

Abstract
Objective:
To report the clinical and radiologic findings of children with NMDA receptor (NMDAR) antibodies and white matter disorders.
Method:
Ten children with significant white matter involvement, with or without anti-NMDAR encephalitis, were identified from 46 consecutive NMDAR antibody–positive pediatric patients. Clinical and neuroimaging features were reviewed and the treatment and outcomes of the neurologic syndromes evaluated.
Results:
Three distinct clinicoradiologic phenotypes were recognized: brainstem encephalitis (n = 3), leukoencephalopathy following herpes simplex virus encephalitis (HSVE) (n = 2), and acquired demyelination syndromes (ADS) (n = 5); 3 of the 5 with ADS had myelin oligodendrocyte glycoprotein as well as NMDAR antibodies. Typical NMDAR antibody encephalitis was seen in 3 patients remote from the first neurologic syndrome (2 brainstem, 1 post-HSVE). Six of the 7 patients (85%) who were treated acutely, during the original presentation with white matter involvement, improved following immunotherapy with steroids, IV immunoglobulin, and plasma exchange, either individually or in combination. Two patients had escalation of immunotherapy at relapse resulting in clinical improvement. The time course of clinical features, treatments, and recoveries correlated broadly with available serum antibody titers.
Conclusion:
Clinicoradiologic evidence of white matter involvement, often distinct, was identified in 22% of children with NMDAR antibodies and appears immunotherapy responsive, particularly when treated in the acute phase of neurologic presentation. When observed, this clinical improvement is often mirrored by reduction in NMDAR antibody levels, suggesting that these antibodies may mediate the white matter disease.
NMDA receptor antibody (NMDAR-Ab) encephalitis is characterized by seizures, movement disorders, and psychiatric symptoms.1 It is now recognized that NMDAR-Ab–mediated neurologic syndromes can range from monosymptomatic and partial phenotypes to the full-blown “anti-NMDAR encephalitis.”1 Despite the severity of the disease in many patients, conventional MRI of the brain is frequently normal, often referred to as the clinical-radiologic paradox.1 When abnormal, MRI usually reveals discrete lesions that are predominantly subtle and nonenhancing, often reverse spontaneously, and are not usually restricted to the white matter1; thus, they appear clinically and radiologically different from demyelinating syndromes such as multiple sclerosis (MS).
Recently, we observed that 1 patient, initially reported in the UK cohort of NMDAR-Ab encephalitis,2 developed MS, and a further patient with MS subsequently developed NMDAR-Ab encephalitis. In addition, in a systematic evaluation of glial, myelin, and neuronal-directed antibodies in children with a range of demyelination syndromes, we identified 2 patients with NMDAR-Ab.3 These observations, together with existing case reports of NMDAR-Ab in patients with neuromyelitis optica spectrum disorder,4 acute demyelinating encephalomyelitis (ADEM),5 or MS,6 raise questions regarding the clinical relevance, potential pathogenic effect, and direct contribution of NMDAR-Abs to neurologic syndromes that involve the white matter.
Here, we report a group of children with NMDAR-Abs and clinical and/or radiologic evidence of a white matter disorder, identified from a larger cohort of children with NMDAR-Abs, and describe their clinical course in relation to (or independent of) features of NMDAR-Ab encephalitis.
METHODS
Ten children with a clinical and/or radiologic evidence of white matter involvement were identified from a cohort of 46 pediatric patients, 18 years of age and younger, with a range of NMDAR-Abs–associated neurologic syndromes. Between 2009 and 2013, patients were referred, based on clinician decision, from 6 pediatric neurology centers to the Clinical Neuroimmunology Service at the Oxford Radcliffe Hospital Trust for serum and/or CSF NMDAR antibody testing and/or advice on further antibody testing. As antibodies to myelin oligodendrocyte glycoprotein (MOG) and aquaporin-4 (AQP4) have been associated with demyelinating disorders,7 sera from all 10 cases and the other 36 NMDAR-Ab–positive patients were tested for both these antibodies. NMDAR, AQP4, and MOG antibodies were measured using cell-based assays in routine clinical use (sera at 1:20 dilution for NMDAR and AQP4 and 1:160 for MOG; CSF at 1:2 dilution for NMDAR) as previously described.2,8,9 For these cell-based assays, the binding of serum immunoglobulin G to the surface of human embryonic kidney cells, transfected with complementary DNA encoding the auto-antigens (MOG courtesy of M. Reindl, Innsbruck), was visualized using a fluorescence-labeled Alexafluor 568 secondary antibody (Molecular Probes, Eugene, OR), and the results were assessed by at least 2 independent observers (Y.H., L.J., P.W.). Clinical information and neuroimaging were reviewed (Y.H., M.A., A.S., M.L.). The outcomes, as measured by the range of difficulties the patients were experiencing, were retrieved from the patient's medical records or, if unavailable, were obtained directly from the patient's primary pediatric neurologist. Full recovery was defined by the absence of medical, educational, and social concerns reported from parents, school, and the patient's primary clinician.
Standard protocol approvals, registrations, and patient consents.
Ethical approval for this study was from the Oxfordshire Regional Ethical Committee A (07/Q1604/28).
RESULTS
Out of a total of 46 patients identified consecutively from 6 centers, 10 patients (cases 1–10; 5 male; age range 1.3–18 years, median 8.5 years; see table 1) were identified with a significant white matter involvement. They were classified as having 3 neurologic syndromes: brainstem encephalitis (n = 3), herpes simplex virus encephalitis (HSVE) relapsing with a distinct leukoencephalopathy (n = 2), and acquired demyelination syndromes (ADS) (n = 5). Their clinical features, imaging, and treatment responses are described below and summarized in table 1 and figures 1 and 2. The remaining 36 NMDAR-Ab–positive patients had minimal or absent white matter involvement; 28 of these had typical NMDAR-Ab encephalitis (cases 11–38; 8 male; age range 2–17 years, median 11 years; see table 2). Of the remaining 8 patients, 1 presented with a neurologic relapse following HSVE, and 7 had a partial phenotype (cases 39–46; 2 male; age range 1–18 years, median 11 years; see table 3). Only 1 patient, a 15-year-old girl with classical NMDAR-Ab encephalitis, had an ovarian teratoma. NMDAR-Abs were positive in the CSF in 10 of 11 patients tested with typical NMDAR-Ab encephalitis, in 2 of 3 tested with the partial phenotype, and in all 3 tested with white matter syndromes; serum levels were higher than CSF in all paired samples. All patients were empirically investigated by their physicians to exclude a range of infective, alternative inflammatory, and neurometabolic etiologies, of which none were identified. Cases 11–23,10 case 39,11 and cases 40–4212 have been previously described.
Table 1.
Clinical and paraclinical features of 10 NMDAR antibody–positive patients with distinct clinicoradiologic white matter syndrome
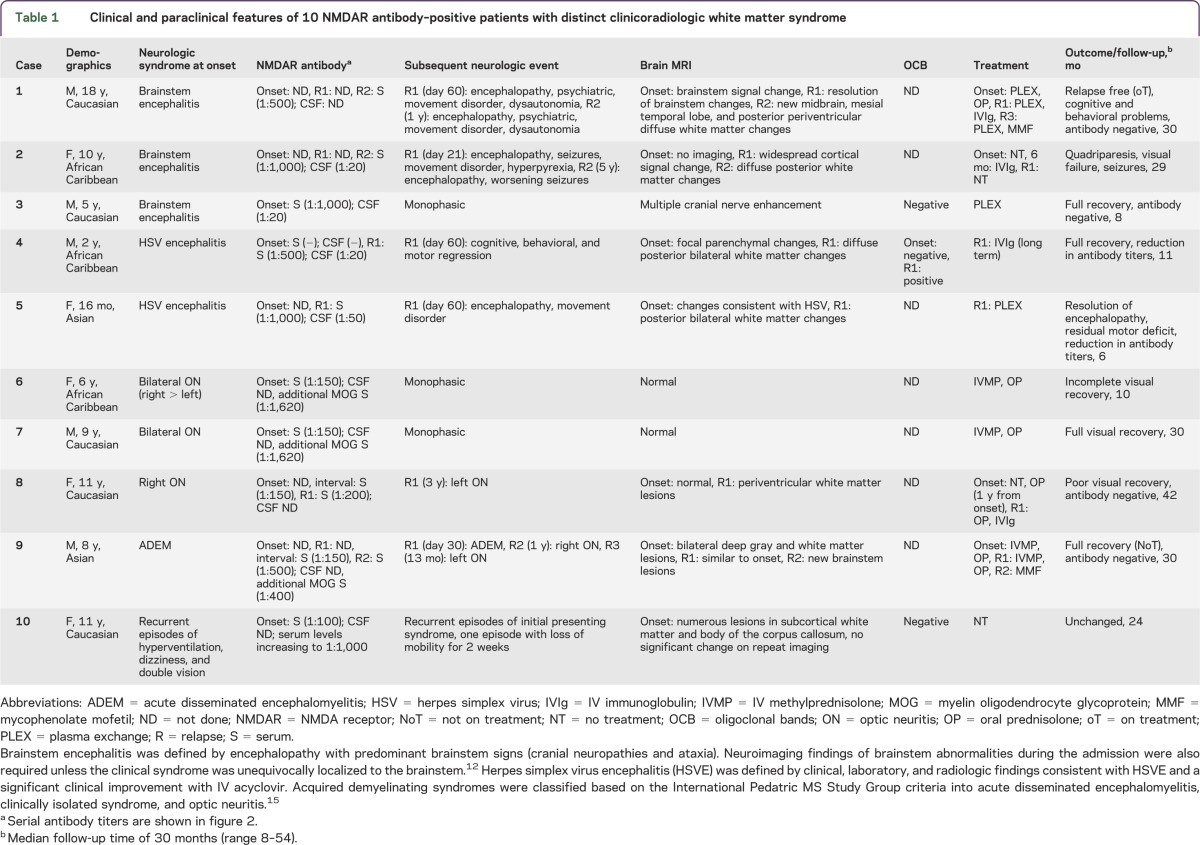
Figure 1. Radiologic features of patients with white matter syndromes in association with NMDA receptor antibodies.

(A, B) Case 1, who presented with a brainstem syndrome. Axial T2 fluid-attenuated inversion recovery (FLAIR) images showing hyperintensity in the midbrain and mesial temporal lobe (arrows in A), which subsequently resolved on follow-up imaging performed at 2 months when the patient had relapsed with a polysymptomatic encephalopathy (B). (C, D) Case 2, who presented initially with a brainstem syndrome followed by periods of recurrent encephalopathy. Axial T2-weighted images 6 months after her initial brainstem syndrome revealed diffuse global cortical atrophy (C). Imaging during an encephalopathic episode 5 years later revealed new white matter changes (arrow in D) and further atrophy. This radiologic feature of leukoencephalopathy progressed on subsequent imaging (not shown). (E–H) Two patients with neurologic syndromes following herpes simplex virus encephalitis (HSVE). Serial axial T2 FLAIR images of case 4 (E–G) showing localized cortical changes in the left thalamus and occipital lobe (arrows in E) on initial imaging that progressed, demonstrating significant bilateral white matter signal changes in the parieto-occipital region 2 months later (arrows in F) when the patient presented with significant worsening of cognitive, behavioral, and motor regression. Following treatment and improvement of symptoms, follow-up neuroimaging 2 months later demonstrated a significant resolution of this white matter change (G). Axial T2-weighted image of case 5 at relapse 2 months after HSVE showing extensive bilateral but asymmetrical cystic encephalomalacia centered on the temporo-insular regions (only parietal changes shown) characteristic of HSVE, but in addition demonstrating the characteristic leukoencephalopathic changes seen globally (arrows in H). (I–L) Patients presenting with acquired demyelinating syndrome (ADS). At time of relapse with left optic neuritis, brain imaging in case 8 showed subtle periventricular white matter signal change on a T2-weighted image (arrow demonstrating one periventricular lesion in I). Imaging of case 9 showing midbrain (arrow in J), capsular (arrow in K), and thalamic changes (not shown) on axial T2 FLAIR. Axial T2 FLAIR image of case 10 demonstrating patchy subcortical white matter changes (L).
Figure 2. NMDAR-Ab levels, clinical syndromes, and therapy in 8 informative patients with white matter syndromes in association with NMDAR-Ab.

(A–C) Cases 1–3 presented initially with brainstem encephalitis; Cases 1 and 2 relapsed with encephalopathy, psychiatric features, movement disorder, and dysautonomia. Case 2 had additional seizures. In both, the diagnosis of NMDAR-Ab encephalitis was made at the time of relapse. Case 3 had a monophasic illness and did not have any of the clinical characteristics of NMDAR-Ab encephalitis. (D, E) Cases 4 and 5 presented with herpes simplex virus encephalitis (HSVE) and then had a neurologic relapse, which correlated with raised NMDAR-Abs in both serum and CSF and demonstrated a clinical response to immunotherapy with reduction of antibody levels. (F) Case 8 presented with optic neuritis and poor visual recovery, which prompted a neuroinflammatory screen and the identification of the antibody positivity. She only received a course of steroids 1 year into her illness and 3 years later had a neurologic relapse. (G) Case 9 presented initially with 2 episodes of acute disseminated encephalomyelitis (ADEM) and relapsed at 1 year with optic neuritis. He was also MOG-Ab positive, which remain detectable even when NMDAR-Abs are no longer detectable and when the patient had clinically recovered. (H) Case 10 had recurrent episodes of hyperventilation, dizziness, and double vision; did not receive any treatment; and both her clinical and radiologic features remained unchanged. Ab = antibody; CSF Ab × 10 = NMDAR antibody titers in CSF (all between 1:20 and 1:50) multiplied by 10 to provide visibility in comparison to serum levels; IVIg = IV immunoglobulin; IVMP = IV methylprednisolone; LeukoE = radiologic leukoencephalopathy; Lt = left; MMF = mycophenolate mofetil; NMDAR = NMDA receptor; ON = optic neuritis; PLEX = plasma exchange; Rt = right; Serum Ab titer = titer measured by endpoint dilution.
Table 2.
Clinical and paraclinical features in patients with NMDAR-Ab encephalitis
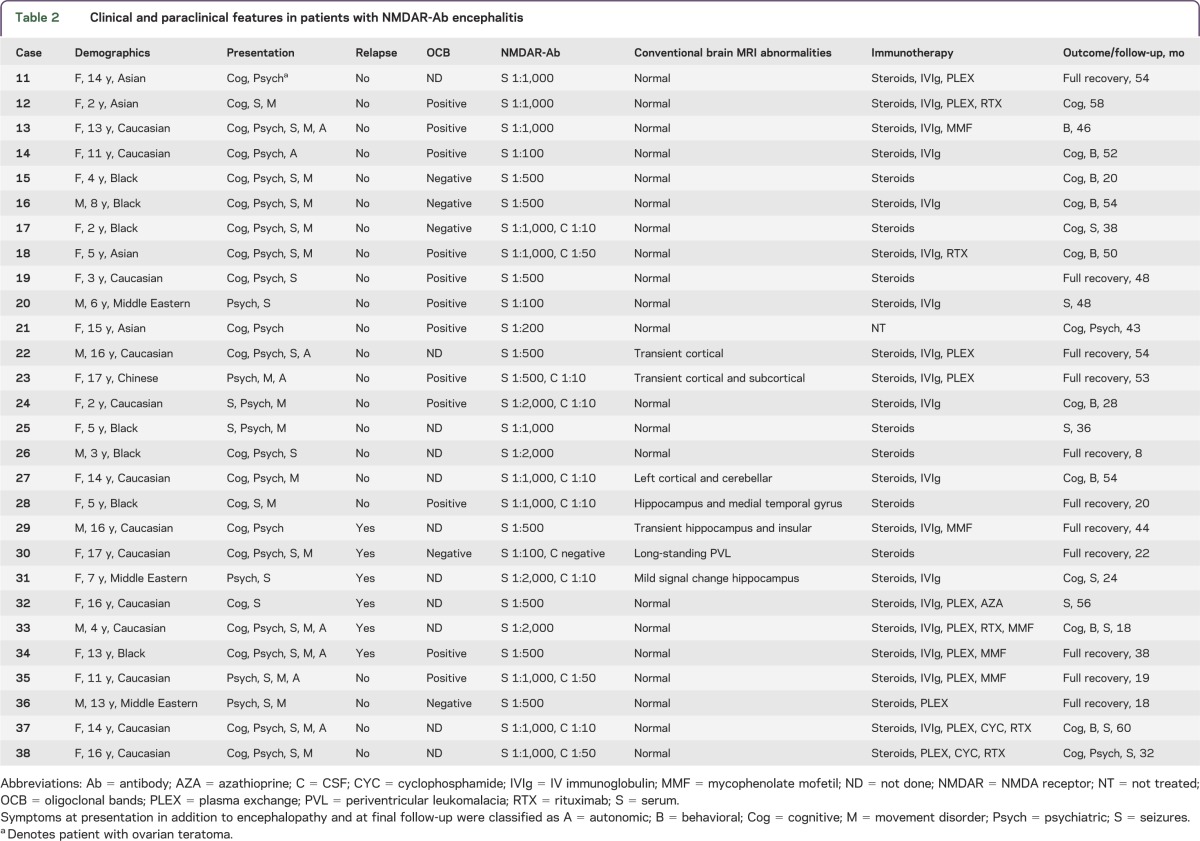
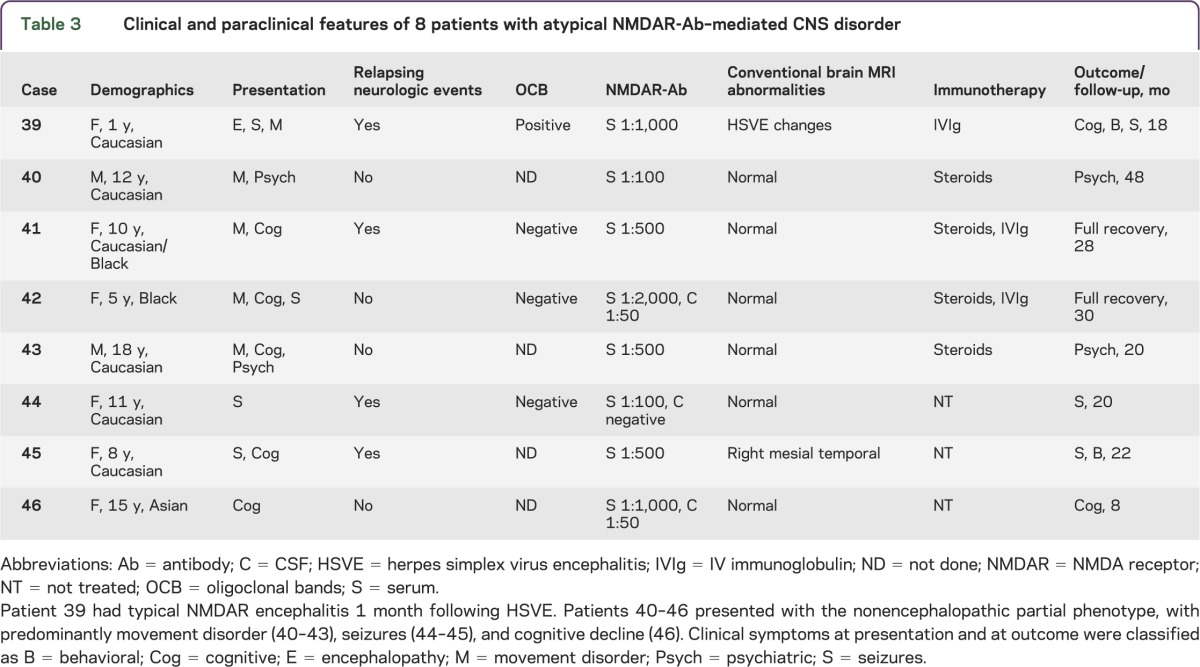
Table 3.
Clinical and paraclinical features of 8 patients with atypical NMDAR-Ab–mediated CNS disorder
Brainstem encephalitis.
Three patients had encephalopathy, predominant brainstem signs (cranial neuropathies and ataxia), and neuroimaging findings of brainstem abnormalities, or a clinical syndrome unequivocally localized to the brainstem.13 Case 1 (M, age 18) had brainstem changes at presentation (figure 1A) but evolved to a polysymptomatic encephalopathy, with resolution of the brainstem changes (figure 1B). NMDAR-Abs were only detected a year later after a relapse but fell with immunotherapies with a good clinical response (figure 2A). Case 2 (F, age 10) presented 3 weeks after first symptoms when her encephalopathy worsened with intractable seizures, movement disorder, and hyperpyrexia. She had recurrent periods of encephalopathy (figure 2B) associated with radiologic features of a leukoencephalopathy (figure 1D) that progressed over the following 5 years (figure 2B), when serum and CSF NMDAR-Abs were first identified. She was not given immunotherapies and is substantially impaired (table 1). Case 3 (M, age 5), the most recent patient, presented with brainstem signs and cranial nerve enhancement; NMDAR-Abs were identified within a few weeks of onset. He made a full recovery with steroids and plasma exchange (PLEX) (figure 2C). All 3 patients were negative for GQ1b antibodies.
HSVE relapse.
Since we and others recognized a relationship between HSVE relapses and NMDAR-Abs,11,14–16 we have identified 2 additional patients, cases 4 and 5 (M, age 2; F, age 16 months), with NMDAR-Abs in serum and CSF (table 1) during neurologic relapses with substantial leukoencephalopathy on imaging (figure 1, E–H) occurring within 2 months of HSVE (headache, fever, seizures, typical imaging, and CSF herpes simplex virus [HSV] PCR positivity). Case 4 presented with cognitive, behavioral, and motor decline, and case 5 presented with a further encephalopathic illness with new-onset movement disorder. Both patients had negative HSV PCRs in the CSF at relapse. Both were treated with IV immunoglobulin (IVIg) or PLEX, which resulted in clinical improvement and reduction of NMDAR-Ab titers (figure 2, D and E). A significant radiologic improvement was observed in case 4 (figure 1G). Other potential causes of leukoencephalopathies, such as neurometabolic conditions, were excluded.
Acquired demyelination syndromes.
Five patients presented with ADS classified according to the International Pediatric MS Study Group criteria.17 Three initially had optic neuritis. Case 6 (F, age 6) and case 7 (M, age 9), with bilateral optic neuritis, were treated with IV methylprednisolone followed by a weaning course of oral prednisolone. Case 8 (F, age 11) presented with right optic neuritis and poor visual recovery; she only received a course of oral steroids 1 year later and relapsed 3 years later with left optic neuritis and new brain lesions (figures 1I and 2F). Case 9 (M, age 8) presented with ADEM, relapsed at 1 month with a second encephalopathic illness and imaging evidence of new brainstem lesions (figure 1J), and developed right and left optic neuritis at 12 and 13 months, respectively. At this stage, he was treated with mycophenolate mofetil with good visual recovery, no further relapse, and a reduction in NMDAR-Ab levels (figure 2G). Case 10 (F, age 10) had an unusual presentation of recurrent episodes of hyperventilation, dizziness, and double vision lasting from 2 days to more than 2 weeks (figure 2H). Her brain imaging revealed periventricular and subcortical white matter changes (figure 1L). She has not been treated as the relevance of the NMDAR-Ab in her clinical and radiologic syndrome remains unclear.
General features of cases 1–10 and differences from the remaining 36 patients with NMDAR-Abs.
The mean follow-up time in the cohort was 34 months (median 30, range 8–60), and this was not significantly different between the patients with white matter syndromes (mean 27, median 30, range 8–54) and the other patients with NMDAR-Abs (mean 36, median 37, range 8–60). Overall in the white matter patients, 6 of the 7 patients (85%) who were treated acutely improved following immunotherapy with steroids, IVIg, and PLEX, either individually or in combination. At relapse, 2 patients had escalation of immunotherapy resulting in improvement of clinical syndrome. Patients who were treated late (n = 2) or untreated (n = 1) had either a poorer outcome or persistence of symptoms. In 6 of the 7 patients with serial samples available, there were relationships between NMDAR-Ab titers and severity of clinical syndrome (figure 2, A–H). Of interest, MOG antibodies did not disappear after treatment in case 9, even when NMDAR-Abs became undetectable and after clinical recovery (figure 2G).
Serum levels of NMDAR-Abs ranged from 1:100 to 1:1,000 at the earliest sample available, with CSF levels (cases 2–5) from 1:20 to 1:50. It is important that both serum and CSF NMDAR-Ab titers were negative during the HSVE that preceded NMDAR-Ab relapse in case 4 (figure 2D); the relevant samples were not available for case 5. After immunotherapies, antibody levels fell in all patients and became undetectable in cases 1, 3, 8, and 9. Follow-up CSFs were not available. MOG antibodies were found in only 3 of the 5 patients with ADS (cases 6, 7, and 9) (table 1). All patients were negative for AQP4 antibodies.
Cases 1–10 presented with distinct white matter changes, either radiologic changes compatible with the respective clinical syndromes (brainstem encephalitis and ADS group) or a leukoencephalopathy in addition to the radiologic changes commonly observed within the relevant phenotype (brainstem encephalitis and HSVE). These patients were radiologically distinct from the NMDAR-Ab encephalitis cases (table 1), where only 6 of 28 (21%) had abnormal neuroimaging, all sparing the white matter (p < 0.0001, Fisher exact test). Cases 1–10 were also distinct clinically. Although 3 patients had classical NMDAR encephalitis, in cases 1 and 2, these episodes were remote from the first neurologic syndrome (brainstem encephalitis), and in case 5, the NMDAR-Ab encephalitis occurred following HSVE. In addition, 7 of the 10 cases (70%) had recurrent neurologic episodes, compared to only 6 of 28 patients (21%) with typical NMDAR-Ab encephalitis or 10 of 36 cases (28%) without white matter involvement (p = 0.003 and 0.02, respectively; Fisher exact test). Oligoclonal bands were positive in the CSF in 1 of 3 patients tested with white matter disorder compared to 14 of 18 with typical NMDAR-Ab encephalitis, although this was not statistically significant (p = 0.18, Fisher exact test). Finally, those patients with white matter disorders were more likely to have MOG antibodies (30% vs 0%; p = 0.008, Fisher exact test) than those without white matter involvement.
DISCUSSION
Three distinct white matter clinicoradiologic syndromes—brainstem encephalitis, leukoencephalopathy following HSVE, and CNS demyelination—were identified in 10 of 46 (22%; 95% confidence interval 11%–36%) consecutive children from 6 centers in the UK in whom NMDAR-Abs were detected. The antibodies were present at high titers (up to 1:1,000), and all available CSFs were positive (up to 1:50). Collectively, relapses were seen in 63% (5/8) of our cohort (excluding the 2 HSVE cases), a rate higher than that seen in our classical NMDAR encephalitis patients (28%; 6/28) or than previously reported.1,2,18 This included 2 of the 3 patients with brainstem encephalitis, usually a monophasic disease,13,19 and 3 of the 5 patients (60%) in the ADS group, in which relapses are seen in 25% of patients.20
Nevertheless, there was a good response to immunotherapies once the antibodies were detected, and improvement broadly paralleled serum NMDAR-Ab levels over the following months.
The brainstem parenchyma could be particularly susceptible to systemic autoantibodies. It is well known that antibodies can diffuse into the area postrema, as in neuromyelitis optica,21 and other brainstem regions may also be permeable, as inferred recently from rodent studies.22 However, although brainstem encephalitis is assumed to be due to autoimmune and inflammatory etiologies,13 no disease-defining autoantibodies have been defined to date. Brainstem encephalitis does share clinical features with NMDAR-Ab encephalitis, and it was suggested that one of the original reported cases of Bickerstaff brainstem encephalitis had features of NMDAR-Ab encephalitis.23 Hypoventilation, probably due to disruption of the medullary-pontine respiratory network1,2 and leading to ventilator dependency despite the improvement of the encephalopathy, was seen in case 3. Typically, however, brainstem involvement in NMDAR-Ab encephalitis manifests as autonomic symptoms such as hyperthermia, blood pressure liability, arrhythmias, hypersalivation, and urinary incontinence,1,2 and is usually seen more frequently in adults than in children.1
Distinct episodes of classical NMDAR-Ab encephalitis were observed in 2 of the 3 patients with brainstem encephalitis. Although the NMDAR-Ab positivity in both cases (cases 1 and 2) could only be determined in sera taken during relapses, when they were polysymptomatic, the identification of NMDAR-Abs (serum and CSF) in case 3 during the acute phase of brainstem encephalitis suggests that the initial brainstem event in all 3 patients could represent a localized form of NMDAR-Ab–mediated brainstem inflammation. Early and optimal treatment, as in case 3, may prevent the subsequent evolution to a more generalized NMDAR-Ab encephalitis.
There have been recent reports that NMDAR-Abs, often with choreoathetosis, can be identified before2 and/or during neurologic relapses following HSVE.11,14–16,24 An infective etiology causing brainstem encephalitis may trigger similar brain autoimmunity. The distinct white matter changes seen in both the HSVE relapse patients (figure 1, E–H) are atypical for NMDAR encephalitis, although we have previously described this radiologic feature in 1 child who relapsed following HSVE,11 and these striking leukoencephalopathic changes were also evident in a patient in the brainstem encephalitis group (case 2, figure 1, C and D). Following immunotherapy, both patients with HSVE improved clinically, and, in the 1 patient imaged after treatment, significant radiologic improvement was demonstrated. The relevance of leukoencephalopathy in these syndromes requires further evaluation; its detection by MRI could be a useful biomarker for patients at risk of relapses after HSVE or brainstem encephalitis, with obvious treatment implications.
There is a growing interest in the role of CNS antibodies in demyelination, stemming from the discovery of AQP4 antibodies in neuromyelitis optica25 and detection of MOG antibodies in a range of ADS,26–28 although the clinical use of MOG antibodies as a marker of disease activity and prognosis is still being evaluated.26 Of interest, 3 of the 5 patients in the ADS group, 2 with bilateral optic neuritis (cases 6 and 7) and 1 (case 9) who presented with ADEM followed by recurrent optic neuritis, had antibodies to MOG as well as NMDAR. Both these demyelinating phenotypes are recognized in children with MOG antibodies,27,28 suggesting that these antibodies may have contributed to the clinical picture in addition to the NMDAR-Abs.
An association between NMDAR antibodies and CNS demyelination has been recognized in individual cases with both monophasic3,5 and relapsing syndromes,4,6 but it is unknown whether these antibodies are causative or secondary to the demyelination. As NMDARs are activated on oligodendrocytes following ischemia,29 the white matter may be a target for antibodies following prior or recurrent neurologic insults; this could be the case in our patients who had either a distinct previous neurologic syndrome (HSVE and brainstem encephalitis) or frequent relapses of demyelination. Furthermore, upregulation of NMDA receptor subunits, in particular highly calcium permeable NR2B-containing and magnesium-insensitive NR3A NMDARs, appears to increase the vulnerability of the white matter to injuries in the developing brain,30 and such age-specific NMDAR subunit composition31 could also predispose children with an actively myelinating CNS to developing leukoencephalopathies, thus explaining the high rate of white matter involvement in this pediatric NMDAR-Ab–positive cohort. Experimental studies looking at the role of NMDARs in white matter suggest their importance in remyelination32 and the regenerating optic nerve,33 which could possibly explain the slow visual recovery seen in cases 6 and 8; if they are induced secondary to the demyelination, the NMDAR-Abs may cause a loss of function and reduce this beneficial response to nerve injury.
It was the leukoencephalopathy, and the less severe but distinct white matter changes, that drew our attention to these patients. In fact, advanced neuroimaging techniques are starting to identify white matter abnormalities in patients with anti-NMDAR encephalitis with no apparent abnormality on conventional imaging,34 suggesting that perturbations in white matter tracts are more widespread than previously thought. These could be mediated by direct antibody binding to NMDAR on oligodendrocytes, as discussed above, and/or secondary to neuronal glutamate-mediated excitotoxicity.31 Although the improvement following immunotherapy in our patients with white matter disorders could simply reflect the natural history of these conditions, the poorer outcomes of those untreated or treated later and the parallel reduction or resolution of antibody levels provide evidence that whatever the origins and roles of the NMDAR-Abs, their removal can be helpful.
Only 3 of 10 patients with white matter syndromes had CSF available for testing. However, in each of the patients reported here, including the 11 with typical NMDAR-Ab encephalitis, serum levels were higher than those in the paired CSFs. Although the added sensitivity of CSF NMDAR-Ab evaluation has recently been reported,35 whether serum analysis without an available paired CSF can result in a “false-positive” result was not evaluated in that study. The definition of a false-positive requires a gold standard clinical diagnosis, which, in the context of an expanding phenotype such as that described here, cannot be established. Moreover, since NMDAR-Abs can remain detected in both serum and CSF35 after the patient has recovered, a positive result does not necessarily reflect “false positivity” but merely that the antibodies are not causing a detectable neurologic syndrome. Our suggestion would be that CSF testing, if unavailable initially, should be performed if the serum is negative with a strongly suggestive clinical phenotype or, conversely, can be used to provide further support for the “clinical relevance” of the serum antibodies in patients in whom the clinical features are less typical.
Other limitations of this study include the retrospective nature of the cohort collection and the small sample size that is further confounded by the heterogeneous clinical phenotypes. Further studies are now required to confirm our observation that children and adults with NMDAR-Abs can present with these distinct clinicoradiologic white matter syndromes and to determine whether, and if so how, NMDAR-Abs bind to white matter, in which patients and clinical syndromes it could be relevant, and the treatment implications.
ACKNOWLEDGMENT
The authors thank Dr. Mark Woodhall for confirming antibody results for AQP4 and MOG. The authors are very grateful for all the patients and the clinicians: Dr. Lucinda Carr, Dr. Carlos de Sousa, Dr. Prab Prabahkar (GOSH, London); Dr. Tammy Hedderly, Dr. Elaine Hughes, Dr. Esse Menson (ECH, London); Dr. Antonia Clarke, Dr. Penny Fallon, Dr. Tim Kerr (SGH, London); Dr. Leena Mewasingh, Dr. Nelly Ninis (SMH, London); Dr. Shakti Agrawal, Dr. Rajat Gupta, Dr. Sunny Philip, Dr. Martin Smith (BCH, Birmingham); Dr. Jackie Palace, Dr. Tony McShane (OUH, Oxford).
GLOSSARY
- Ab
antibody
- ADEM
acute disseminated encephalomyelitis
- ADS
acquired demyelination syndromes
- AQP4
aquaporin-4
- HSV
herpes simplex virus
- HSVE
HSV encephalitis
- IVIg
IV immunoglobulin
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NMDAR
NMDA receptor
- PLEX
plasma exchange
Footnotes
AUTHOR CONTRIBUTIONS
Yael Hacohen: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, study concept or design. Michael Absoud: drafting/revising the manuscript, acquisition of data. Cheryl Hemingway: acquisition of data, drafting/revising the manuscript. Leslie Jacobson: acquisition of data, drafting/revising the manuscript. Jean-Pierre Lin: acquisition of data, drafting/revising the manuscript. Michael G. Pike: acquisition of data, drafting/revising the manuscript. Sunil Pullaperuma: acquisition of data, drafting/revising the manuscript. Ata Siddiqui: acquisition of data, drafting/revising the manuscript. Evangeline Wassmer: acquisition of data, drafting/revising the manuscript. Patrick Waters: acquisition of data, drafting/revising the manuscript. Sarosh Irani: drafting/revising the manuscript, study concept or design. Camilla Buckley: acquisition of data, drafting/revising the manuscript, study concept or design. Angela Vincent: analysis or interpretation of data, drafting/revising the manuscript, study concept or design, study supervision. Ming Lim: analysis or interpretation of data, drafting/revising the manuscript, study concept or design, study supervision.
STUDY FUNDING
This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust and the University of Oxford (Y.H., P.W., and A.V.); the Oxford University Clinical Academic Graduate School (Y.H.); the NHS specialized services for rare diseases (neuromyelitis optica; P.W. and A.V.); and ERUK, UK-US Fulbright commission and MS Society (S.R.I.).
DISCLOSURE
Yael Hacohen, Leslie Jacobson., Sunil Pullaperuma, and Ata Siqddqui report no disclosures relevant to the article. Michael Absoud is funded by the MS Society and Action Medical Research charities. Cheryl Hemingway has received travel grants from Merck Serono and Bayer. Jean-Pierre Lin is chair of the British Paediatric Neurology Association Movement Disorders Special Interest Group and is medical advisor to the European Dystonia Federation and Faculty of Dystonia Europe; has received research grants from Action Medical Research, The Dystonia Society (UK), and Guy's and St Thomas' Charity; and has acted as a consultant and received funding for travel and speaker honoraria from Medtronic Ltd. Michael G. Pike receives research grants from Action Medical Research and MS Society and has received a meeting support grant from Euroimmun. Evangeline Wassmer receives research grants from Action Medical Research and MS Society; has received travel grants from UCB, Shire, and Biogen Idec; has received educational grants to organize meetings from Merck Serono, Novartis, Bayer, and Biogen Idec; and has received speaker's fees from Merck Serono and consultancy fees from Genzyme. Patrick Waters is supported by the NHS National Specialized Commissioning Group for Neuromyelitis Optica and by the NIHR Oxford Biomedical Research Centre. Sarosh Irani serves on the scientific advisory board for Encephalitis Society and has received an NIHR Fellowship, Department of Health, UK. Camilla Buckley receives research support from the Medical Research Council, UK. Angela Vincent serves/has served on scientific advisory boards for the Patrick Berthoud Trust, the Brain Research Trust, and the Myasthenia Gravis Foundation of America; has received funding for travel and a speaker honorarium from Baxter International, Inc. and Biogen, Inc.; serves as an associate editor for Brain; receives royalties from the publication of Clinical Neuroimmunology (Blackwell Publishing, 2005) and Inflammatory and Autoimmune Disorders of the Nervous System in Children (Mac Keith Press, 2010); receives/has received research support from the European Union, NIHR Biomedical Research Centre Oxford, Euroimmun AG, and the Sir Halley Stewart Trust; and has received Musk antibody royalties and consulting fees from Athena Diagnostics, Inc. Ming Lim receives research grants from Action Medical Research and MS Society; receives research support grants from the London Clinical Research Network and Evelina Appeal; has received consultation fees from CSL Behring; received travel grants from Merck Serono; and was awarded educational grants to organize meetings by Novartis, Biogen Idec, Merck Serono, and Bayer. Sarosh Irani, Patrick Waters, Angela Vincent, and the University of Oxford hold patents and/or receive royalties and payments for antibody assays in neurologic diseases. Go to Neurology.org/nn for full disclosures.
REFERENCES
- 1.Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011;10:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010;133:1655–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hacohen Y, Absoud M, Woodhall M, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry. Epub 2013 Oct 16. 10.1136/jnnp-2013-306411 [DOI] [PubMed]
- 4.Kruer MC, Koch TK, Bourdette DN, et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology 2010;74:1473–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lekoubou A, Viaccoz A, Didelot A, et al. Anti-N-methyl-D-aspartate receptor encephalitis with acute disseminated encephalomyelitis-like MRI features. Eur J Neurol 2012;19:e16–e17 [DOI] [PubMed] [Google Scholar]
- 6.Takeda A, Shimada H, Tamura A, et al. A case of anti-N-methyl-D-aspartate receptor encephalitis with multiple sclerosis-like demyelinated lesions. Mult Scler Relat Disord. Epub 2013 Oct 25. 10.1016/j.msard.2013.09.005 [DOI] [PubMed]
- 7.Derfuss T, Meinl E. Identifying auto antigens in demyelinating diseases: valuable clues to diagnosis and treatment? Curr Opin Neurol 2012;25:231–238 [DOI] [PubMed] [Google Scholar]
- 8.Di Pauli F, Mader S, Rostasy K, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol 2011;138:247–254 [DOI] [PubMed] [Google Scholar]
- 9.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hacohen Y, Wright S, Waters P, et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry 2013;84:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hacohen Y, Deiva K, Pettingill P, et al. N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord 2014;29:90–96 [DOI] [PubMed] [Google Scholar]
- 12.Hacohen Y, Dlamini N, Hedderly T, et al. N-methyl-D-aspartate receptor antibody-associated movement disorder without encephalopathy. Dev Med Child Neurol 2014;56:190–193 [DOI] [PubMed] [Google Scholar]
- 13.Tan IL, Mowry EM, Steele SU, et al. Brainstem encephalitis: etiologies, treatment, and predictors of outcome. J Neurol 2013;260:2312–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohammad SS, Sinclair K, Pillai S, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-Methyl-D-aspartate receptor or dopamine-2 receptor. Mov Disord 2014;29:117–122 [DOI] [PubMed] [Google Scholar]
- 15.Armangue T, Titulaer MJ, Málaga I, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain Autoimmunity. Ann Neurol. Epub 2013 Dec 8. 10.1002/ana.24083 [DOI] [PMC free article] [PubMed]
- 17.Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 2013;19:1261–1267 [DOI] [PubMed] [Google Scholar]
- 18.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013;84:576–583 [DOI] [PubMed] [Google Scholar]
- 20.Banwell B, Bar-Or A, Arnold DL, et al. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 2011;10:436–445 [DOI] [PubMed] [Google Scholar]
- 21.Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One 2011;6:e27412; 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang T, Alexopoulos H, Pettingill P, et al. Immunization against GAD Induces antibody binding to GAD-independent antigens and brainstem GABAergic neuronal loss. PLoS One 2013;8:e72921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merwick A, Dalmau J, Delanty N. Insights into antibody-associated encephalitis—Bickerstaff's 1950's papers revisited. J Neurol Sci 2013;334:167–168 [DOI] [PubMed] [Google Scholar]
- 24.Prüss H, Finke C, Höltje M, et al. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol 2012;72:902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112 [DOI] [PubMed] [Google Scholar]
- 26.Reindl M, Di Pauli F, Rostásy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 2013;9:455–461 [DOI] [PubMed] [Google Scholar]
- 27.Rostasy K, Mader S, Schanda K, et al. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 2012;69:752–756 [DOI] [PubMed] [Google Scholar]
- 28.Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler 2013;19:941–946 [DOI] [PubMed] [Google Scholar]
- 29.Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005;438:1162–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jantzie LL, Talos DM, Jackson MC, et al. Developmental expression of n-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb Cortex. Epub 2013 Sep 17. 10.1093/cercor/bht246 [DOI] [PMC free article] [PubMed]
- 31.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013;14:383–400 [DOI] [PubMed] [Google Scholar]
- 32.Li C, Xiao L, Liu X, et al. A functional role of NMDA receptor in regulating the differentiation of oligodendrocyte precursor cells and remyelination. Glia 2013;61:732–749 [DOI] [PubMed] [Google Scholar]
- 33.Kolls BJ, Meyer RL. N-methyl-D-aspartate receptors strongly regulate postsynaptic activity levels during optic nerve regeneration. J Neurosci Res 2013;91:1263–1279 [DOI] [PubMed] [Google Scholar]
- 34.Finke C, Kopp UA, Scheel M, et al. Functional and structural brain changes in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. Epub 2013 May 20. 10.1002/ana.23932 [DOI] [PubMed]
- 35.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]