

Abstract
We describe the metagenomics-derived feline enteric virome in the faeces of 25 cats from a single shelter in California. More than 90 % of the recognizable viral reads were related to mammalian viruses and the rest to bacterial viruses. Eight viral families were detected: Astroviridae, Coronaviridae, Parvoviridae, Circoviridae, Herpesviridae, Anelloviridae, Caliciviridae and Picobirnaviridae. Six previously known viruses were also identified: feline coronavirus type 1, felid herpes 1, feline calicivirus, feline norovirus, feline panleukopenia virus and picobirnavirus. Novel species of astroviruses and bocaviruses, and the first genome of a cyclovirus in a feline were characterized. The RNA-dependent RNA polymerase region from four highly divergent partial viral genomes in the order Picornavirales were sequenced. The detection of such a diverse collection of viruses shed within a single shelter suggested that such animals experience robust viral exposures. This study increases our understanding of the viral diversity in cats, facilitating future evaluation of their pathogenic and zoonotic potentials.
Introduction
Zoonotic diseases have been emerging worldwide, with increasing frequency (Jones et al., 2008). Cats share habitats with humans, domesticated animals and wildlife, and can transmit several viruses which are capable of infecting humans, including rabies virus (Gunn-Moore & Reed, 2011), pandemic H1N1 influenza virus (Sponseller et al., 2010) and cowpox virus (Schulze et al., 2007). Some viruses that infect domestic cats are genetically close to viruses from humans, such as feline astrovirus strain 1637F (Lau et al., 2013) and Felis domesticus papillomavirus type 1 (Tachezy et al., 2002). A feline rotavirus strain cat2 was shown recently to originate from multiple reassortment events involving canine, feline, human and bovine rotaviruses (Tsugawa & Hoshino, 2008).
Cats in rescue shelters are temporarily kept awaiting rescue by a legal owner or a new guardian. In most shelters, there is high animal density, a continuous turnover of animals, and mixing and relocation of animals. This causes a high level of stress in sheltered animals, so that circulating pathogens have the opportunity to thrive in immunocompromised host where opportunities for treatment are often limited. Stress also contributes to higher levels of viral shedding (Pesavento & Murphy, 2014; Pusterla et al., 2009), increasing exposure to other cats in the shelter.
In this study, next-generation sequencing was used to develop a more comprehensive view of the viruses shed in the faeces of sheltered cats. We describe complete or partial viral genomes, including those of astroviruses, bocaviruses, cycloviruses and other viral species. This study increases the number of known feline viruses, and provides candidate genomes for future studies of their transmission, epidemiology and association with diseases.
Results
Virome of cat faeces
An Illumina MiSeq library was constructed using random reverse transcription (RT)-PCR with viral nucleic acids enriched from 25 faecal samples from cats in a shelter that generated 15 852 036 sequence reads (see Methods). Sequence reads were de novo assembled, and contigs and singletons were compared to GenBank’s viral database using blastx with an E value cut-off 10−5. A total of 319 526 sequence reads showed detectable similarity to viral sequences, of which 91.32 % matched eukaryotic viral sequences and the others matched bacteriophage sequences. The majority of bacteriophage sequences in cat faeces belonged to the dsDNA order Caudovirales and to the ssDNA family Microviridae, which was similar to previous studies on the faeces of horses, humans, California sea lions, pine martens and European badgers (Breitbart et al., 2003; Cann et al., 2005; Li et al., 2011a; van den Brand et al., 2012). As the 25 faecal specimens were analysed in six pools of four to five specimens, the viruses identified are reported per pool rather than for individual specimens (Fig. 1). The most highly represented mammalian viruses were, in order of sequence read abundance, mamastroviruses (63.7 % of all viral reads) from the family Astroviridae, coronaviruses (8.05 % of all viral reads) from the family Coronaviridae and bocaviruses (2.6 % of all viral reads) from the family Parvoviridae. We detected known viruses (with very high sequence similarities to genomes in GenBank) belonging to six viral families known to infect mammals (Astroviridae, Coronaviridae, Parvoviridae, Herpesviridae, Caliciviridae and Picobirnaviridae). Viruses with lower levels of similarity to known genomes belonged to four viral families (Astroviridae, Parvoviridae, Circoviridae and Anelloviridae). These more divergent genomes were further characterized.
Fig. 1.
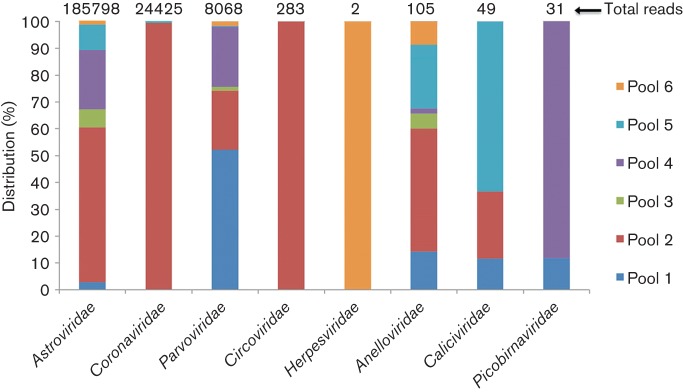
Distribution of sequence reads to different viral families in six cat faecal pools.
Feline astroviruses
The family Astroviridae consists of positive ssRNA viruses whose genomes range in length from 6.4 to 7.3 kb and contain three ORFs, designated ORF1a (non-structural proteins), ORF1b [RNA-dependent RNA polymerase (RdRp)] and ORF2 [capsid (Cap)]. Astroviruses can cause gastroenteritis in mammalian and avian species, and have been identified in numerous mammalian species (De Benedictis et al., 2011). Astroviruses have also been associated recently with encephalitis in a child with agammaglobulinemia (Quan et al., 2010), with a shaking syndrome in farmed minks (Blomström et al., 2010) and with neurological disease in cattle (Li et al., 2013).
Astroviruses were present in all faecal sample pools. Sequence assembly within each pool generated near-complete genome sequences of three different astrovirus strains including complete coding sequences of structural and non-structural proteins, but missing the 5′-most extremity of the genomes. The nearly complete genomes of the feline astroviruses FastV-D1 (GenBank accession number KM017741), FastV-D2 (GenBank accession number KM017742) and FastV-D3 (GenBank accession number KM017743) were 6598, 6813 and 6748 bases, respectively [excluding their 3′ poly(A) tails acquired by 3′ RACE], each encoding three ORFs (Fig. 2a). Despite overall genomic similarities to other astroviruses described to date, slight differences were identified in FastV-D1. The RdRp of astroviruses is typically translated following ribosome frameshifting at the boundary of the 1a and 1b ORFs due to a ‘slippery’ AAAAAC sequence resulting in a readthrough of the 1a step codon. Atypically, the ORF1b encodes a methionine codon near its N terminus only found in the genome of one other astrovirus strain (GenBank accession number KC692365) from a fox (Bodewes et al., 2013). That methionine codon therefore has the potential to act as an internal translation initiator codon, although the usual frameshift mechanism is likely to be operational.
Fig. 2.

(a) Genomic organization of feline astroviruses (FAstV-D1, -D2 and -D3) (distance in nt). (b, c) Neighbour-joining phylogenetic analysis based on amino acid sequences of Cap (b) and RdRp (c) regions of feline astroviruses. Feline astroviruses identified in this study are marked with black diamonds.
To determine the divergence in sequence between the feline astroviruses found here and other astroviruses, amino acid sequence alignments of Cap and RdRp of the three feline astroviruses in the present study and 23 representative astrovirus strains were performed, and neighbour-joining trees generated. Both trees revealed that the three strains of astrovirus in this study fell into two genetic lineages within the genus Mamastrovirus. The tree for the Cap region (Fig. 2b) indicated that FastV-D1 was related to the clade composed of mink astroviruses and a California sea lion astrovirus, sharing 58 and 56 % amino acid similarity, respectively, in the Cap region, suggesting that FastV-D1 is a novel astrovirus species. The tree based on the RdRp region showed that FAstV-D1 was more closely related to the fox astrovirus (GenBank accession number KC692365) (Bodewes et al., 2013) (Fig. 2c), sharing 80 % amino acid identity. FastV-D2 and FastV-D3 shared >90 % amino acid sequence similarities with recently characterized feline astroviruses, Viseu and 1637F, which are the closest currently known relatives of the classic human astroviruses (mamastrovirus 1). RT-PCR testing of individual specimens indicated that eight of 25 faecal samples were positive for the novel FAstV-D1 (32 %).
Feline bocavirus
Bocaviruses are small, non-enveloped, ssDNA viruses belonging to the family Parvoviridae and consist of linear positive-strand DNA genomes 4–6 kb in length. Bocaviruses also encode a third ORF of unknown function (NP) in the middle of their genomes. Recently, bocaviruses have been identified in humans and numerous mammals, such as dogs, cattle, pigs, sea lions, and cats (Allander et al., 2005; Kapoor et al., 2012; Lau et al., 2012; Li, et al., 2011a; Shan et al., 2011a). Bocaviruses have been associated with diverse symptoms, most notably respiratory problems and diarrhoea (Jartti et al., 2012; Manteufel & Truyen, 2008).
In the present study, bocaviruses were identified in five of the six cat faecal pools. Sequence assembly revealed two near-complete genomes with complete ORFs that we named feline bocaviruses FeBoV3-FBD1 (GenBank accession number KM017744) and FeBoV1-FBD2 (GenBank accession number KM017745) (Fig. 3a). The complete genome of FBD1 was 5407 bases with 304 bases in the 5′ UTR and 136 bases in the 3′ UTR. The complete genome of FBD2 was 5466 bases, of which the 5′ and 3′ UTRs were 309 and 211 bases, respectively. Phylogenetic analysis based on the amino acid sequences of NS1 and VP1 both indicated that FBD1 and FBD2 clustered with the other four feline bocavirus strains with complete genomes available in GenBank (Fig. 3b, c). FBD1 shared amino acid sequence identities of 68–76 % with the other feline bocavirus strains in the NS1 and VP1 proteins, respectively, suggesting that FBD1 belonged to a novel species based on the proposed species-defining criterion of >15 % NS1 divergence to its closest relative (Cotmore et al., 2014). Phylogenetically, FBD2 belonged to known feline bocavirus type 1 (carnivore bocaparvovirus 1 species) (Cotmore et al., 2014), clustering closely with three feline bocaviruses from Hong Kong, sharing >96 % sequence identities in the NS1 and VP1 proteins. Another species of cat bocavirus (FBoV2-POR1) was described recently and is shown in Fig. 3(b, c) as GenBank accession number KF792837 (Ng et al., 2014). RT-PCR was then used to test individual specimens for FeBoV3-FBD1. Two of the 25 samples were positive for this bocavirus species (8 %).
Fig. 3.

(a) Genomic organization of feline bocaviruses (distance in nt). (b, c) Neighbour-joining phylogenetic analysis based on amino acid sequences of NS1 (b) and VP1 (c) regions of feline bocavirus. Feline bocaviruses identified in this study are marked with black diamonds.
Cyclovirus
The family Circoviridae is composed of non-enveloped viruses containing a circular ssDNA genome. Cycloviruses belong to the family Circoviridae and have recently been found in different sample types from different hosts, including mammals and insects (Dayaram et al., 2013; Ge et al., 2011; Li et al., 2010a, b; Padilla-Rodriguez et al., 2013; Phan et al., 2014; Rosario et al., 2011). Cycloviruses have a slightly smaller genome than that of circoviruses, encoding a smaller Rep and Cap protein, and with shorter or no 3′ intergenic regions between the stop codons of the two major ORFs and a longer 5′ intergenic region between the start codons of the two major ORFs. Based on the phylogenetic analysis, cycloviruses were suggested to be grouped into a new genus of the family Circoviridae (Li et al., 2010a).
In this study, cycloviruses were present in only one pool of cat faecal samples with a total of 283 sequence reads. By sequence assembly, a complete genome of cyclovirus was acquired and named CyCVs-FD (GenBank accession number KM017740). The genome of CyCVs-FD consisted of 1773 bp and had the typical genome organization with two main ORFs arranged in opposite directions, encoding the putative Rep and Cap proteins (Fig. 4a). CyCVs-FD had a 5′ intergenic region of 275 bp and a 3′ intergenic region of 7 bp. A potential stem–loop structure with a nonanucleotide motif (5′-TAGTATTAC-3′) was located between the 5′ ends of the two ORFs, which is required to initiate the replication of the viral genome. Typical of cycloviruses and unlike that of the related circoviruses, the nonanucleotide motif was not on the Rep-encoding strand of the genome. The Rep proteins of CyCVs-FD and of representative strains of circoviruses and cycloviruses were used for phylogenetic analysis. The resulting tree revealed that CyCVs-FD formed a deep branch within the cyclovirus clade (Fig. 4b), suggesting that this virus represents a novel species. The putative Rep protein of CyCVs-FD was 272 aa and shared 39–47 % sequence identity with those of previously reported cycloviruses.
Fig. 4.

(a) Genomic organizations of the novel feline cylcovirus. The locations of putative Rep and Cap genes are indicated by arrows. The stem–loop is also shown. (b) Neighbour-joining phylogenetic analysis based on amino acid sequences of Rep of feline bocavirus. CSF, cerebrospinal fluid. Feline cyclovirus identified in this study is marked with a black diamond.
RdRp sequence from divergent viruses
Sequences encoding the RdRp gene are amongst the most conserved genes in positive-strand RNA viruses. In the present study, we found four different sequence contigs showing low identity on the protein level to RdRp of RNA viruses. The first contig was 471 bases and the putative amino acid sequence shared 30 % identity with the foot-and-mouth disease virus (FMDV), a member of the family Picornaviridae. Using sequence assembly and genome walking, a 6218 base sequence of this virus was acquired, encoding a single 1473 aa polyprotein including the RdRp region and Cap protein (Rhv-like domains) (Fig. 5). However, blastp with the non-structural and Rhv-like regions both produced best matches with cricket paralysis RNA virus instead of with FMDV, with amino acid sequence identity of 23 %. The second contig was 940 bases encoding a nearly complete RdRp with a closest match to hepatitis E virus, sharing amino acid sequence identity of 30 %. The third contig was 1030 bp, also encoding a nearly complete RdRp region. blastp indicated that this amino acid sequence shared the highest sequence identity (28 %) with calhevirus-1, a genome found in human faeces but predicted to be of insect host origin based on nucleotide composition (Kapoor et al., 2010). The fourth contig was 4554 bp, and contained two large ORFs encoding non-structural and structural proteins (Fig. 5). The putative amino acid sequence encoded a complete RdRp region and showed the highest sequence identity (41 %) with tetnovirus-2 which was discovered in human faeces and also predicted based on nucleotide composition to be of insect host origin (Kapoor et al., 2010). We provisionally named the four sequences fesavirus-1, -2, -3 and -4, respectively (feline stool-associated RNA viruses 1, 2, 3 and 4).
Fig. 5.
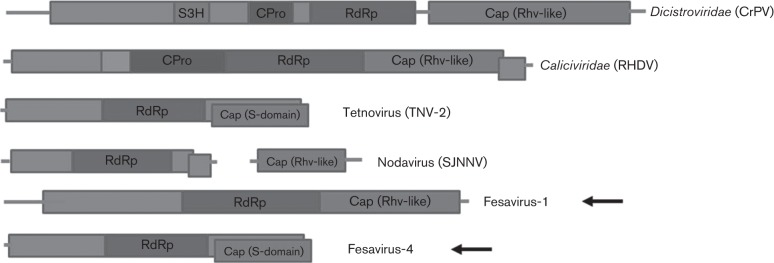
Genomic organization showing the ORFs and conserved protein domains of fesavirus-1 and -4, and other RNA viruses with related genomic organization. CPro, cysteine protease; S3H, superfamily 3 helicase. CrPV, cricket paralysis virus; RHDV, rabbit haemorrhagic disease virus; SJNNV, striped jack nervous necrosis virus. Fesavirus-1 and -4 in this study are marked with arrows.
In order to identify sequence relationships of these four RdRp sequences, phylogenetic analysis based on the most conserved RdRp region of the picorna-like virus supergroup was performed by adding to an alignment generated in a previous report (Koonin et al., 2008). A maximum-likelihood phylogenetic tree was reconstructed by use of mega5.0. The resulting tree was almost identical to that described previously (Koonin et al., 2008) (Fig. 6). The four RdRp sequences in this study fell into four different groups. Fesavirus-1 (GenBank accession number KM017736) RdRp grouped with sequences from plant viruses and was most closely related to apple latent spherical virus and cherry rasp leaf virus, suggesting that this virus might infect a plant. Fesavirus-2 sequence (GenBank accession number KM017737) clustered with those of five viruses from the Hepeviridae clade. Fesavirus-3 (GenBank accession number KM017738) and fesavirus-4 sequences (GenBank accession number KM017739) were related, and closest to those of calhevirus-1 and tetnovirus, respectively, two viral genome sequences of uncertain tropism, possibly infecting insects (Kapoor et al., 2010).
Fig. 6.

Maximum-likelihood phylogenetic analysis based on the divergent amino acid sequences of RdRp in the present study and the members of the picorna-like virus supergroup presented in a previous study (Koonin et al., 2008). The best blastp hits of the RdRp sequences in this study and five RdRp representative sequences from the family Hepeviridae were also included in the alignment. The six main clades identified and abbreviations of viruses are labelled according to their previous designations (Koonin et al., 2008). Bootstrap values >50 are indicated on the tree.
Known mammalian viruses
Table 1 presents the mammalian viruses in the cat faecal virome that showed high sequence similarity to known viruses in GenBank. Feline coronaviruses accounted for ~8.05 % of the recognized cat faecal virome, 99 % of which were in a single pool, likely reflecting a single heavily shedding cat. Feline coronaviruses cause infections in wild and domestic felids, and are widespread, with seropositivity of 20–60 % in domestic cats and up to 90 % in animal shelters or households with multiple cats. Infected animals often develop persistent infection and shed the virus over extended periods of time (Addie et al., 2003). Feline coronavirus can be separated into type I and type II, and 80–90 % of the naturally occurring infections are caused by type I (Addie et al., 2003; Kummrow et al., 2005). Sequence analysis indicated that the coronavirus in the present study also belonged to type I, and all the sequences reads shared high sequence similarities with several closely related feline coronaviruses strains, suggesting a single feline coronaviruses strain was prevalent in this cat population. Feline herpesvirus type 1 (FHV-1) is an important pathogenic agent that causes feline viral rhinotracheitis. There were 19 sequence reads related to FHV-1 in the cat faecal virome, all of which were from a single pool sharing 76–79 % protein sequence identity with the FHV-1 prototype strain C-27 (Tai et al., 2010). There were two types of calicivirus sequences in the cat faecal virome, one type (total 48 reads in two pools) was genetically related to feline calicivirus and the other type (total 12 reads in a different pool) was related to feline norovirus, both with high sequence similarities. Feline calicivirus infection is one of the most common and widespread viral diseases of cats, inducing chronic infection of the upper respiratory tract, pneumonia and lower urinary tract disease (Stiles, 2014), whilst feline norovirus has been reported in domestic cats with gastroenteritis (Pinto et al., 2012). Picobirnaviruses were detected in three pools of cat faeces. Sequence analysis revealed that these feline picobirnavirus sequences shared high identities (>90 %) with the previously reported feline picobirnavirus (Ng et al., 2014), which also showed a close relationship with a human picobirnavirus strain 3-GA-91 (Rosen et al., 2000). Lastly, feline panleukopenia virus belongs to the feline parvovirus subgroup within the genus Parvovirus, and was originally identified in domestic cats and later on other large felids, such as tigers, panthers, cheetahs and lions (Steinel et al., 2001; Truyen & Parrish, 2013). In young wild and domestic carnivores, feline panleukopenia virus infection usually causes severe gastroenteritis, frequently haemorrhagic. In the current study, a total of 504 feline panleukopenia virus sequence reads were identified in two sample pools, sharing high sequence similarities (98–100 %) to several closely related feline panleukopenia virus strains. The sequence reads of known viruses reported here are available in the online Supplementary Material.
Table 1. Characterization of the viral sequence reads with high sequence identities to known viruses.
| Coronaviridae | Herpesviridae | Caliciviridae 1 | Caliciviridae 2 | Picobirnaviridae | Parvoviridae | |
| Match | Feline coronavirus | Felid herpesvirus 1 | Feline calicivirus | Norovirus cat | Feline/human picobirnavirus | Feline panleukopenia virus |
| GenBank accession no(s). | DQ160294EU186072FJ938051JQ408981 | FJ478159 | AAB23553AY560114Z11536 | JF781268 | KF792838,AF246941 | X55115EU659112JN867595 |
| Amino acid identity (%) | >93 | 76–79 | 92–100 | 96–98 | 78–100 | 98–100 |
| Total reads | 24 425 | 2 | 47 | 2 | 31 | 499 |
| Presence in pool no(s). | 2, 3, 5 | 6 | 2, 5 | 1 | 1, 4, 6 | 3, 4 |
Insect, plant and fish viruses
About 1.2 % of the cat faecal virome (3786 sequence reads) was related to plant viruses. DNA viruses were predominant with 73 % of these reads, all of which were from the family Phycodnaviridae. RNA viruses accounted for the rest, with the majority related to ssRNA viruses in the family Tombusviridae (55 %), followed by the families Alphaflexiviridae and Secoviridae, genus Umbravirus, and family Rhabdoviridae.
About 0.2 % of the cat faecal virome (602 sequence reads) was related to insect viruses, 87 % of which belonged to RNA viruses from the families Nodaviridae and Dicistroviridae, whilst 13 % were related to the DNA virus family Ascoviridae. The majority of the insect virus-like sequences shared protein similarities of <70 % with annotated insect viral proteins.
Likely fish viruses (588 sequence reads) accounted for ~0.2 % of the cat faecal virome reads; 87 % belonged to the family Iridoviridae, and included virus sequences related to invertebrate iridovirus and frog virus 3 (FV3), sharing about 60 and 95 % protein sequence identities, respectively. FV3 is the type species of the genus Ranavirus. In past decades, FV3 infections have resulted in considerable morbidity and mortality in wild and cultivated amphibian species (Williams et al., 2005). The remaining 13 % of reads were related to viruses from the family Nodaviridae, some members of which can infect both insects and fishes.
Discussion
Studies characterizing viromes in animal faeces have been performed for different mammals, starting with humans (Breitbart et al., 2003; Finkbeiner et al., 2008; Kapoor et al., 2008), horses (Cann et al., 2005), bats (Donaldson et al., 2010; Li et al., 2010b; Wu et al., 2012), pigs (Sachsenröder et al., 2014; Shan et al., 2011b; Zhang et al., 2014), rodents (He et al., 2013; Phan et al., 2011), California sea lions (Li et al., 2011a), pine martens (van den Brand et al., 2012) and others. In the present study, we used metagenomics to describe the composition of the viral community in the faeces of cats in an animal shelter. The virome showed a majority of eukaryotic viruses with fewer recognizable (using blastx) prokaryotic viruses. In addition to the mammalian viruses, the eukaryotic viruses detected also included possible insect, fish and plant viruses, originating from the cat diet.
Astroviruses were detected in all of the sample pools analysed and made up the highest number of viral reads detected. Astroviruses can cause diarrhoeal and occasionally extra-intestinal disease in various species (De Benedictis et al., 2011). The main mode of astrovirus transmission is faecal–oral. Such transmission may be particularly effective within a crowded animal shelter environment. Phylogenetic analysis indicated the presence of three different astrovirus strains. Although two of the three astrovirus strains clustered with previously identified feline astroviruses, the third belonged to a novel species clustering with astroviruses from other carnivores. Not all astroviruses infecting a given animal host species phylogenetically cluster together, leading to the conclusion that astrovirus cross-species transmission occurs frequently (De Benedictis et al., 2011; Finkbeiner et al., 2009; Kapoor et al., 2009). Our detection of a feline astrovirus highly divergent from the other (closely related) feline astroviruses further supports this conclusion.
Although human bocaviruses have been discovered in respiratory and faecal samples of children worldwide, their ability to cause enteric disease is still under debate because of their frequent detection in healthy children and co-detection with other pathogens (Allander et al., 2005; Hao et al., 2013; Söderlund-Venermo et al., 2009). Bocaviruses have been identified in both healthy and diseased animals, including dogs, cattle, pigs, sea lions and cats (Allander et al., 2005; Kapoor et al., 2012; Lau et al., 2012; Li et al., 2011a; Shan et al., 2011a). Feline bocavirus was first discovered in stray cats in Hong Kong where it was detected in multiple tissues (Lau et al., 2012). In the current study, two different bocavirus strains were prevalent in the cat population analysed, one of them sharing >96 % sequence identity with the feline bocavirus from Hong Kong. The other feline bocavirus may represent a novel species due to an NS1 divergence of >15 % to the next closest species. Phylogenetic trees based on NS1 and VP1 protein sequences available from GenBank and this study therefore indicate that together with the POR1 strain (Ng et al., 2014) that there are currently three known species of feline bocavirus.
Diverse cycloviruses have been found in both mammals and insects (Dayaram et al., 2013; Ge et al., 2011; Li et al., 2010a, 2011b; Padilla-Rodriguez et al., 2013; Phan et al., 2014; Rosario et al., 2011). The detection of cyclovirus DNA in different human samples, including faeces (Li et al., 2010a), nasopharyngeal aspirates (Phan et al., 2014) and cerebrospinal fluid (Smits et al., 2013; Tan et al., 2013), and in the muscle tissues of farm animals (Li et al., 2011b), suggests that cycloviruses may infect mammals. In the present study, a novel species of cyclovirus was detected in cat faeces. Phylogenetic analysis based on the protein sequence of Rep revealed that this feline cyclovirus fell between the circoviruses and cylcoviruses, whilst its genomic features were more akin to cycloviruses. Whether this cyclovirus replicates in feline cells and is associated with cat disease will require further studies.
In conclusion, our study provides an overview of the feline faecal virome which we expect to further expand as more animals from different geographical regions are sampled. Cats from Portugal were shown recently to shed a novel picornavirus not detected in these Californian cats (Ng et al., 2014). Whether these viruses are commensals or can on occasion or in concert cause disease in susceptible cats will require further studies, including in situ hybridization of tissues showing pathologies and case-control studies of specific feline diseases.
Methods
Samples and viral metagenomics.
Twenty-five faecal specimens were collected from clinically normal cats in a shelter in Davis, California, in October 2013 and stored at −80 °C. These cats had been in the shelter between a few days and 2 weeks. They were kept in separate cages in a single large room, where the cages were stacked three high and 10 across. The cats were fed the same food, which changed depending on the donations received. Stool samples were resuspended in 10 vols PBS and vortexed vigorously for 5 min. Supernatant (400 µl) was collected after centrifugation (10 min, 15 000 g), and filtered through a 0.45 µm filter (Millipore) to remove eukaryotic and bacterial cell-sized particles. The filtrates, enriched in viral particles, were treated with a mixture of DNases [Turbo DNase (Ambion), Baseline-ZERO (Epicentre) and benzonase (Novagen)] and RNase (Fermentas) to digest unprotected nucleic acid at 37 °C for 90 min (Victoria et al., 2009). Viral nucleic acids protected from digestion within viral capsids and other small particles were then extracted using magnetic beads of the MagMAX Viral RNA Isolation kit (Ambion) according to the manufacturer’s instructions. Six separate pools of nucleic acids from 25 faecal specimens were generated randomly, of which five contained four faecal specimens and the other one contained five faecal specimens. These six viral nucleic acid pools, containing both DNA and RNA viral sequences, were then subjected to RT reactions with SuperScript III reverse transcriptase (Invitrogen) and 100 pmol of a random hexamer primer, followed by a single round of DNA synthesis using Klenow fragment polymerase (New England BioLabs). A library was then constructed using the Nextera XT DNA Sample Preparation kit (Illumina) and sequenced using the MiSeq Illumina platform with 250 bp paired ends with a distinct molecular tag for each pool.
Bioinformatics analysis.
Paired-end reads of 250 bp generated by MiSeq were debarcoded using vendor software from Illumina. An in-house analysis pipeline running on a 32-node Linux cluster was used to process the data. Clonal reads were removed and low-sequencing-quality tails were trimmed using Phred quality score 10 as the threshold. Adaptors were trimmed using the default parameters of VecScreen, which is National Center for Biotechnology Information (NCBI) blastn (Altschul et al., 1997) with specialized parameters designed for adaptor removal. The cleaned reads were de novo assembled by SOAPdenovo2 version r240 using Kmer size 63 with default settings (Luo et al., 2012). The assembled contigs, along with singlets, were aligned to an in-house viral proteome database using blastx. The significant hits to virus were then aligned to an in-house NVNR (non-virus non-redundant) universal proteome database using blastx. Hits with a more significant adjusted E value to NVNR than to virus were removed.
PCR screening.
DNA and RNA were also directly extracted from centrifuged stool supernatant using the magnetic beads of the MagMAX Viral RNA Isolation kit (Ambion) according to the manufacturer’s instructions. For FAstV-D1-related sequence screening, primers FastvL1 (5′-GGAGGTGGCTAAGGAGATAGT-3′) and FastvR1 (5′-CCTCTCTGAAGACGCCATGACT-3′) were used for the first round of nested PCR, and primers FastvL2 (5′-CCCTCGAAGCGCTGGCACAA-3′) and FastvR2 (5′-CACCGAGCCCACCCCAGCTA-3′) were used for the second round of nested PCR, resulting in the amplification of a 433 bp fragment of the Cap gene. FeBoV3-FBD1-related sequences were detected using primers FBD1L1 (5′-TGACTCGTCTGTGGCGGGCT-3′) and FBD1R1 (5′-TCGTTCGTGAGACGCTGCCA-3′) for the first round of nested PCR, and primers FBD1L2 (5′-CAAAGGATCGGGAGCGGGCG-3′) and FBD1R2 (5′-TGCCCATGGTGTTGTGATTCCTATCCA-3′) for the second round of nested PCR, amplifying a 388 bp fragment of the VP1 gene. PCR products were Sanger sequence-confirmed.
Phylogenetic analysis.
Phylogenetic analyses were performed based on predicted viral amino acid sequences together with their closest viral relatives (best BLASTX hits) and representative members of related viral species or genera. Sequence alignment was performed using clustal w (version 2.1) with the default settings (Larkin et al., 2007). Aligned sequences were trimmed to match the genomic regions of the viral sequences obtained in the study. A phylogenetic tree with 100 bootstrap resamples of the alignment datasets was reconstructed using the neighbour-joining method based on the Jones–Taylor–Thornton matrix-based model in mega5.0 (Tamura et al., 2011). Bootstrap values (based on 100 replicates) for each node are given. Putative ORFs in the genome were predicted by NCBI ORF finder.
Acknowledgements
The work was supported by grants from the Blood Systems Research Institute and National Institutes of Health (R01 HL105770) to E. D., and China Scholarship Council (201208320503) to W. Z.
Footnotes
Supplementary material is available with the online version of this paper.
References
- Addie D. D., Schaap I. A. T., Nicolson L., Jarrett O. (2003). Persistence and transmission of natural type I feline coronavirus infection. J Gen Virol 84, 2735–2744. 10.1099/vir.0.19129-0 [DOI] [PubMed] [Google Scholar]
- Allander T., Tammi M. T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. (2005). Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A 102, 12891–12896. 10.1073/pnas.0504666102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997). Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomström A.-L., Widén F., Hammer A.-S., Belák S., Berg M. (2010). Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol 48, 4392–4396. 10.1128/JCM.01040-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodewes R., van der Giessen J., Haagmans B. L., Osterhaus A. D. M. E., Smits S. L. (2013). Identification of multiple novel viruses, including a parvovirus and a hepevirus, in feces of red foxes. J Virol 87, 7758–7764. 10.1128/JVI.00568-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Hewson I., Felts B., Mahaffy J. M., Nulton J., Salamon P., Rohwer F. (2003). Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol 185, 6220–6223. 10.1128/JB.185.20.6220-6223.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann A. J., Fandrich S. E., Heaphy S. (2005). Analysis of the virus population present in equine faeces indicates the presence of hundreds of uncharacterized virus genomes. Virus Genes 30, 151–156. 10.1007/s11262-004-5624-3 [DOI] [PubMed] [Google Scholar]
- Cotmore S. F., Agbandje-McKenna M., Chiorini J. A., Mukha D. V., Pintel D. J., Qiu J., Soderlund-Venermo M., Tattersall P., Tijssen P. & other authors (2014). The family Parvoviridae. Arch Virol 159, 1239–1247. 10.1007/s00705-013-1914-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayaram A., Potter K. A., Moline A. B., Rosenstein D. D., Marinov M., Thomas J. E., Breitbart M., Rosario K., Argüello-Astorga G. R., Varsani A. (2013). High global diversity of cycloviruses amongst dragonflies. J Gen Virol 94, 1827–1840. 10.1099/vir.0.052654-0 [DOI] [PubMed] [Google Scholar]
- De Benedictis P., Schultz-Cherry S., Burnham A., Cattoli G. (2011). Astrovirus infections in humans and animals – molecular biology, genetic diversity, and interspecies transmissions. Infect Genet Evol 11, 1529–1544. 10.1016/j.meegid.2011.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson E. F., Haskew A. N., Gates J. E., Huynh J., Moore C. J., Frieman M. B. (2010). Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol 84, 13004–13018. 10.1128/JVI.01255-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S. R., Allred A. F., Tarr P. I., Klein E. J., Kirkwood C. D., Wang D. (2008). Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog 4, e1000011. 10.1371/journal.ppat.1000011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S. R., Holtz L. R., Jiang Y., Rajendran P., Franz C. J., Zhao G., Kang G., Wang D. (2009). Human stool contains a previously unrecognized diversity of novel astroviruses. Virol J 6, 161. 10.1186/1743-422X-6-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X., Li J., Peng C., Wu L., Yang X., Wu Y., Zhang Y., Shi Z. (2011). Genetic diversity of novel circular ssDNA viruses in bats in China. J Gen Virol 92, 2646–2653. 10.1099/vir.0.034108-0 [DOI] [PubMed] [Google Scholar]
- Gunn-Moore D. A., Reed N. (2011). CNS disease in the cat: current knowledge of infectious causes. J Feline Med Surg 13, 824–836. 10.1016/j.jfms.2011.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao R., Ni K., Xia Q., Peng C., Deng Y., Zhao X., Fu Z., Liu W., Liu E. (2013). Correlation between nucleotide mutation and viral loads of human bocavirus 1 in hospitalized children with respiratory tract infection. J Gen Virol 94, 1079–1085. 10.1099/vir.0.047472-0 [DOI] [PubMed] [Google Scholar]
- He B., Li Z., Yang F., Zheng J., Feng Y., Guo H., Li Y., Wang Y., Su N. & other authors (2013). Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel mammalian viruses. PLoS ONE 8, e61950. 10.1371/journal.pone.0061950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jartti T., Hedman K., Jartti L., Ruuskanen O., Allander T., Söderlund-Venermo M. (2012). Human bocavirus – the first 5 years. Rev Med Virol 22, 46–64. 10.1002/rmv.720 [DOI] [PubMed] [Google Scholar]
- Jones K. E., Patel N. G., Levy M. A., Storeygard A., Balk D., Gittleman J. L., Daszak P. (2008). Global trends in emerging infectious diseases. Nature 451, 990–993. 10.1038/nature06536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Victoria J., Simmonds P., Slikas E., Chieochansin T., Naeem A., Shaukat S., Sharif S., Alam M. M. & other authors (2008). A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proc Natl Acad Sci U S A 105, 20482–20487. 10.1073/pnas.0807979105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Li L., Victoria J., Oderinde B., Mason C., Pandey P., Zaidi S. Z., Delwart E. (2009). Multiple novel astrovirus species in human stool. J Gen Virol 90, 2965–2972. 10.1099/vir.0.014449-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Simmonds P., Lipkin W. I., Zaidi S., Delwart E. (2010). Use of nucleotide composition analysis to infer hosts for three novel picorna-like viruses. J Virol 84, 10322–10328. 10.1128/JVI.00601-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Mehta N., Dubovi E. J., Simmonds P., Govindasamy L., Medina J. L., Street C., Shields S., Lipkin W. I. (2012). Characterization of novel canine bocaviruses and their association with respiratory disease. J Gen Virol 93, 341–346. 10.1099/vir.0.036624-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Wolf Y. I., Nagasaki K., Dolja V. V. (2008). The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat Rev Microbiol 6, 925–939. 10.1038/nrmicro2030 [DOI] [PubMed] [Google Scholar]
- Kummrow M., Meli M. L., Haessig M., Goenczi E., Poland A., Pedersen N. C., Hofmann-Lehmann R., Lutz H. (2005). Feline coronavirus serotypes 1 and 2: seroprevalence and association with disease in Switzerland. Clin Diagn Lab Immunol 12, 1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A. & other authors (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Lau S. K. P., Woo P. C. Y., Yeung H. C., Teng J. L. L., Wu Y., Bai R., Fan R. Y. Y., Chan K.-H., Yuen K.-Y. (2012). Identification and characterization of bocaviruses in cats and dogs reveals a novel feline bocavirus and a novel genetic group of canine bocavirus. J Gen Virol 93, 1573–1582. 10.1099/vir.0.042531-0 [DOI] [PubMed] [Google Scholar]
- Lau S. K. P., Woo P. C. Y., Yip C. C. Y., Bai R., Wu Y., Tse H., Yuen K.-Y. (2013). Complete genome sequence of a novel feline astrovirus from a domestic cat in Hong Kong. Genome Announc 1, e00708-13. 10.1128/genomeA.00708-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Kapoor A., Slikas B., Bamidele O. S., Wang C., Shaukat S., Masroor M. A., Wilson M. L., Ndjango J.-B. N. & other authors (2010a). Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol 84, 1674–1682. 10.1128/JVI.02109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Victoria J. G., Wang C., Jones M., Fellers G. M., Kunz T. H., Delwart E. (2010b). Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol 84, 6955–6965. 10.1128/JVI.00501-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Shan T., Wang C., Côté C., Kolman J., Onions D., Gulland F. M. D., Delwart E. (2011a). The fecal viral flora of California sea lions. J Virol 85, 9909–9917. 10.1128/JVI.05026-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Shan T., Soji O. B., Alam M. M., Kunz T. H., Zaidi S. Z., Delwart E. (2011b). Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J Gen Virol 92, 768–772. 10.1099/vir.0.028704-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Diab S., McGraw S., Barr B., Traslavina R., Higgins R., Talbot T., Blanchard P., Rimoldi G. & other authors (2013). Divergent astrovirus associated with neurologic disease in cattle. Emerg Infect Dis 19, 1385–1392. 10.3201/eid1909.130682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q. & other authors (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18. 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manteufel J., Truyen U. (2008). Animal bocaviruses: a brief review. Intervirology 51, 328–334. 10.1159/000173734 [DOI] [PubMed] [Google Scholar]
- Ng T. F. F., Mesquita J. R., Nascimento M. S. J., Kondov N. O., Wong W., Reuter G., Knowles N. J., Vega E., Esona M. D. & other authors (2014). Feline fecal virome reveals novel and prevalent enteric viruses. Vet Microbiol 171, 102–111. 10.1016/j.vetmic.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Rodriguez M., Rosario K., Breitbart M. (2013). Novel cyclovirus discovered in the Florida woods cockroach Eurycotis floridana (Walker). Arch Virol 158, 1389–1392. 10.1007/s00705-013-1606-x [DOI] [PubMed] [Google Scholar]
- Pesavento P. A., Murphy B. G. (2014). Common and emerging infectious diseases in the animal shelter. Vet Pathol 51, 478–491. 10.1177/0300985813511129 [DOI] [PubMed] [Google Scholar]
- Phan T. G., Kapusinszky B., Wang C., Rose R. K., Lipton H. L., Delwart E. L. (2011). The fecal viral flora of wild rodents. PLoS Pathog 7, e1002218. 10.1371/journal.ppat.1002218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. G., Luchsinger V., Avendaño L. F., Deng X., Delwart E. (2014). Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J Gen Virol 95, 922–927. 10.1099/vir.0.061143-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto P., Wang Q., Chen N., Dubovi E. J., Daniels J. B., Millward L. M., Buonavoglia C., Martella V., Saif L. J. (2012). Discovery and genomic characterization of noroviruses from a gastroenteritis outbreak in domestic cats in the US. PLoS ONE 7, e32739. 10.1371/journal.pone.0032739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusterla N., Mapes S., Madigan J. E., Maclachlan N. J., Ferraro G. L., Watson J. L., Spier S. J., Wilson W. D. (2009). Prevalence of EHV-1 in adult horses transported over long distances. Vet Rec 165, 473–475. 10.1136/vr.165.16.473 [DOI] [PubMed] [Google Scholar]
- Quan P. L., Wagner T. A., Briese T., Torgerson T. R., Hornig M., Tashmukhamedova A., Firth C., Palacios G., Baisre-De-Leon A. & other authors (2010). Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg Infect Dis 16, 918–925. 10.3201/eid1606.091536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario K., Marinov M., Stainton D., Kraberger S., Wiltshire E. J., Collings D. A., Walters M., Martin D. P., Breitbart M., Varsani A. (2011). Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J Gen Virol 92, 1302–1308. 10.1099/vir.0.030338-0 [DOI] [PubMed] [Google Scholar]
- Rosen B. I., Fang Z. Y., Glass R. I., Monroe S. S. (2000). Cloning of human picobirnavirus genomic segments and development of an RT-PCR detection assay. Virology 277, 316–329. 10.1006/viro.2000.0594 [DOI] [PubMed] [Google Scholar]
- Sachsenröder J., Twardziok S. O., Scheuch M., Johne R. (2014). The general composition of the faecal virome of pigs depends on age, but not on feeding with a probiotic bacterium. PLoS ONE 9, e88888. 10.1371/journal.pone.0088888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze C., Alex M., Schirrmeier H., Hlinak A., Engelhardt A., Koschinski B., Beyreiss B., Hoffmann M., Czerny C.-P. (2007). Generalized fatal Cowpox virus infection in a cat with transmission to a human contact case. Zoonoses Public Health 54, 31–37. 10.1111/j.1863-2378.2007.00995.x [DOI] [PubMed] [Google Scholar]
- Shan T., Lan D., Li L., Wang C., Cui L., Zhang W., Hua X., Zhu C., Zhao W., Delwart E. (2011a). Genomic characterization and high prevalence of bocaviruses in swine. PLoS ONE 6, e17292. 10.1371/journal.pone.0017292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan T., Li L., Simmonds P., Wang C., Moeser A., Delwart E. (2011b). The fecal virome of pigs on a high-density farm. J Virol 85, 11697–11708. 10.1128/JVI.05217-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S. L., Zijlstra E. E., van Hellemond J. J., Schapendonk C. M. E., Bodewes R., Schürch A. C., Haagmans B. L., Osterhaus A. D. M. E. (2013). Novel cyclovirus in human cerebrospinal fluid, Malawi, 2010–2011. Emerg Infect Dis 19, 19. 10.3201/eid1909.130404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderlund-Venermo M., Lahtinen A., Jartti T., Hedman L., Kemppainen K., Lehtinen P., Allander T., Ruuskanen O., Hedman K. (2009). Clinical assessment and improved diagnosis of bocavirus-induced wheezing in children, Finland. Emerg Infect Dis 15, 1423–1430. 10.3201/eid1509.090204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sponseller B. A., Strait E., Jergens A., Trujillo J., Harmon K., Koster L., Jenkins-Moore M., Killian M., Swenson S. & other authors (2010). Influenza A pandemic (H1N1) 2009 virus infection in domestic cat. Emerg Infect Dis 16, 534–537. 10.3201/eid1603.091737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinel A., Parrish C. R., Bloom M. E., Truyen U. (2001). Parvovirus infections in wild carnivores. J Wildl Dis 37, 594–607. 10.7589/0090-3558-37.3.594 [DOI] [PubMed] [Google Scholar]
- Stiles J. (2014). Ocular manifestations of feline viral diseases. Vet J 201, 166–173. 10.1016/j.tvjl.2013.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachezy R., Duson G., Rector A., Jenson A. B., Sundberg J. P., Van Ranst M. (2002). Cloning and genomic characterization of Felis domesticus papillomavirus type 1. Virology 301, 313–321. 10.1006/viro.2002.1566 [DOI] [PubMed] [Google Scholar]
- Tai S. H. S., Niikura M., Cheng H. H., Kruger J. M., Wise A. G., Maes R. K. (2010). Complete genomic sequence and an infectious BAC clone of feline herpesvirus-1 (FHV-1). Virology 401, 215–227. 10.1016/j.virol.2010.02.021 [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan V., van Doorn H. R., Nghia H. D. T., Chau T. T. H., Tu T. P., de Vries M., Canuti M., Deijs M., Jebbink M. F. & other authors (2013). Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. MBio 4, e00231–13. 10.1128/mBio.00231-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truyen U., Parrish C. R. (2013). Feline panleukopenia virus: its interesting evolution and current problems in immunoprophylaxis against a serious pathogen. Vet Microbiol 165, 29–32. 10.1016/j.vetmic.2013.02.005 [DOI] [PubMed] [Google Scholar]
- Tsugawa T., Hoshino Y. (2008). Whole genome sequence and phylogenetic analyses reveal human rotavirus G3P[3] strains Ro1845 and HCR3A are examples of direct virion transmission of canine/feline rotaviruses to humans. Virology 380, 344–353. 10.1016/j.virol.2008.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brand J. M. A., van Leeuwen M., Schapendonk C. M., Simon J. H., Haagmans B. L., Osterhaus A. D. M. E., Smits S. L. (2012). Metagenomic analysis of the viral flora of pine marten and European badger feces. J Virol 86, 2360–2365. 10.1128/JVI.06373-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J. G., Kapoor A., Li L., Blinkova O., Slikas B., Wang C., Naeem A., Zaidi S., Delwart E. (2009). Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol 83, 4642–4651. 10.1128/JVI.02301-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T., Barbosa-Solomieu V., Chinchar V. G. (2005). A decade of advances in iridovirus research. Adv Virus Res 65, 173–248. 10.1016/S0065-3527(05)65006-3 [DOI] [PubMed] [Google Scholar]
- Wu Z., Ren X., Yang L., Hu Y., Yang J., He G., Zhang J., Dong J., Sun L. & other authors (2012). Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol 86, 10999–11012. 10.1128/JVI.01394-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Tang C., Yue H., Ren Y., Song Z. (2014). Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J Gen Virol 95, 1603–1611. 10.1099/vir.0.063743-0 [DOI] [PubMed] [Google Scholar]