondary to disease progression.8 Affected patients usually become dependent and confined to wheel-chair or bed at the second decade of their life as a result of ankylosing of all major joints of both axial and appendicular skeleton.2 However, many patients survived till the age of 60 and beyond.9 Many patients were reported to have conductive hearing loss due to fusion of the ear ossicles.2 The prognosis is usually worse if temporomandibular joints are affected. The restriction of jaw movement limits the ability of the patient to eat and sometimes patients can only take fluids orally.10,11
Myositis ossificans progressiva is generally a clinical diagnosis that is based on the presence of congenital anomalies of the great toes, progressive heterotopic ossification and the classical pattern of disease progression. Biopsies are not recommended for the diagnosis because they might worsen the ossification at the site, and diagnosis might be confused with bone malignancies like fibrosarcoma.11
Plain radiographs and computed tomography may play a role in confirming the diagnosis and evaluating its extent. In the plain films characteristic findings might be seen, including deformed shortened great toes with or without absence of a phalanx, ectopic calcifications within soft tissues, shortened broad femoral necks pseudo-exostoses and prominent calcaneal spur. Osseous bridging between the axial and appendicular skeleton might be seen in advanced stages. The lesions can be detected even earlier by bone scan using Gallium 67 citrate and Tc-99 diphosphonate agents rather than plain radiographs.10
In addition to providing accurate anatomical location of the lesions, Computed Tomography (CT) scan can also provide valuable information about the extent of the disease in the preosseous stages, which may manifest as swelling and edema of the muscular fascia planes and muscular bundles.6
MRI has a role in making the diagnosis in the early stage (preosseous stage). The preosseous lesions usually have low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. Adjacent muscle bundles usually show high signal intensity on T2-weighted images probably as secondary changes.6
Many approaches have been tried to treat this disease. There was no one specific approach which showed promising results.2,10 One of the most effective approaches is the prevention of trauma by all means, because trauma is considered as a cause of flare-ups. Falls and different injuries should be avoided. Even intramuscular vaccines should not be given to the patients.2 Many trials of different medications were done, and to date no evidence of their effectiveness.2 Corticosteroids, non-steroidal anti-inflammatory drugs, leukotrienes and Cox-2 inhibitors were tried.2,10 Some studies suggested the use of high doses of bisphosphonate (Etidronate) which showed good results due to its potential effect on inhibiting mineralization of the newly formed cartilage. However, one should consider also its side effects when given in high doses as its action will include the unaffected parts of the skeleton resulting in osteomalacia. Another drug from the aminobisphosphonate group, called Pamidronate, was thought to have promising results as it is more potent compared to Etidronate. The main role of bisphosphonates and corticosteroids is in the treatment of acute flare-ups.2,12 However, disease progression remains unchanged.
Conclusion
Myositis ossificans progressiva is a rare congenital disease of progressive ectopic ossification of soft tissues. Physicians should be able to diagnose this disease in its early stages in order to prevent its disabling progression. It usually presents in a classical pattern and it has characteristic radiological findings in plain films and CT scan. The early stages can be detected using bone scan or MRI. The best treatment is still prevention of trauma with possible role of bisphosphonates and corticosteroids in the acute flare-ups.
Acknowledgements
The authors reported no conflict of interest and no funding was received on this work.
References
- 1.Nucci A, Queiroz LS, Santos AO, Camargo EE, Ribeiro MVLM. Fibrodysplasia ossificans progressiva. Arq. Neuro-Psiquiatr. São Paulo June 2000;58:2A. [DOI] [PubMed]
- 2.Gonçalves AL, Masruha MR, de Campos CC, Delai PL, Vilanova LC. Fibrodysplasia ossificans progressiva: case report. Arq Neuropsiquiatr 2005. Dec;63(4):1090-1093. 10.1590/S0004-282X2005000600032 [DOI] [PubMed] [Google Scholar]
- 3.Khan SA, Zahid M, Asif N, Gogi N. Unusual presentations in myositis ossificans progressiva. A case report. Acta Orthop Belg 2001. Feb;67(1):86-89. [PubMed] [Google Scholar]
- 4.Norman A. Myositis Ossificans & Fibrodysplasia ossificans Progressiva. Chapter 132. In: Taveras J, Ferrucci. JT Radiology (Diagnosis, Imaging -Intervention).Philadelphia: Lippincott. Raven Publishers 1996;5:6-9. [Google Scholar]
- 5.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006. May;38(5):525-527. 10.1038/ng1783 [DOI] [PubMed] [Google Scholar]
- 6.Hagiwara H, Aida N, Machida J, Fujita K, Okuzumi S, Nishimura G. Contrast-enhanced MRI of an early preosseous lesion of fibrodysplasia ossificans progressiva in a 21-month-old boy. AJR Am J Roentgenol 2003. Oct;181(4):1145-1147. 10.2214/ajr.181.4.1811145 [DOI] [PubMed] [Google Scholar]
- 7.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res 1994. Jul;(304):238-247. [PubMed] [Google Scholar]
- 8.Kussmaul WG, Esmail AN, Sagar Y, Ross J, Gregory S, Kaplan FS. Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Relat Res 1998. Jan;(346):104-109. [PubMed] [Google Scholar]
- 9.lllingworth AS. Myositis ossificans progressiva (Munchmeyer’s disease). Arch Dis Child 1971;46:264-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rokni Yazdi H, Rahmani M. Fibrodysplasia ossificans progressiva: Report of a case. Iran J Radiol 2003. Dec;1:97-100 [Google Scholar]
- 11.Thickman D, Bonakdar-pour A, Clancy M, Van Orden J, Steel H. Fibrodysplasia ossificans progressiva. AJR Am J Roentgenol 1982. Nov;139(5):935-941. 10.2214/ajr.139.5.935 [DOI] [PubMed] [Google Scholar]
- 12.Frederick S. Kaplan, Eileen M. Shore, David L. Glaser, Stephen Emerson. The medical management of fibrodysplasia Ossificans Progressiva current treatment considerations. Clin Proc Intl Clin Consort FOP 2011;4:1-100 [Google Scholar]