

Abstract
Importance
Mutations in the gene encoding parkin (PARK2) are the most common cause of autosomal recessive juvenile-onset and young-onset parkinsonism. The few available detailed neuropathologic reports suggest that homozygous and compound heterozygous parkin mutations are characterized by severe substantia nigra pars compacta neuronal loss.
Objective
To investigate whether parkin -linked parkinsonism is a different clinicopathologic entity to Parkinson disease (PD).
Design, Setting, and Participants
We describe the clinical, genetic, and neuropathologic findings of 5 unrelated cases of parkin disease and compare them with 5 pathologically confirmed PD cases and 4 control subjects. The PD control cases and normal control subjects were matched first for age at death then disease duration (PD only) for comparison.
Results
Presenting signs in the parkin disease cases were hand or leg tremor often combined with dystonia. Mean age at onset was 34 years; all cases were compound heterozygous for mutations of parkin. Freezing of gait, postural deformity, and motor fluctuations were common late features. No patients had any evidence of cognitive impairment or dementia. Neuronal counts in the substantia nigra pars compacta revealed that neuronal loss in the parkin cases was as severe as that seen in PD, but relative preservation of the dorsal tier was seen in comparison with PD (P = .04). Mild neuronal loss was identified in the locus coeruleus and dorsal motor nucleus of the vagus, but not in the nucleus basalis of Meynert, raphe nucleus, or other brain regions. Sparse Lewy bodies were identified in 2 cases (brainstem and cortex).
Conclusions and Relevance
These findings support the notion that parkin disease is characterized by a more restricted morphologic abnormality than is found in PD, with predominantly ventral nigral degeneration and absent or rare Lewy bodies.
Autosomal recessive families of juvenile-onset or young-onset parkinsonism were reported from Japan almost 20 years ago.1 These patients had a relatively benign course, sleep benefit, and a sustained response to low doses of levodopa but with early development of interdose choreoathetosis and dystonia. Linkage studies pinpointed a locus on the long arm of chromosome 6 (6q25.2q27) and were followed by identification of a gene, which was named parkin.2,3 From the large number of parkin cases subsequently reported worldwide, a distinctive phenotype has been proposed, characterized by prominent leg tremor,4 foot dystonia,5,6 normosmia,7 and marked behavioral disturbances,5,8 although it has been hypothesized that the early age at onset rather than the presence of a parkin mutation9 is the critical determinant of the clinical picture. In some cases, there may be no detectable family history and rarely has onset in the fifth and sixth decades of life been reported.10-12
The parkin gene has 12 coding exons and the subsequent protein comprises 3 RING fingers13 separated by an in-between domain at the carboxyl terminal. Parkin plays an important role in mitochondrial functioning, as well as the ubiquitin proteasome system, where it acts as a ubiquitin E3 ligase.14 Impaired autophagy and mitophagy, protein accumulation, and mitochondrial dysfunction are the main mechanisms proposed in the development of parkinsonism due to mutations of parkin.15
Parkinson disease (PD) is characterized pathologically by severe loss of dopaminergic neurons in the substantia nigra (SN), with numerous cytoplasmic inclusions containing α-synuclein, known as Lewy bodies (LBs), in the surviving neurons. Autopsy reports of patients with parkinsonism carrying 2 mutations in the parkin gene described localized severe nigral degeneration with gliosis, mild neuronal loss, and depigmentation of the locus coeruleus (LC) but an absence of LBs in most cases (Table 1 and Table 2).16- 27
Table 1. Pathological Reports of Published Parkin Cases.
| Source | Yamamura et al,16 1998 | Mori et al,17 1998 | Hayashi et al,18 2000 | Van de Warrenburg et al,19 2001 | Farrer et al,20 2001 | Mori et al,21 2003 | Gouider-Khouja et al,22 2003 |
|---|---|---|---|---|---|---|---|
| Parkin status | Homozygous | Homozygous | Homozygous | Compound heterozygous | Compound heterozygous | Compound heterozygous | Homozygous |
| Sex | Female | Male | Male | Male | Male | Male | Male |
| Ethnicity | Japanese | Japanese | Japanese | Dutch | North American | Japanese | Tunisian |
| Siblings affected | 2 of 6 | 3 of 5 | 3 of 8 | 2 of 3 | O of 1 (+father) | 0 | 2 of 3 |
| Age at disease onset, y | 20 | 24 | 32 | 18 | 41 | 30 | 34 |
| Age at death, y | 52 | 62 | 70 | 75 | 52 | 47 | 47 |
| Disease duration, y | 33 | 38 | 38 | 57 | 11 | 17 | 13 |
| Cause of death | NA | Ileus | Pneumonia | Cardiac failure | Accident | NA | Cerebral abscess |
| Presenting symptom | Slowness | Foot tremor | Gait difficulty | Leg tremor | Gait difficulty | Gait difficulty | Hand tremor |
| SNpc depigmented | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Neuronal loss in SN | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| SN tier most depleted | VL | VL | VL | NA | NA | VL | NA |
| SN gliosis | Yes | Yes | Yes | Yes | NA | Yes | Yes |
| Neuronal loss in LC | Yes | Yes | Yes | No | Yes | Yes | Yes |
| Lewy bodies present | No | No | No | No | Yes | No | No |
| Tau deposition | NA | Yes | Yes | Yes | No | No | No |
Abbreviations: LC, locus coeruleus; NA, not available; SN, substantia nigra; SNpc, substantia nigra pars compacta; VL, ventrolateral.
Table 2. Additional Pathological Reports of Published Parkin Cases.
| Source | Sasaki et al,23 2004 | Pramstaller et a1,24 2005 | Orimo et al,25 2005 | Orimo et al,25 2005 | Torres et al,26 2011 | Miyakawa et al,27 2013 |
|---|---|---|---|---|---|---|
| Parkin status | Homozygous | Compound heterozygous |
Homozygous | Homozygous | Compound heterozygous |
Homozygous |
| Sex | Female | Male | Male | Male | Female | Female |
| Ethnicity | Japanese | Italian | Japanese | Japanese | Peruvian | Japanese |
| Siblings affected | 1 of 1 | NA | 3 | NA | 2 of 4 | 0a |
| Age at disease onset, y | 33 | 49 | 24 | 28 | About 15 | 61 |
| Age at death, y | 70 | 73 | 73 | 66 | NA | 72 |
| Disease duration, y | 37 | 23 | 49 | 38 | >40 | 11 |
| Cause of death | Pneumonia | Embolus | NA | NA | NA | Pneumonia |
| Presenting symptom | Gait difficulty | Arm tremor | Hand tremor | Tremor | NA | Gait difficulty |
| SNpc depigmented | Yes | Yes | Yes | Yes | NA | Yes |
| Neuronal loss in SN | Yes | Yes | Yes | Yes | Yes | Yes |
| SN tier most depleted | VL | NA | NA | NA | NA | NA |
| SN gliosis | Yes | Yes | Yes | Yes | NA | NA |
| Neuronal loss in LC | Yes | Yes | Yes | Yes | No | Yes |
| Lewy bodies present | Nob | Yes | No | No | No | Yes |
| Tau deposition | No | No | NA | NA | NA | Yes |
Abbreviations: LC, locus coeruleus; NA, not available; SN, substantia nigra; SNpc, substantia nigra pars compacta; VL, ventrolateral.
Parents were first cousins; mother possibly had Parkinson disease.
Basophilic inclusions in the pedunculopontine nucleus (α-synuclein and ubiquitin positive).
We conducted a detailed clinicopathologic study in 5 cases with compound heterozygous mutations of parkin to define the clinical features, late disease course, and pathologic lesion in parkin disease.
METHODS
MATERIALS
Cases with 2 confirmed mutations of parkin with clinical data and pathologic material were included. Three cases were identified from the Queen Square Brain Bank and 2 from the Dublin Brain Bank, where tissue is collected using ethically approved protocols and material stored under a license issued by the Human Tissue Authority. None had received a diagnosis of parkin disease in life, but they were genetically tested following suspicion raised on review of available clinical and family history data. Five PD control cases and 4 normal control cases matched first for age at death then disease duration (PD only) were also selected for comparison.
GENETIC, NEUROPATHOLOGIC, AND CLINICAL METHODS
Genomic DNA was extracted using standard methods from frozen brain tissue in all suspected cases. Parkin coding region and splice sites were screened for point mutations by polymerase chain reaction and subsequent bidirectional sequencing using BigDye Terminator version 3.1 (Applied Biosystems) sequencing chemistry. Exon rearrangements were detected by multiplex ligation-dependent probe amplification using the P051 and P052 Salsa MLPA Parkinson probe sets, according to the manufacturer’s instructions (MRC-Holland).
Brain tissue fixed with 10% buffered formalin was sampled, processed for histology, and stained according to standard protocols. Formalin-fixed paraffin-embedded tissue sections from cortical, subcortical, brainstem, and cerebellar regions were stained using routine histologic stains (hematoxylin and eosin and Luxol fast blue/cresyl violet) and supplemented by immunohistochemical staining using the following primary antibodies: glial fibrillary acidic protein, α-synuclein, tau (AT8), amyloid-β, ubiquitin, p62, neurofilaments, IC2, fused in sarcoma (FUS), and TAR DNA-binding protein 43 (full details are in eAppendix 1). All cases were assessed by an experienced neuropathologist who was blind to the diagnosis and graded regional neuronal loss and gliosis using a 4-point semiquantitative scale (0 = absent, 1 = mild, 2 = moderate, 3 = severe, based on previously published studies).28 α-Synuclein, tau, ubiquitin, p62, and amyloid-β immunoreactive structures were analyzed in selected brain areas. Lewy body pathology was assessed according to the recommendations of the third report of the Dementia with Lewy Bodies Consortium29 and also by Braak stage.30 Concomitant pathologies were assessed (eAppendix 1).
Detailed study of the severity of nigral neuronal loss was carried out in the PD and parkin cases only. A single, transverse 7-μm thick section of midbrain was taken at the level where the fascicles of the third cranial nerve emerge from the midbrain, allowing evaluation of pertinent nuclear groups at a defined level. These sections were stained with the Luxol fast blue/cresyl violet method; single-section counts of all neuromelanin-containing neurons, with or without a visible nucleus, in the SN pars compacta were obtained by using Image-Pro Plus software package (Media Cybernetics). All the counts were performed twice by the same investigator blinded to clinical data. Each SN pars compacta was outlined and further divided into ventral and dorsal tiers as described in detail elsewhere.31 Pars lateralis was excluded from the analysis because, in this region, there is a considerable mixing of neuronal types. The software automatically divided each examined area into a number of nonoverlapping counting squares of equal size of 300 μm × 300 μm, where all pigmented neurons were counted at ×200 magnification. The number of these squares was used to determine the surface area and, finally, the neuronal density was expressed as neurons per square millimeter. Single-section counting has been shown to be as reliable as the dissector method in evaluating the neuronal loss from SN.32
Clinical record review was undertaken in the parkin, PD, and control cases for details of disease presentation, response to medication, progression, and late features. Full case descriptions were summarized for the parkin cases.
STATISTICAL METHODS
Using a semiquantitative approach, we first compared the severity (ie, none, mild, moderate, or severe) of neuronal loss and gliosis among the 3 groups: parkin disease (5 cases), PD (5 cases), and control (4 cases). In each case, 9 brain areas were selected for examination. The grades of neuronal loss (or gliosis) in each brain area were pooled and an ordinal logistic regression model with a robust variance estimator was used to assess the increased odds of having more severe neuronal loss (or gliosis) and take into account clustering among individuals.
In the second analysis, only parkin and PD groups were compared with regard to the neuronal densities in the ventral, dorsal, and total (ventral and dorsal combined) nigral tiers. Neuronal counts in the SN were performed twice on each case (5 parkin and 5 PD). Intrarater reliability for the nigral neuronal counts was assessed with intraclass correlation. Intraclass correlation coefficients were greater than 0.80 for all the ratings performed, reflecting high reliability. The means of the 2 counts for each given nigral area were then plotted and assessed for normality. Independent-sample t tests or Mann-Whitney U tests were used depending on the distribution. P values of less than .05 were considered to indicate a significant trend. Linear regression was used to assess what proportion of the variation of each outcome variable was explained by each predictor variable (R2) and to quantify differences between individuals with parkin disease and those with PD. SPSS version 19 (SPSS Inc) and STATA version 11.2 (StataCorp) statistical programs were used to analyze the data.
RESULTS
The clinical features and genetic mutations in the 5 parkin cases are summarized in Table 3. The clinical description of the first parkin case is detailed here (descriptions of cases 2-5 are available in eAppendix 2 and Video 1 and Video 2 of case 5 in the online-only material).
Table 3. Clinical and Genetic Details of Parkin Disease Cases.
| Case | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Nationality | British | British | Irish | Irish | British |
| Sex | Female | Female | Female | Male | Male |
| Age at onset, y | 36 | 25 | 33 | 32 | 46 |
| Age at death, y | 86 | 62 | 60 | 68 | 82 |
| Disease duration, y | 50 | 37 | 27 | 36 | 36 |
| Presenting symptoms | Toes turning up, leg pain | Hand spasm and tremor | Hand and leg tremor | Tremor, gait difficulties | Leg tremor |
| Presenting signs | Foot dystonia, leg tremor | Hand tremor and dystonia | Hand and arm tremor | Tremor | Bilateral leg tremor |
| Posture | Kyphoscoliotic | Kyphoscoliotic | Stooped | Normal | Normal |
| Visual hallucinations | Yes, brief | No | No | No | No |
| Dementia | No | No | No | No | No |
| Late disease features | Falls, freezing of gait | Painful dystonia | Painful dystonia | Rapid on/off, dystonia | Falls, freezing of gait, pain |
| Initial diagnosis | Dystonia | YOPD | ET | YOPD | BTP |
| Final clinical diagnosis | PD | YOPD | PD | YOPD | PD |
| Siblings affected (No.) | No | Yes (1 of 5) | Yes (1) | Yes (2 of 7) | No |
| Mutation 1 | R275W | R275W | R275W | G430D | R275W |
| Mutation 2 | Del exon 6 | Pro113fs | G430W | Pro113fs | Del exon 6 |
Abbreviations: BTP, benign tremulous parkinsonism; ET, essential tremor; PD, Parkinson disease; YOPD, young-onset PD.
CASE 1: CLINICAL SUMMARY
At age 36 years, this woman noticed the toes on her left foot turning up. Initial examination revealed involuntary dorsiflexion of her left great toe associated with a coarse tremor of her left foot and leg. Focal dystonia was diagnosed, and she was treated with anticholinergic medication to good effect. Ten years later, a diagnosis of PD was considered when reduced arm swing and arm tremor were noted; however, levodopa treatment was not started until the age of 56 years, when she had an excellent response. While in her 70s, she began to experience falls and freezing of gait, but she still walked unaided when taking medication. Examination in the year she died revealed occasional tremor of both hands with bilateral rigidity and bradykinesia, and she reported that her medication (combined levodopa/carbidopa/entacapone, 150/37.5/200 mg, 4 times per day, and selegiline hydrochloride, 5 mg, twice daily) still improved her symptoms. In her last year of life, she had an episode of confusion and disorientation and experienced some visual hallucinations, but these were short lived. She was not demented and denied any memory problems. She died of ischemic heart disease 4 days following repair of a fractured neck of femur at age 86 years.
GENETIC ANALYSIS
Four different parkin mutations were detected: 2 missense changes (c.823C>T p.R275W; c.1289G>A, p.G430D), 1 frameshift deletion causing the premature introduction of a stop codon (c.337_376del; p.Pro113fs), and 1 whole-exon rearrangement (exon 6 deletion). All the identified mutations have been previously described and shown (when homozygous or compound heterozygous) to be associated with early-onset parkinsonism.
NEUROPATHOLOGY
Parkin -Linked Cases
Macroscopically, the changes in the parkin cases appeared identical to that of PD, with marked nigral (all cases) and LC (2 of the 5 cases) pallor. Table 4, Figure 1, and Figure 2 provide a summary of the histologic findings in the parkin cases, the most striking feature being the loss of pigmented neurons in the SN (moderate to severe); in all cases, the pattern was one of the ventral tier being most severely affected (Figure 1A and B). This was accompanied by mild to moderate cell loss in the LC with pigment incontinence. Other structures affected by neuronal loss included the dorsal motor nucleus of the vagus (DMV) (mild in 2 of 3 cases examined) and the cerebellar cortex (examined at the level of the dentate nucleus and the superior cerebellar peduncle), which showed mild Purkinje cell loss with empty baskets. In no other region examined was any appreciable neuronal loss evident including the nucleus basalis of Meynert (NBM) (4 cases examined; Figure 2A). Mild to moderate gliosis accompanied the neuronal loss previously described, but there was also evidence of gliosis (in the absence of detectable neuronal loss) in the raphe nucleus, NBM, striatum, globus pallidus, dentate nucleus, amygdala, hippocampus, and cerebral cortices (Figure 2B). None of the parkin disease cases looked similar to PD when immunochemistry for α-synuclein was performed. The results from 2 cases were negative for α-synuclein (cases 1 and 2): 1 had sparse Lewy neurites in the SN but no LBs (case 4) and 1 had sparse Lewy neurites and a total of 2 LBs (midbrain periaquaductal gray matter and transentorhinal cortex) (case 3). Case 5 had brainstem-predominant LB disease according to McKeith et al criteria29 but was unusual for PD because the severe loss of pigmented nigral neurons was accompanied by only very sparse LBs (Figure 1C and D). Mild neuronal loss in the LC was associated with moderate numbers of LBs (Figure 1E and F). Only 1 or 2 LBs were identified in the NBM, transentorhinal cortex, amygdala, and cingulate cortex sections, and the pattern of pathology did not conform well to the Braak PD staging scheme as the density of brainstem LBs did not show the expected increase when LB pathology extended beyond the brainstem.30
Table 4. Neuropathology of Parkin Disease Cases.
| Case | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Neuronal loss in SN | Moderate | Moderate | Severe | Severe | Severe |
| Neuronal loss in LC | Mild | Mild | Moderate | Moderate | Mild |
| Lewy bodies present | No | No | Yes | No | Yes |
| VNN density, neurons/mm2 | 5.55 | 12.83 | 2.60 | 7.67 | 11.11 |
| DNN density, neurons/mm2 | 16.72 | 21.55 | 20.88 | 15.45 | 21.23 |
| Total nigral neuronal density, neurons/mm2 | 12.21 | 18.25 | 12.53 | 12.63 | 16.13 |
| Ratio of VNN/DNN density | 0.33 | 0.60 | 0.12 | 0.50 | 0.52 |
| Aβ diffuse deposits | Severe | None | None | Severe | Mild |
| Aβ mature deposits | Mild | None | None | Severe | None |
| CAA | None | None | Mild | None | None |
| Braak & Braak AD stage | II | None | None | I | I |
| Small-vessel disease | Mild | Mild | None | None | Mild |
| TDP-43–positive inclusions | None | None | None | None | None |
Abbreviations: Aβ, amyloid-β; AD, Alzheimer disease; CAA, cerebral amyloid angiopathy; DNN, dorsal nigral neuronal; LC, locus coeruleus; SN, substania nigra; TDP-43, TAR DNA-binding protein 43; VNN, ventral nigral neuronal.
Figure 1. Histologic findings in the parkin cases.

Case 2 substantia nigra illustrates the more severe neuronal loss in the ventral tier (white arrow) compared with the dorsal tier (black arrow) (A), accompanied by severe gliosis (white arrow) in the ventral tier compared with the dorsal tier (black arrow) (B). Case 5 showed severe neuronal loss in the ventrolateral substantia nigra (C) and only sparse α-synuclein pathology (arrows) (D), with mild cell dropout in the locus coeruleus (E) accompanied by moderate numbers of Lewy bodies (arrows) and Lewy neurites (F). The scale bar represents 260 μm in parts A and B, 50 μm in parts C and D, 100 μm in parts E and F, and 25 μm in the inset in part F. Hematoxylin-eosin staining in parts A, C, and E; glial fibrillary acidic protein in part B; and α-synuclein in parts D and F.
Figure 2. Regional neuronal loss (A) and gliosis (B) in parkin, Parkinson disease (PD), and control cases.

A, The severity of neuronal loss in various brain regions in the 5 parkin, 5 PD, and 4 control cases (not all structures were available for examination in all cases [eg, the raphe was only available for examination in 3 parkin, 2 PD, and 3 control cases]). B, The severity of gliosis in various brain regions in the parkin, PD, and control cases. GP indicates globus pallidus; DMV, dorsal motor nucleus of the vagus; LC, locus coeruleus; NBM, nucleus basalis of Meynert; SN, substantia nigra.
There was either no (2 cases) or only mild (3 cases) deposition of hyperphosphorylated tau, which did not exceed Braak and Braak stage II pathology. Sparse tau immunoreactive neuropil threads were identified in the SN in 2 cases (cases 1 and 4) and in the LC in case 5. Tau pathology characteristic of other tauopathies, such as tufted astrocytes or astrocytic plaques, was not observed.
Ubiquitin and p62 stains highlighted small irregular neuronal intracytoplasmic inclusions in the cytoplasm of pigmented nigral neurons in 3 cases. These inclusions were not recognized in immunohistochemical preparations for α-synuclein, tau, IC2 (recognizing polyglutamine repeat–containing proteins), neurofilaments, TAR DNA-binding protein 43, or FUS, and they were also observed in 3 of 4 control cases, indicating that they are not disease specific.
PD and Control Cases
The pathologically confirmed PD cases were retrospectively reviewed, and the demographics and mean SN neuronal density measurements are given in Table 5. The clinical details were consistent with PD and none of the cases had a positive family history of parkinsonism. The pathologic findings were also classic for PD in all cases without the finding of other significant pathology.
Table 5. Comparison of Parkin and Parkinson Disease Cases.
| Mean (Range) |
||||
|---|---|---|---|---|
| Parkin | Parkinson Disease | P Value | Test | |
| Cases | 5 | 5 | ||
| Sex | ||||
| Male | 2 | 5 | ||
| Female | 3 | 0 | ||
| Age at onset, y | 33.6 (25-46) | 45.2 (36-64) | .09 | t test |
| Disease duration, y | 38.3 (27-50) | 26.1 (21-31) | .03a | MWU |
| Age at death, y | 71.9 (60-86) | 71.3 (61-81) | .84 | MWU |
| VNN density, neurons/mm2 | 8 (3-13) | 7.7 (6-11) | .89 | t test |
| DNN density, neurons/mm2 | 19.2 (15-22) | 14.3 (10-20) | .04a | t test |
| Total nigra neuronal density, neurons/mm2 | 14.3 (12-18) | 11.7 (9-14) | .12 | t test |
| Ratio of VNN/DNN | 0.41 (0.12-0.60) | 0.55 (0.39-0.77) | .26 | t test |
Abbreviations: DNN, dorsal nigral neuronal; MWU, Mann-Whitney U test; VNN, ventral nigral neuronal.
Significant at the P < .05 level.
The causes of death in the control cases were metastatic bowel cancer in 2 and myocardial infarction in 2. None had experienced any symptoms suggestive of parkinsonism or other neurodegenerative diseases. One control case was found to have sparse cortical and brainstem LBs without significant SN or LC neuronal loss. There was no clinical correlate to these findings; the patient was 81 years old at death and the pathologic diagnosis of incidental LB disease was made.
STATISTICAL ANALYSES
Figure 2 shows the distribution and severity of neuronal loss and gliosis in the 3 groups. The PD group had the greatest severity of neuronal loss and the control group had the least severe: after adjusting for age at death, the odds ratios of having an increased severity of neuronal loss were 1.2 (95% CI, 0.8-1.8) and 0.5 (95% CI, 0.4-0.7) for PD and control cases, respectively, relative to parkin disease (global P < .001, averaged over all brain areas examined). Gliosis also differed significantly between the 3 diagnoses (P = .02), with a 1.4 increased odds ratio (95% CI, 0.6-3.1) of having more severe gliosis in PD compared with parkin disease cases and a 0.4 decreased odds ratio (95% CI, 0.1-0.9) for control compared with parkin disease cases. Detailed statistical analysis in each region was limited by small case number, but the largest differences in severity were observed in the SN, LC, and DMV for neuronal loss (Figure 2A), as well as in the SN, LC, and NBM for gliosis (Figure 2B).
Table 5 and Figure 3 illustrate the neuronal densities in the different tiers of the SN in the PD and parkin disease cases. Analysis revealed a significantly greater mean cell density in the dorsal tier of the parkin disease cases (19.16 neurons/mm2) than in the PD cases (14.28 neurons/mm2) (P = .04; Table 5). The neuronal density of the ventral nigra did not differ between parkin and PD cases (P = .89). An unadjusted standard linear regression model found that diagnosis (parkin or PD) predicted a mean 4.88 neurons/mm2 (95% CI, 0.36-9.41; P = .04) higher dorsal SN density in parkin compared with PD cases.
Figure 3. Clustered box plot illustrating neuronal density in ventral, dorsal, and combined (total) nigral tiers in the parkin and Parkinson disease (PD) cases.
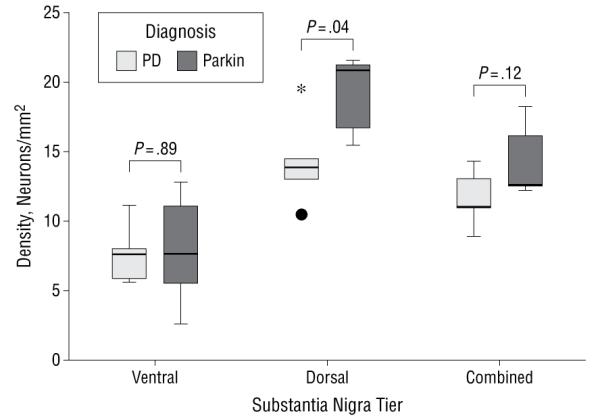
The asterisk indicates an extreme score (ie, the value is more than 3 box lengths from the upper quartile) and the black dot indicates an outlier (ie, the value is more than 1.5 box lengths from the lower quartile).
COMMENT
Parkin disease has recently been described as a nigropathy owing to its restricted pathology.33 Previous neuropathologic reports described marked SN neuronal loss (5 noted that the ventrolateral tier was most severely affected16- 18,21,23), mild to moderate neuronal loss of the LC (11 of 13 cases), limited tau pathology27 (few neurofibrillary tangles17,18 and occasional tau-positive astrocytes19), and LBs staining positively with α-synuclein in only 3 cases.20,24,27 To our knowledge, this is the largest and most detailed neuropathologic study of parkin disease to date, and our comparison with PD and control cases led us to consider parkin disease as a ventral-predominant nigropathy with additional involvement of the LC. In contrast to PD, marked neuronal loss of the DMV, NBM, and the midbrain raphe was not found; LBs were rare and, when observed, were sparse.
Lewy bodies have been observed in other genetic forms of parkinsonism: neurodegeneration with brain iron accumulation associated with the PLA2G6 mutation (NBIA type 2 or PARK14)34 and recently in a young patient (age, 39 years) with compound heterozygous PINK1 mutations.35 In PARK8 (leucine-rich repeat kinase 2), LBs appear to be more commonly found than in parkin disease36 (especially in those with the G2019S mutation); however, limited nigral and LC neuronal loss in the absence of other pathology is also described. Tau pathology has been reported in a large proportion of leucine-rich repeat kinase 2 cases,37 whereas our series combined with existing reported cases would suggest there is much less tau burden in parkin disease.
The disparity between the severity of nigral loss with sparse or no LBs that we observed supports the notion that abnormal α-synuclein deposition is not an integral component of the pathology of parkin disease; therefore, it is innately different from PD. It can be argued that the LBs we identified were incidental owing to their paucity in comparison with PD. Case 5 died at age 82 years, an age at which the finding of incidental LB disease post mortem is known to occur in at least 15% of normal control subjects,38- 41 and case 3 died at age 61 years and only 2 LBs were identified after thorough study. Incidental LBs were also identified in 1 of our control cases with age at death of 81 years. Incidental LB disease may also explain 2 of the LB-positive parkin cases described in the literature.24,27 Age at disease onset is another possible factor that has been linked to the finding of LBs post mortem in parkin disease (Christine Klein, MD, oral communication, June 2012). Combining our 5 cases with those previously reported and comparing those that had LBs at post mortem with those that did not, there was a significant difference in age at disease onset between the groups (mean age at onset: LB-positive cases, 46 years; LB-negative cases, 27 years; P < .001; t test). Parkin cases with LBs discovered at pathologic examination were on average 19 years older at presentation.
An incomplete loss of ubiquitin ligase activity, recognized to occur with certain missense mutations of parkin,42 might permit the formation of LBs. In vitro studies have shown that different mutant parkin isoforms have different levels of enzymatic activity, with certain point mutations exhibiting only partial loss of function or possibly toxic gain of function.42 This is exemplified by the p.R275W mutation, which retains a functional RING domain (critical for E3 ligase activity) and has preserved ubiquitylation activity42 and the capacity to produce aggresomes.43 Alternatively, it has been proposed that parkin mutations resulting in loss of RING domain function are unable to form LBs as α-synuclein cannot be ubiquitinated.44 The p.R275W missense mutation was present in 4 of our cases (2 of which had LBs) and also in the case with LBs reported by Farrer and colleagues.20 We postulate that if the p.R275W-mutated protein is able to ubiquitinate some of its substrates to form an aggresome, then LBs could occur.
Another consideration is that 2 of the LB-positive cases20,24 descended from pedigrees not reflective of simple autosomal recessive inheritance—parkinsonism was seen in consecutive generations.
All of our cases had the characteristic clinical features felt to be particular for parkin disease—early-onset tremor often combined with dystonia and sustained response to dopaminergic medication. All experienced troublesome severe l-dopa–induced motor fluctuations. Freezing of gait and painful off-period dystonia were other common features. Three patients were described as having abnormal postures, either stooped forward or laterally flexed (scoliotic), which may reflect the chronicity of truncal dystonia throughout their disease course. The lack of cognitive and psychiatric features experienced in our patients might reflect sparing of the NBM and cerebral cortex. Only 1 patient experienced brief visual hallucinations at the end of her life (while taking levodopa and selegiline therapy), and no patients were reported as having delirium, amnesia, or dementia, despite their long disease duration (range, 27-50 years). This is important for clinical practice and may allay some patient fears in considering the long-term aspects of their parkin disease and its management.
In conclusion, parkin disease appears clinically and pathologically distinct from PD. It can be conceptualized as an early-onset, slowly progressive, levodopa-responsive parkinsonism without hallucinations or dementia due to neuronal loss in the ventral SN.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported by the Reta Lila Weston Trust for Medical Research, the Dublin Brain Bank, the Wellcome Trust/MRC Joint Call in Neurodegeneration award WT089698, Parkinson’s UK, the Multiple System Atrophy Trust, and Alzheimer’s Research UK, as well as partly supported by the National Institute for Health Research Biomedical Research Unit based at University College London Hospitals, University College London.
Additional Contributions: We thank Catherine Strand, MSc, Tammaryn Lashley, PhD, and Karen Shaw, RMN, for their assistance in this project.
Footnotes
Conflict of Interest Disclosures: Dr Doherty’s work is funded by the Reta Lila Weston Trust for Medical Research. She is the beneficiary of an innovation grant from Parkinson’s UK, and in the past 3 years, she has received travel compensation to attend scientific meetings from Teva Pharmaceuticals, Ipsen, Novartis, GlaxoSmithKline, and Orion. Dr Silveira-Moriyama has received honoraria from Britannia Pharmaceuticals; grants from Parkinson’s UK, the Parkinson Disease Foundation, the Reta Lila Weston Trust, FAPESP, and the University of Campinas; and travel support from UCB Pharmaceuticals, Genus Pharmaceuticals, and Abbott Laboratories. She is employed by the Reta Lila Weston Institute of Neurological Studies and the University of Campinas. Dr Parkkinen is a career development fellow funded by the Monumental Trust Award from Parkinson’s UK. Dr Lynch has received honorarium from Abbott Laboratories, Boehringer Ingelheim, Lundbeck, and Orion; educational grants from Bayer-Schering, Biogen Idec, Lundbeck, and Medtronic; and grants from the Irish Institute of Clinical Neuroscience, and Mater College, as well as PRTL1 funding. He serves on advisory boards for Abbott Laboratories, Novartis, UCB Pharmaceuticals, Teva Pharmaceuticals, Merck Serono, and Biogen Idec. Dr Revesz’s work is supported by grants from the Multiple System Atrophy Trust, Parkinson’s UK, and Alzheimer’s Research UK. Dr Lees serves as a consultant for Genus Pharmaceuticals and advisory board member for Novartis, Teva Pharmaceuticals, Boehringer Ingelheim, GlaxoSmithKline, Ipsen, Lundbeck, Allergan, Orion, BIAL Pharmaceuticals, Noscira, and Roche. Dr Lees has received honoraria from Novartis, Teva Pharmaceuticals, Meda, Boehringer Ingelheim, GlaxoSmithKline, Ipsen, Lundbeck, Allergan, Orion, BIAL Pharmaceuticals, Noscira, and Roche, as well as grants from the PSP Association and the Reta Lila Weston Trust for Medical Research. Dr Holton’s work is supported by the Reta Lila Weston Trust for Medical Research, Parkinson’s UK, the Multiple System Atrophy Trust, Alzheimer’s Research UK, and the National Institute for Health Research Biomedical Research Unit at University College London Hospitals, University College London.
Disclaimer: The views expressed are those of the authors and not necessarily those of the UK National Health Service, the National Institute for Health Research, or the Department of Health.
Previous Presentation: This paper was presented at the 16th International Congress of Parkinson’s Disease and Movement Disorders; June 20, 2012; Dublin, Ireland.
REFERENCES
- 1.Ishikawa A, Tsuji S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile parkinsonism. Neurology. 1996;47(1):160–166. doi: 10.1212/wnl.47.1.160. [DOI] [PubMed] [Google Scholar]
- 2.Matsumine H, Saito M, Shimoda-Matsubayashi S, et al. Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25.2-27. Am J Hum Genet. 1997;60(3):588–596. [PMC free article] [PubMed] [Google Scholar]
- 3.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 4.Khan NL, Brooks DJ, Pavese N, et al. Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain. 2002;125(pt 10):2248–2256. doi: 10.1093/brain/awf237. [DOI] [PubMed] [Google Scholar]
- 5.Khan NL, Graham E, Critchley P, et al. Parkin disease: a phenotypic study of a large case series. Brain. 2003;126(pt 6):1279–1292. doi: 10.1093/brain/awg142. [DOI] [PubMed] [Google Scholar]
- 6.Khan NL, Horta W, Eunson L, et al. Parkin disease in a Brazilian kindred: manifesting heterozygotes and clinical follow-up over 10 years. Mov Disord. 2005;20(4):479–484. doi: 10.1002/mds.20335. [DOI] [PubMed] [Google Scholar]
- 7.Khan NL, Katzenschlager R, Watt H, et al. Olfaction differentiates parkin disease from early-onset parkinsonism and Parkinson disease. Neurology. 2004;62(7):1224–1226. doi: 10.1212/01.wnl.0000118281.66802.81. [DOI] [PubMed] [Google Scholar]
- 8.Lohmann E, Periquet M, Bonifati V, et al. French Parkinson’s Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson’s Disease. How much phenotypic variation can be attributed to parkin genotype? Ann Neurol. 2003;54(2):176–185. doi: 10.1002/ana.10613. [DOI] [PubMed] [Google Scholar]
- 9.Kim HJ, Kim HJ, Lee JY, et al. Phenotype analysis in patients with early onset Parkinson’s disease with and without parkin mutations. J Neurol. 2011;258(12):2260–2267. doi: 10.1007/s00415-011-6110-1. [DOI] [PubMed] [Google Scholar]
- 10.Lücking CB, Dürr A, Bonifati V, et al. French Parkinson’s Disease Genetics Study Group European Consortium on Genetic Susceptibility in Parkinson’s Disease. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 11.Klein C, Pramstaller PP, Kis B, et al. Parkin deletions in a family with adultonset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol. 2000;48(1):65–71. [PubMed] [Google Scholar]
- 12.Nichols WC, Pankratz N, Uniacke SK, et al. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson’s disease families. J Med Genet. 2002;39(7):489–492. doi: 10.1136/jmg.39.7.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hristova VA, Beasley SA, Rylett RJ, Shaw GS. Identification of a novel Zn2_-binding domain in the autosomal recessive juvenile Parkinson-related E3 ligase parkin. J Biol Chem. 2009;284(22):14978–14986. doi: 10.1074/jbc.M808700200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Z, Wood NW. Cell death pathways in Parkinson’s disease: role of mitochondria. Antioxid Redox Signal. 2009;11(9):2135–2149. doi: 10.1089/ars.2009.2624. [DOI] [PubMed] [Google Scholar]
- 15.Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta. 2011;1813(4):623–633. doi: 10.1016/j.bbamcr.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamura Y, Kuzuhara S, Kondo K, et al. Clinical, pathologic and genetic studies on autosomal recessive early-onset parkinsonism with diurnal fluctuation. Parkinsonism Relat Disord. 1998;4(2):65–72. doi: 10.1016/s1353-8020(98)00015-7. [DOI] [PubMed] [Google Scholar]
- 17.Mori H, Kondo T, Yokochi M, et al. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology. 1998;51(3):890–892. doi: 10.1212/wnl.51.3.890. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi S, Wakabayashi K, Ishikawa A, et al. An autopsy case of autosomal recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. 2000;15(5):884–888. doi: 10.1002/1531-8257(200009)15:5<884::aid-mds1019>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 19.van de Warrenburg BPC, Lammens M, Lücking CB, et al. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology. 2001;56(4):555–557. doi: 10.1212/wnl.56.4.555. [DOI] [PubMed] [Google Scholar]
- 20.Farrer M, Chan P, Chen R, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50(3):293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- 21.Mori H, Hattori N, Mizuno Y. Genotype-phenotype correlation: familial Parkinson disease. Neuropathology. 2003;23(1):90–94. doi: 10.1046/j.1440-1789.2003.00476.x. [DOI] [PubMed] [Google Scholar]
- 22.Gouider-Khouja N, Larnaout A, Amouri R, et al. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family: clinical, genetic and pathological study. Parkinsonism Relat Disord. 2003;9(5):247–251. doi: 10.1016/s1353-8020(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology. 2004;63(4):678–682. doi: 10.1212/01.wnl.0000134657.25904.0b. [DOI] [PubMed] [Google Scholar]
- 24.Pramstaller PP, Schlossmacher MG, Jacques TS, et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58(3):411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- 25.Orimo S, Amino T, Yokochi M, et al. Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord. 2005;20(10):1350–1353. doi: 10.1002/mds.20594. [DOI] [PubMed] [Google Scholar]
- 26.Torres LCM, Mata I, Mazzetti PE, et al. A novel PARK2 mutation in a Peruvian family: clinical and pathological characteristics. Mov Disord. 2011;26(suppl 2):S311–S312. doi: 10.1016/j.parkreldis.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyakawa S, Ogino M, Funabe S, et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov Disord. doi: 10.1002/mds.25346. doi:10.1002/mds.25346. [DOI] [PubMed] [Google Scholar]
- 28.Ozawa T, Paviour D, Quinn NP, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004;127(pt 12):2657–2671. doi: 10.1093/brain/awh303. [DOI] [PubMed] [Google Scholar]
- 29.McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 30.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 31.Halliday G. Substantia nigra and locus coeruleus. In: Paxinos G, Juergen KM, editors. The Human Nervous System. 2nd ed. Elsevier Academic Press; London, England: 2004. pp. 449–463. [Google Scholar]
- 32.Ma SY, Collan Y, Röyttä M, Rinne JO, Rinne UK. Cell counts in the substantia nigra: a comparison of single section counts and disector counts in patients with Parkinson’s disease and in controls. Neuropathol Appl Neurobiol. 1995;21(1):10–17. doi: 10.1111/j.1365-2990.1995.tb01023.x. [DOI] [PubMed] [Google Scholar]
- 33.Ahlskog JE. Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson’s disease. Parkinsonism Relat Disord. 2009;15(10):721–727. doi: 10.1016/j.parkreldis.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paisán-Ruiz C, Li A, Schneider SA, et al. Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol Aging. 2012;33(4):814–823. doi: 10.1016/j.neurobiolaging.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samaranch L, Lorenzo-Betancor O, Arbelo JM, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133(pt 4):1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- 36.Cookson MR, Hardy J, Lewis PA. Genetic neuropathology of Parkinson’s disease. Int J Clin Exp Pathol. 2008;1(3):217–231. [PMC free article] [PubMed] [Google Scholar]
- 37.Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord. 2012;27(7):831–842. doi: 10.1002/mds.24962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibb WRG, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51(6):745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 40.Parkkinen L, Soininen H, Laakso M, Alafuzoff I. Alpha-synuclein pathology is highly dependent on the case selection. Neuropathol Appl Neurobiol. 2001;27(4):314–325. doi: 10.1046/j.0305-1846.2001.00342.x. [DOI] [PubMed] [Google Scholar]
- 41.Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009;68(7):816–822. doi: 10.1097/NEN.0b013e3181ac10a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sriram SR, Li X, Ko HS, et al. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14(17):2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- 43.Cookson MR, Lockhart PJ, McLendon C, O’Farrell C, Schlossmacher M, Farrer MJ. RING finger 1mutations in Parkin produce altered localization of the protein. Hum Mol Genet. 2003;12(22):2957–2965. doi: 10.1093/hmg/ddg328. [DOI] [PubMed] [Google Scholar]
- 44.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6(7):652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.