

Abstract
Radiotherapy (XRT) delivered with the antibody cetuximab is a standard treatment option for squamous cell carcinomas of head and neck (SCCNH). Cetuximab acts by blocking epidermal growth factor receptor (EGFR) signaling to inhibit cancer progression. However, a significant percentage of patients will not respond to XRT and cetuximab. Statins reduce the synthesis of cholesterol and isoprenoid derivates that may be required for efficient EGFR signaling. We assessed whether the statin simvastatin could improve this combined therapy. In vitro, simvastatin enhanced the effects of XRT alone and in combination with cetuximab in wound healing, cell proliferation, and clonogenic assays in FaDu cells. These results were reflected in xenoimplanted tumors growing into subcutaneous tissue of athymic mice where concomitant treatment with simvastatin decreased tumor growth. Consistently, lower levels of phosphorylated extracellular signal–regulated kinases 1 and 2, phosphatidylinositol 3-kinase/AKT–protein kinase B, and signal transducer and activator of transcription 3 oncoproteins and higher levels of caspase-3 and apoptosis in cell cultures and xenografts were observed. The EGFR-overexpressing A431 cell line was used to reproduce these antitumor effects of simvastatin. Our findings suggest that simvastatin may improve the efficiency of concomitant XRT and cetuximab. Further investigation in the treatment of SCCNH is warranted.
Introduction
Radiotherapy (XRT) delivered concomitantly with monoclonal antibody cetuximab (C225) is a standard treatment option for locally advanced head and neck cancer [1], [2]. C225 acts by binding to the epidermal growth factor receptor (EGFR) to counteract downstream signals that drive cancer cells' aberrant proliferation and resistance to radiation-induced cell killing. However, although C225 leads to improved clinical outcomes in many cases, it appears to be partially or wholly inactive in others due to either intrinsic resistance or acquired resistance to EGFR inhibition [3].
Statins act by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase, which reduces the synthesis rate of endogenous mevalonate, a compound that is necessary for the biosynthesis of cholesterol and isoprenoid derivates such as farnesyl and geranylgeranyl residues. The addition of isoprenoid derivates (prenylation) to small GTP-binding proteins (e.g., RAS and RAS-homologous GTPases) is an essential posttranslational modification for the normal activity of these proteins. This prenylation allows the correct localization and function of small GTP-binding proteins in the inner leaflet of the plasma membrane [4]. In particular, the decreased farnesylation rate of the RAS proteins reduces the efficiency with which these proteins convey signals from growth factor receptors (including EGFR) to downstream effectors, thus interfering with cell survival [5]. In addition to decreasing protein prenylation, statins may also reduce plasma membrane fluidity, particularly in cholesterol-rich rafts, thus interfering with molecular interactions (receptor dimerization) involved in cell signaling emission [6]. A mutated tumor-suppressor protein p53 has been found to upregulate the mevalonate pathway, an observation that suggests that statins may help revert the malignant phenotype of p53-mutated cancer cells [7].
We hypothesized that the statin simvastatin would contribute to C225 radiosensitization by weakening EGFR cell signaling, interfering with the repair of radiation-induced DNA damage and cell proliferation. Simvastatin would participate in the cancer cell killing due to XRT and C225 and eventually would improve tumor control. The principal aim of our study was to preclinically evaluate whether the addition of simvastatin could increase the antitumor effects of concomitant XRT and C225 in xenografted tumors derived from head and neck squamous carcinoma cells.
Materials and Methods
Cancer Cell Lines
Because in this work we explored EGFR inhibition by C225 in head and neck cancer, our study was carried out with the FaDu cell line, derived from a human squamous cell carcinoma of the hypopharynx that overexpresses EGFR, a common trait of human squamous cell carcinomas of head and neck (SCCHN). In addition, we repeated most of the experiments using the EGFR-overexpressing cell line A431 derived from a human vulvar squamous carcinoma, which has been typically used to underpin the biology of treatment with C225. Although these two cell lines are different in their origin, they are, interestingly, highly dependent on EGFR [8], and potentially sensitive to C225, and its modulation by simvastatin. These cells were purchased from the American Type Cell Collection (LGC Promochem, Barcelona, Spain) and were maintained under standard cell culture conditions free of mycoplasma contamination. FaDu and A431 cells are wild type according to the DNA sequencing analysis we performed on codons 12 and 13 of exon 2 in the KRAS gene.
XRT and Pharmacological Treatments
XRT was administered at room temperature using 6-MV X-rays at a dose rate of 2.7 Gy/min from a Varian Clinac 2100 linear accelerator. Mice received local irradiation as described elsewhere [9]. C225 (Merck, Darmstadt, Germany) was directly administered to cell cultures at doses ranging from 10 to 30 nM and to mice at 1 mg/mouse per week. Simvastatin was dissolved in DMSO for cell culture experiments and used at doses ranging from 1 to 25 μM depending on the type of experiment, while it was dissolved in 1,2-propanediol in distilled water 1:1 (vol/vol) for animal treatments and used at a fixed dose of 50 mg/kg per day (simvastatin, DMSO, and propanediol were purchased from Sigma-Aldrich, St Louis, MO). Drug doses were based on preliminary studies from our group and previously published works [10], [11], [12], [13], [14], [15]. We have varied doses and timings to adapt them to the type of assay to examine the effect of concomitancy between drugs for at least a period of 48 hours. Equivalent mock irradiations and treatments with appropriate vehicles were carried out as controls.
Wound Healing Assay
FaDu cells were seeded in 60-mm dishes and cultured until confluence. Then, the cell cultures were pretreated for 48 hours with 15 μM simvastatin, 30 nM C225, or 30 nM C225 plus 15 μM simvastatin before being irradiated with 3-Gy dose. Immediately thereafter, monolayers were scratched (three different locations per dish) with a 200-μl pipette tip to simulate a wound and cultured in the presence of the drugs. Distances between the wound margins were measured at 0, 1, 2, 4, 8, and 24 hours under a Leica DMIL LED light microscope with the Leica Application Suite LAS v.2.6 software (Leica, Wetzlar, Germany).
Cell Proliferation Assay
A total of 300,000 cells were seeded in 60-mm dishes and cultured for 3 days until semi-confluence. Then, corresponding cell cultures were treated with 10 nM C225 for FaDu cells or 30 nM for A431 cells alone—in preliminary experiments, FaDu cells were found to be more sensitive to C225 than A431 cells (data not shown)—or combined with 15 μM simvastatin, both drugs added 2 hours before XRT (3 Gy) and during the assays. The number of cells present in the cell cultures was counted using a hemocytometer at 0, 24, 48, and 72 hours.
Clonogenic Assay
Two thousand single cells were seeded in 60-mm dishes and allowed to attach for 24 hours. Then, cell cultures were pretreated for 24 hours before XRT with 1 μM simvastatin alone, C225 alone (10 nM C225 for FaDu cells or 30 nM for A431 cells), or with the two drugs. Next, cell cultures were either irradiated (2 Gy) or subjected to mock irradiation in the presence or absence of the drugs. Colonies were stained with crystal violet. Clonogenic cell survival was calculated as the ratio between the number of colonies presented after irradiation and the number of cells plated, which was then normalized by the clonogenic efficiency of the untreated controls. Note that when XRT was applied, clonogenic cell survival was the survival after 2 Gy, which is the most useful clinical marker of intrinsic radiosensitivity.
Xenografts and In Vivo Treatments
To generate tumor xenografts, 106 cells suspended in 100 μl of medium were injected into subcutaneous tissues on the right hind limb of 6- to 8-week-old female athymic Swiss nu/nu mice (Harlan, Gannat, France). Cells were injected on a Monday and left to grow for 7 days, moment when the treatments began. Tumor growth was measured—π/6 × (large diameter) × (small diameter)2—twice weekly. Mice were killed when the tumor volume reached 1200 mm3, when the mice showed moderate to severe toxicities, or when significant differences between groups were observed. All experimental procedures were approved by the Institutional Animal Care and Ethics Committee.
The mice received fractionated XRT, C225, and simvastatin. XRT was selectively delivered from Monday to Friday for 2 weeks using the 6-MV X-ray beams at doses of 20 to 30 Gy depending on type of experiment, in 10 fractions, 1 fraction each day. On the first day of treatment, C225 was intraperitoneally injected 6 hours before irradiation at doses of 1 mg per animal to allow the antibody to have time to saturate the EGFR. Next, C225 was administered on days 3, 7, and 10 at doses of 0.5 mg per animal 2 hours (together with simvastatin or its vehicle) before irradiation as a maintenance C225 dose. Simvastatin (50 mg/kg) was administered orally on a daily basis for 12 days 2 hours before irradiation. Mice were randomly allocated to receive XRT plus C225 or XRT, C225, and simvastatin as well as to receive single treatments with XRT, C225, or simvastatin alone. In addition, a group of mice treated in parallel was killed on day 4 to obtain tumor samples for immunofluorescence.
Western Blot Analysis
Semi-confluent cell cultures were pretreated for 48 hours with C225 and simvastatin in FBS-free medium and then irradiated with a single dose of 5 Gy. Twenty minutes after irradiation, cell cultures were rinsed in ice-cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay buffer with protease and phosphatase inhibitors. Vehicle and mock irradiation were provided as controls. Protein concentration in the lysates was determined by the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). Proteins (35 μg) were resolved in the SDS-PAGE system (Bio-Rad Laboratories, Hercules, CA) and blotted onto Hybond nitrocellulose membrane (GE Healthcare–Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). We used the following primary antibodies: mouse anti-EGFR total at a 1:200 dilution, rabbit anti–phosphorylated EGFR-Y1086 at a 1:1000 dilution, mouse anti–phosphorylated extracellular signal–regulated kinases 1 and 2 (ERK1/2) at a 1:5000 dilution, and mouse anti–α-tubulin at a 1:5000 dilution (all of the aforementioned antibodies were purchased from Sigma-Aldrich); rabbit anti–phosphatidylinositol 3-kinase/AKT–protein kinase B (AKT) total at a 1:500 dilution, rabbit anti–phosphorylated AKT-S473 at a 1:500 dilution, rabbit anti–signal transducer and activator of transcription 3 (STAT3) total at a 1:1000 dilution, rabbit anti–phosphorylated STAT3-Y705 at a 1:500 dilution, and anti–cleaved caspase-3 antibody at a 1:500 dilution, all from Cell Signaling Technology (Danvers, MA); rabbit anti-ERK1/2 total [16]; rabbit anti–caspase-3 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA) at a 1:1000 dilution. The nitrocellulose-bound primary antibodies were incubated with anti-mouse IgG or anti-rabbit IgG HRP-linked antibody (GE Healthcare) and were detected by enhanced chemiluminescence staining ECL/ECL Plus (GE Healthcare). Chemiluminescence staining was transformed to arbitrary units of optical density using a digital imaging analysis system (GelDoc 2000 and Quantity One software; Bio-Rad Laboratories), and the results were represented on histograms.
Immunofluorescence for Cleaved Caspase-3
Cleavage of caspase-3, used as an apoptotic marker, was determined by a standard immunofluorescence process on cells cultured on sterilized coverslips and on 3-μm cryostat sections of the xenografts scheduled on the fourth day of treatment. Regardless of the origin, the samples were fixed, permeabilized (0.1% Triton in PBS for 10 minutes), and incubated for 1 hour with a protein-blocking solution (20% goat and 20% horse sera in PBS). Next, the samples were incubated overnight with a rabbit anti–cleaved caspase-3 monoclonal antibody (Cell Signaling Technology) at a 1:100 dilution at 4°C. To detect primary antibodies, the samples were incubated with a goat anti-rabbit Alexa Fluor 594 antibody (red fluorescence) (Invitrogen, Carlsbad, CA) at a 1:200 dilution for 1 hour at room temperature. Then, slices were mounted using Vectashield (Vector Laboratories Inc, Burlingame, CA) mounting medium with 4′-6-diamidino-2-phenylindole DNA staining fluorochrome (blue fluorescence). Fluorescence images were captured using a Nikon Eclipse 80i epifluorescence microscope (Nikon Instruments, Kanagawa, Japan) and then analyzed with the Nis-Elements, Basic Research (Nikon) software. The apoptosis index was calculated as the ratio between red fluorescence (from detection of cleaved caspase-3) and blue fluorescence from nuclei.
Statistics
Results were expressed as means ± SEM. Statistically significant differences in between-group comparisons were defined by a two-tailed significance level of P < .05 using the Mann-Whitney test. The Statistical Package for Social Sciences, version 13.0 (IBM, Madrid, Spain) was used for data analysis.
Results
Simvastatin Decreased Wound Healing, Cell Proliferation, and Clonogenic Survival of Cells Treated with XRT and Cetuximab
The principal aim of this study was to test whether simvastatin may increase the therapeutic effect of XRT and C225 on tumor growth in xenograft models derived from human squamous cell carcinomas. In the first instance, an in vitro approach was undertaken to evaluate whether this statin could influence cell viability of cell cultures treated with XRT and C225. The range of doses for XRT (single dose of 2-3 Gy) and for C225 (10-30 nM) that were used in this study were based on previous reports [13], [15], whereas the dose for simvastatin was based on dose-response preliminary results (data not shown), and data from the literature [11], and varied according to the length of each assay. We examined immediate effects of treatments by means of wound healing assay in FaDu cell line. Wound size decreased progressively, as wounds were repaired, over a 24-hour period. All treatments slowed down wound healing, but the rate of healing was lower in cultures that received simvastatin (Table 1). At 2, 4, and 8 hours after creating the wound, we found that the presence of simvastatin was involved in the higher inhibitory effects of the treatments, although differences were not significant. However, when the observation was extended to 24 hours, differences became more apparent and statistically significant, suggesting that inhibition of cell proliferation rather than cell migration—the latter being an early event—could have been implicated in this observation. It is important to note that triple treatment with XRT, C225, and simvastatin was more cytotoxic than XRT and C225 without the statin, which indicates a potential role for simvastatin (Table 1).
Table 1.
Wound Healing Assays in FaDu Cell Line
| Wound Size (Percentage of Healed Wound) |
|||||
|---|---|---|---|---|---|
| 2 Hours | 4 Hours | 8 Hours | 24 Hours | P Value at 24 Hours | |
| Nontreatment | 5.80 ± 1.20 | 10.59 ± 1.68 | 15.55 ± 1.85 | 36.80 ± 3.23 | |
| XRT (3 Gy) | 5.61 ± 0.87 | 9.09 ± 1.37 | 13.11 ± 2.16 | 25.27 ± 2.19 | .077 (1) |
| C225 (30 nM) | 5.17 ± 1.15 | 7.83. ± 1.47 | 11.07 ± 2.15 | 18.78 ± 3.55 | .001 (1) |
| Simvastatin (15 μM) | 2.04 ± 0.46 | 6.37 ± 1.73 | 9.32 ± 1.52 | 12.75 ± 2.37 | .001 (1) |
| XRT + C225 | 5.14 ± 1.05 | 8.93 ± 1.52 | 12.46 ± 1.73 | 21.66 ± 2.35 | .001 (1) |
| XRT + C225 + simvastatin | 4.20 ± 0.80 | 6.98 ± 1.36 | 9.43 ± 1.58 | 12.36 ± 1.78 | .001 (1) |
| .005 (2) | |||||
Cell cultures were treated as indicated in the Materials and Methods section. Values are means ± SE of three independent experiments, with each experiment duplicated. Doses used in the combined treatments are the same as in the single treatments. P value (Mann-Whitney test): (1) compared to nontreatment condition and (2) compared to XRT + C225.
To investigate the effects of simvastatin on cell proliferation in FaDu cells, as well as in A431 cells, we subjected cell cultures to the different treatments for longer periods of time of 24, 48, and 72 hours (Table 2). In both cell lines, cell number increased as a function of time, but FaDu cells showed higher proliferation rates. Likewise, in both cell types, cell proliferation was inhibited by all the therapeutic schemes, an effect that was more obvious as time increased. For individual treatments, XRT and simvastatin alone had the highest effect. Regarding combined treatments, it is of note that the addition of C225 to XRT was not reflected in a significant decrease of proliferation although these cells were sensitive to C225 alone. On the contrary, we found that the addition of simvastatin to XRT plus C225 effectively resulted in a significant inhibition of proliferation, leading at 72 hours to a decrease of 2.7-fold for FaDu cells and 5.5-fold for A431 cells compared to XRT alone and 1.93-fold and 4.3-fold, respectively, compared to XRT and C225 (Table 2). Interestingly, the effect of C225 and simvastatin on proliferation was smaller than the effect of XRT, C225, and simvastatin together, in particular at 72 hours after the beginning of treatments. These observations suggest that C225 and simvastatin in collaboration may contribute to weaken cell recovery from XRT resulting in higher cell killing.
Table 2.
Cell Proliferation Assays in FaDu and A431 Cell Lines
| Cell Number (106) |
||||||
|---|---|---|---|---|---|---|
| 24 Hours | P Value | 48 Hours | P Value | 72 Hours | P Value | |
| FaDu cell line | ||||||
| Nontreatment | 2.63 ± 0.24 | – | 4.39 ± 0.35 | – | 5.88 ± 0.55 | – |
| XRT (3 Gy) | 1.46 ± 0.50 | .062 (1) | 2.36 ± 0.26 | .007 (1) | 2.93 ± 0.33 | .017 (1) |
| C225 (10 nM) | 2.32 ± 0.14 | .156 (1) | 3.48 ± 0.34 | .121 (1) | 4.26 ± 0.28 | .071 (1) |
| Simvastatin (15 μM) | 2.34 ± 0.60 | .497 (1) | 3.05 ± 0.35 | .042 (1) | 3.09 ± 0.42 | .017 (1) |
| C225 + simvastatin | 1.24 ± 0.29 | .027 (1) | 1.63 ± 0.31 | .007 (1) | 2.02 ± 0.50 | .007 (1) |
| XRT + C225 | 2.11 ± 0.11 | .093 (1) | 2.62 ± 0.12 | .003 (1) | 2.94 ± 0.08 | .002 (1) |
| XRT + C225 + simvastatin | 1.97 ± 0.11 | .071 (1) | 2.10 ± 0.14 | .002 (1) | 1.52 ± 0.09 | .002 (1) |
| .631 (2) | .025 (2) | .004 (2) | ||||
| A431 cell line | ||||||
| Nontreatment | 1.89 ± 0.33 | – | 2.19 ± 0.15 | – | 2.97 ± 0.38 | – |
| XRT (3 Gy) | 1.18 ± 0.07 | .136 (1) | 1.56 ± 0.04 | .010 (1) | 0.95 ± 0.04 | .010 (1) |
| C225 (30 nM) | 0.81 ± 0.02 | .011 (1) | 1.35 ± 0.07 | .010 (1) | 1.28 ± 0.09 | .010 (1) |
| Simvastatin (15 μM) | 1.49 ± 0.07 | .497 (1) | 1.14 ± 0.06 | .010 (1) | 0.92 ± 0.03 | .010 (1) |
| C225 + simvastatin | 1.01 ± 0.03 | .011 (1) | 0.53 ± 0.06 | .010 (1) | 0.42 ± 0.04 | .010 (1) |
| XRT + C225 | 1.29 ± 0.19 | .055 (1) | 1.07 ± 0.14 | .004 (1) | 0.98 ± 0.19 | .006 (1) |
| XRT + C225 + simvastatin | 0.92 ± 0.03 | .004 (1) | 0.34 ± 0.07 | .004 (1) | 0.23 ± 0.08 | .004 (1) |
| .150 (2) | .004 (2) | .004 (2) | ||||
Cell cultures were treated as indicated in the Materials and Methods section. Values are means ± SE of three independent experiments, with each experiment duplicated. Doses used in the combined treatments are the same as in the single treatments. P value (Mann-Whitney test): (1) compared to nontreatment condition and (2) compared to XRT + C225 without simvastatin.
To further verify and extend the results of these wound healing and cell proliferation assays, the effect of treatments on clonogenic cell survival was evaluated (Table 3). All conditions were evaluated by performing assays under two types of drug exposures in combination with the same type of irradiation and period of colony formation: drug exposure maintained for 14 days or drug exposure for only 48 hours (24 hours pre-XRT and 24 hours post-XRT). These two different strategies were aimed to discriminate a possible effect of drugs on cell proliferation from an early clonogenic cell killing effect, which can be properly assessed without the presence of drugs during the complete period of colony formation. We observed that the effect of drugs was dependent on duration of exposure. The baseline plating efficiency for FaDu and A431 cells were comparable, 16.76 ± 2.48% and 14.29 ± 0.63%, respectively (Table 3). Regarding single treatments, FaDu cells displayed higher radiosensitivity than A431 cells and were clearly more sensitive to C225, as previously noted. One micromolar simvastatin was definitely less effective than the doses of simvastatin used in wound healing and proliferation assays. However, it is interesting to note that simvastatin administered at a dose of 1 μM (as used in the clonogenic assays) is closer to blood levels of simvastatin that were achieved in clinical settings [17]. However, higher doses of simvastatin precluded colony growth at all, because zero colonies grew. With respect to the effect of drugs on XRT, the addition of simvastatin enhanced radiation cell killing as reported by others [14], although in FaDu and A431 cells our findings were not consistent regarding duration of simvastatin exposure (Table 3). The addition of C225 also enhanced the effect of XRT alone as described previously in SCCHN [18]. In FaDu cells, clonogenic survival was dramatically decreased by C225, whereas it was moderately diminished in A431 cells (Table 3).
Table 3.
Clonogenic Assays in FaDu and A431 Cell Lines
| Clonogenic Cell Survival (%) |
||||
|---|---|---|---|---|
| Drugs for 14 Days | P Value | Drugs for 48 Hours | P Value | |
| FaDu cell line | ||||
| Nontreatment | 100 ± 0.00 | – | 100 ± 0.00 | – |
| XRT (2 Gy) | 71.7 ± 1.37 | .002 (1) | 80.6 ± 3.47 | .002 (1) |
| C225 (10 nM) | 28.2 ± 1.92 | .002 (1) | 33.1 ± 1.31 | .002 (1) |
| Simvastatin (1 μM) | 93.1 ± 2.01 | .040 (1) | 87.3 ± 3.48 | .040 (1) |
| XRT + simvastatin | 69.7 ± 1.88 | .002 (1) | 62.1 ± 3.15 | .002 (1) |
| C225 + simvastatin | 23.0 ± 2.20 | .002 (1) | 68.0 ± 6.46 | .002 (1) |
| XRT + C225 | 22.4 ± 1.76 | .002 (1) | 47.1 ± 1.78 | .002 (1) |
| XRT + C225 + simvastatin | 13.0 ± 2.11 | .002 (1) | 33.0 ± 1.05 | .002 (1) |
| .004 (2) | .004 (2) | |||
| A431 cell line | ||||
| Nontreatment | 100 ± 0.00 | – | 100 ± 0.00 | – |
| XRT (2 Gy) | 78.5 ± 2.57 | .001 (1) | 89.8 ± 0.84 | .037 (1) |
| C225 (30 nM) | 74.3 ± 4.09 | .001 (1) | 88.5 ± 1.02 | .037 (1) |
| Simvastatin (1 μM) | 75.6 ± 3.47 | .001 (1) | 87.5 ± 1.30 | .037 (1) |
| XRT + simvastatin | 64.7 ± 2.21 | .001 (1) | 90.1 ± 1.95 | .037 (1) |
| C225 + simvastatin | 62.5 ± 3.50 | .001 (1) | 93.7 ± 0.52 | .037 (1) |
| XRT + C225 | 58.0 ± 3.46 | .001 (1) | 92.5 ± 1.14 | .037 (1) |
| XRT + C225 + simvastatin | 44.7 ± 4.27 | .001 (1) | 76.9 ± 3.63 | .037 (1) |
| .009 (2) | .050 (2) | |||
Cell cultures were treated as indicated in the Materials and Methods section. Values are means ± SE. For FaDu and A431 cells, two and four independent experiments, with each experiment duplicated, were performed for 14-day and 48-hour schemes, respectively. Doses used in the combined treatments are the same as in the single treatments. P value (Mann-Whitney test): (1) compared to nontreatment condition and (2) compared to XRT + C225 without simvastatin.
As our objective was to evaluate the role of simvastatin in XRT treatment combined with C225, it was interesting to observe that triple combination including simvastatin had the most inhibitory effect on clonogenic survival in both cell lines irrespectively of the fact that the drugs were applied for 14 days or for 48 hours. Triple treatment augmented XRT alone cell killing by a factor of 5.5 (71.7% vs 13.0%) and 2.4 (80.6% vs 33.0%), respectively, for FaDu cells and 1.75 (78.5% vs 44.7%) and 1.16 (89.8% vs 76.9%), respectively, for A431 cells. Second, and more importantly, the impact of simvastatin on the triple treatment was clearly significant as indicated by the outcomes showing decreases in clonogenic survival by a factor of 1.72 (22.4% vs 13.0%) and 1.42 (47.1% vs 33%), respectively, for FaDu cells and 1.3 (58.0% vs 44.7%) and 1.2 (92.5% vs 76.9%), respectively, for A431 cells compared to double treatment of XRT with C225 (Table 3). Moreover, in both cell lines, double treatment with 48-hour C225 exposure was less effective than C225 alone, an observation that suggests the participation of an early acceleration of cell proliferation, a radiation-induced reaction already described in A431 cell line by Schmidt-Ullrich and co-workers [19]. Interestingly, this possible adaptive response was not observed after the triple treatment, perhaps counteracted by simvastatin (Table 3). Taken together, the in vitro results suggest that simvastatin could decrease cell proliferation in combination with XRT and C225, being its effect potentiated in long-term drug exposures, and provide new insights about the triple combination.
Simvastatin Slowed the Growth of Xenografts Treated with XRT and Cetuximab
Because of preliminary in vitro findings indicating a possible activity of simvastatin as cell proliferation inhibitor in combination with C225 and XRT, this study was continued to investigate simvastatin role in xenografts. In tumors derived from FaDu and A431 cell lines, single treatment with simvastatin alone had no effect on tumor growth. On the contrary, treatment with C225 or XRT significantly reduced tumor growth compared to untreated tumors, XRT being the most effective treatment (Figures 1A and 2A). FaDu tumors were more sensitive to XRT and C225 than A431 ones as was also seen in clonogenic assays (Table 3).
Figure 1.
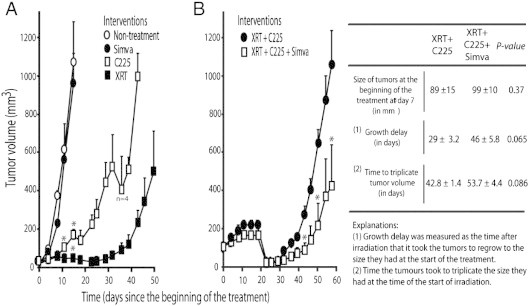
Effect of simvastatin on the growth of FaDu xenografts treated with radiation (XRT) and cetuximab (C225). Mice received XRT (30 Gy), C225, or simvastatin as single treatments (A) or XRT (30 Gy) plus C225 or XRT (30 Gy) plus C225 plus simvastatin (B), as specified in the Materials and Methods section. Values are the means ± SE of five tumors per group (unless otherwise specified). P values were calculated by Mann-Whitney test. *P < .05 compared to nontreatment group in A or XRT + C225 without simvastatin group in B.
Figure 2.
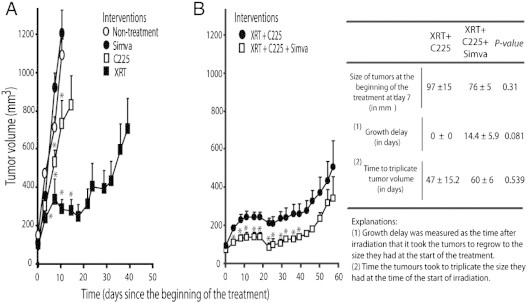
Effect of simvastatin on the growth of A431 xenografts treated with radiation (XRT) and cetuximab (C225). Mice received XRT (30 Gy), C225, or simvastatin as single treatments (A) or XRT (20 Gy) plus C225 or XRT (20 Gy) plus C225 plus simvastatin (B), as specified in the Materials and Methods section. Values are the means ± SE of five tumors per group. P values were calculated by Mann-Whitney test. *P < .05 compared to nontreatment group in A or XRT + C225 without simvastatin group in B.
To focus on the main interest of this study, we started experiments irradiating FaDu tumors with 3 Gy per day for 10 days in combination with C225 in the presence or absence of simvastatin. Irrespectively of simvastatin, XRT plus C225 induced a transitory complete regression of tumors that lasted around 7 days (Figure 1B). After that, tumor growth rebounded but showed lower rates of regrowth when the animals received simvastatin. The time that the tumors took to achieve the size they had at the start of the treatment experienced a considerable delay when simvastatin was added to XRT + C225. The delay in mice that received simvastatin was 46 ± 5.8 days compared to 29 ± 3.2 days in the absence of simvastatin (a difference of 17 days; P value = .065). From the start of XRT, the time for the tumor volume to triple in size was 53.7 ± 4.4 days versus 42.8 ± 1.4 days depending on the presence of simvastatin or not, respectively (a difference of 11 days; P value = .086).
In A431-tumors, to prevent a complete response, XRT dose was lowered to 2 Gy per day for 10 days. Contrary to the FaDu xenografts, A431 tumors did not achieve a complete disappearance, but similarly it was found that the mice treated with simvastatin showed A431 tumors with lower rates of regrowth (Figure 2B). Consistently with a simvastatin-induced enhancement in tumor growth inhibition, the growth delay after irradiation for the tumors treated with simvastatin was 14.4 ± 5.9 days in contrast with the mice that did not receive an additional dose of simvastatin, in which tumor size never decreased below the size the tumors had at the time of irradiation (P value = .081). In these mice, the time to triplicate the initial tumor volume was increased if they received simvastatin from 47 ± 15.2 to 60 ± 6 days (a difference of 13 days; P value = .539). Although these experiments were not statistically significant, they were suggestive of an antitumor effect, in line with the results we observed for FaDu tumors. Regarding animals' global health status, no differences were observed in between groups related to mouse weight and physical or clinical appearance.
Because in vivo, and in vitro, findings were compatible with the notion that simvastatin could enhance the antitumor effect of XRT and C225 in FaDu and A431 cell–derived tumors, we decided to evaluate if simvastatin could have a negative influence on the biology of these tumors.
Simvastatin Induced Apoptosis after XRT plus Cetuximab
We hypothesized that the effect of simvastatin might be related to apoptosis activation. To evaluate this possibility, we determined the cleaved caspase-3, a surrogate marker that indicates irreversible cell death through apoptosis. In cultured cells, we found that levels of cleaved caspase-3 increased in simvastatin-treated cells in a dose-dependent manner, while the levels of pro-caspase-3 remained unchanged (Figure 3). To validate these in vitro findings and establish whether apoptosis was increased by simvastatin in FaDu and A431 cells treated with XRT and C225, xenograft tumors were sampled as previously described. Although the tumors received only 3 days of treatment and the percentages of apoptotic cells were relatively low, we already found that the number of cleaved caspase-3–positive cells was significantly higher in FaDu-derived tumors treated with triple treatment at this time point (1.99 ± 0.20% vs 5.96 ± 0.56%; P = .0001; Figure 4A). The same observation was made in A431-derived tumors (4.40 ± 0.62% vs 8.83 ± 1.46%; P = .005; Figure 4B).
Figure 3.
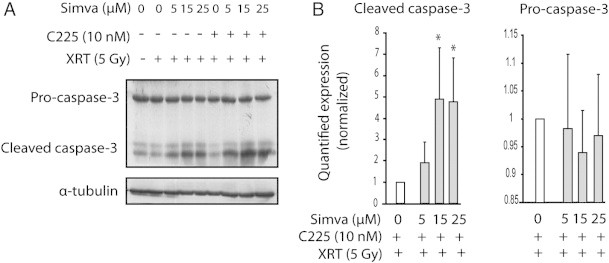
Effect of the addition of simvastatin to XRT and C225 on the cleaved caspase-3. After a 48-hour period of exposure to 10 nM C225 and 5, 15, or 25 μM simvastatin in FBS-free medium, FaDu cells were irradiated with a dose of 5 Gy as indicated. (A) Western blot analysis was used to determine the levels of proteins as it is shown. α-Tubulin was included as a loading control. (B) Optical densitometry quantifications (in arbitrary units) of the protein levels are indicated as fold relative to the XRT + C225 condition; bars show SEM. Three independent lysates, each blotted in triplicate, were used. *P < .05 compared to XRT plus C225 without simvastatin.
Figure 4.
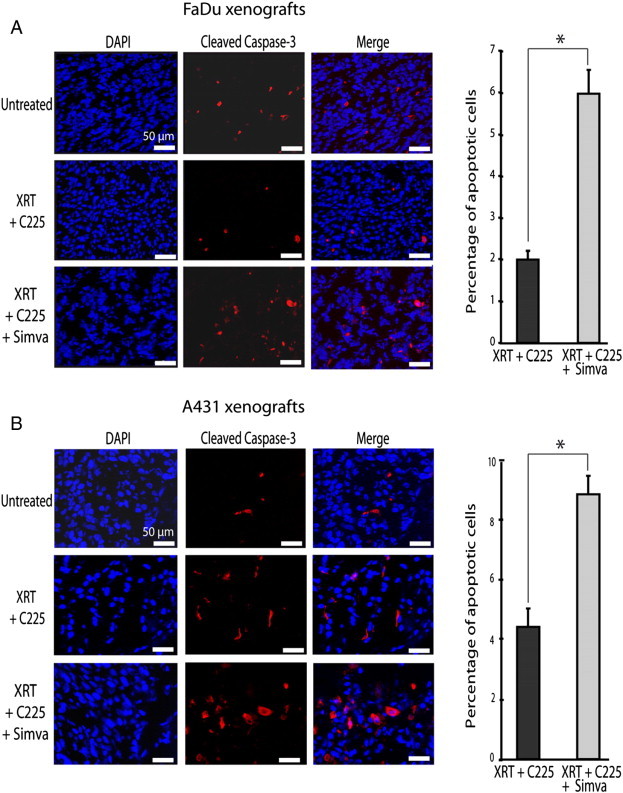
Simvastatin was associated with apoptosis in xenografts. Mice were treated with XRT and C225 or with XRT, C225, and simvastatin as described in Figure 1. Tumors were extirpated on the fourth day, 24 hours after the last treatment. Representative pictures showing cleaved caspase-3 (red fluorescence) and cell nucleus (blue fluorescence) are shown for FaDu- (A) and A431-derived (B) tumors. Columns show the mean values and bars show the SEM of apoptotic cells relative to the total number of cells. Six randomly selected field microscopic images were analyzed per slice. On average, 200 cells were counted per field. Data were obtained from two independent experiments from each treatment scheme. *P < .05 compared to XRT plus C225.
Simvastatin Interfered with Oncogenic Signaling Pathways in Cells Treated with XRT and Cetuximab
We also investigated whether simvastatin could affect crucial cellular signaling pathways involved in the malignant phenotype of cancers. We found that the ionizing radiation elicited the phosphorylation of EGFR on tyrosine 1086. However, the addition of simvastatin to XRT did not modify phosphorylated levels of EGFR (Figure 5). In contrast, C225 had an inhibitory effect on the radiation-induced phosphorylation of EGFR, which was neither changed in the presence of simvastatin, indicating that simvastatin had little effect on EGFR (at least on phosphorylated tyrosine 1086). Although simvastatin was inactive on EGFR, we observed a noticeable reduction of the phosphorylation of ERK1/2. Simvastatin has a weak effect on the activation of phosphorylated AKT and phosphorylated STAT3 and lacked of a dose-response inhibitory effect compared to ERK1/2 protein. No effect on the levels of total EGFR, ERK1/2, AKT, and STAT3 were found (Figure 5).
Figure 5.
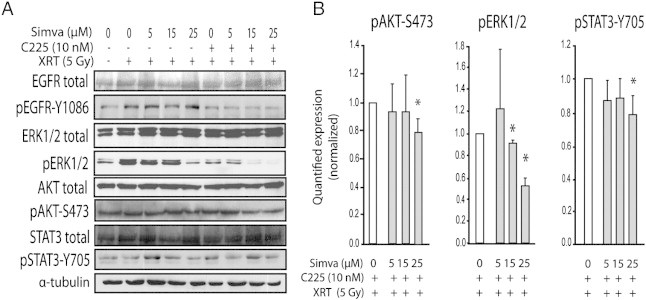
Effects of simvastatin on the levels of phosphorylated ERK, AKT, and STAT3. FaDu cells were treated with XRT, C225, and simvastatin as described in Figure 3. (A) Western blot analysis was used to determine the levels of proteins as it is shown. α-Tubulin was included as a loading control. (B) Optical densitometry quantifications (in arbitrary units) of the protein levels are indicated as fold relative to the XRT + C225 condition; bars show SEM. Three independent lysates, each blotted in triplicate, were used. *P < .05 compared to XRT plus C225 without simvastatin.
Discussion
In this study, we preclinically explored whether a treatment regime involving the addition of simvastatin to XRT and C225 merits further research. Given the fact that XRT and concurrent C225 is a common treatment for locally advanced SCCHN, we believe that this is a relevant question. Although early time points explored in scratch and proliferation assays did not provide a clear clue on the effectiveness of simvastatin, it was shown that the addition of simvastatin for 48 hours or more significantly decreased proliferation and clonogenic survival of cells treated with XRT and C225. Moreover, we used an experimental model with tumor cells derived from squamous cell carcinoma of the hypopharynx that suggests that simvastatin may increase the antitumor effect of XRT plus C225—at doses and fractions of XRT that mimic doses administered in the clinical setting. The effects of simvastatin were recapitulated using A431 cell line validating the notion that simvastatin may have a role in combination with XRT and C225. The addition of simvastatin was associated with an increase in apoptosis and a decrease in the levels of activated ERK1/2, AKT, and STAT3 oncoproteins, a set of observations that provide support to the higher antitumor effects produced by the triple treatment.
The role of statins in cancer therapy has been reviewed previously elsewhere [17], [20], [21], [22]. In noncancerous tissues, statins reduce the proliferation of the atherosclerotic plaque and the chronic inflammatory process associated with atheromatosis [23]. Similarly, simvastatin represses the proliferation of glomerular mesangial cells, suggesting a preventive role in diabetic nephropathy, an effect mediated by its interference with isoprenylation of small GTP-binding proteins [24]. In addition to the antiproliferative and anti-inflammatory properties of statins in non-neoplastic tissues, increasing evidence supports a role for statins in cancer through the inhibition of cancer cell proliferation, angiogenesis, and metastatic potential. These effects have been proven in numerous different cell lines derived from myeloid and lymphoblastic leukemia, neuroblastoma, rhabdomyosarcoma, medulloblastoma squamous cell carcinoma of the cervix, melanoma, high-grade glioma, and cancer of the kidney, testis, breast, stomach, prostate, and small cell lung cancer [11], [25], [26], [27]. Published data indicate that statins can sensitize cancer cells to chemical drugs such as doxorubicin, nitrosourea, cisplatin, and 5-fluorouracil [28], [29]. Recently, it was reported that the combination of simvastatin and C225 sensitize colon cancer cells bearing RAS mutations [12]. In combination with XRT, the statin lovastatin has also been found to have a radiosensitizing effect in lung cancer and osteosarcoma cell lines that express mutated RAS [14], [30]. Interestingly, several randomized controlled trials and case-control studies have found that statins used to lower cholesterol levels may exert a protective effect against cancer [31], [32], [33]. In addition, a recent epidemiological study found evidence suggesting that statin use can reduce cancer-related mortality [34]. A number of clinical trials have investigated the antitumor effect of statins. In one trial, the combination of 5-fluorouracil and the statin pravastatin was associated with a higher tumor response and better survival than chemotherapy alone in patients with unresectable hepatocarcinoma [35]. Similarly, a review carried out by Hindler et al. described the promising results for statin use in SCCHN and other types of cancer [21].
To our knowledge, this is the first in vivo study of combined XRT, C225, and statins in an experimental model that suggests that simvastatin may increase antitumor effects, providing new translational data to sustain clinical investigation of statins in radiation oncology. The results from tumor growth and cell death analysis of tumor samples from the two cell lines give support to the increased antitumor effect of triple combination. The findings we report are consistent with the mechanism of anticancer action of simvastatin described previously as monotherapy or in combination with radiation or classic chemotherapies. However, this is the first report in which simvastatin has been successfully assessed in combination with an anti-EGFR therapy using xenoimplanted tumors. We have observed that statins have antiproliferative effects [20], [22] and that they can contribute to cancer cell killing by apoptosis [11], [12], [14], [27]. We have also observed that the levels of ERK1/2, AKT, and STAT3 proteins that promote cancer progression were reduced by simvastatin, a finding that correlated with a loss of cell viability and with apoptosis. In addition to increasing apoptosis, this decrease in activated ERK1/2, AKT, and STAT3 levels—oncoproteins known to have a role in repairing radiation-induced damage and in promoting the development of aggressive malignant phenotypes [13], [15], [36]—could impair the ability of cancer cells to recover from XRT and C225.
We believe that the evidence in the present report warrants further clinical investigation, although we have to add some comments that deserve a particular mention. We and others have found significant antitumor activity at concentration levels ranging from 1 to 25 μM. However, the typical plasma levels to treat hypercholesterolemia are approximately 10 times lower [37]. This observation raises additional concerns about statin-induced liver and muscle toxicity, especially given that only a few clinical trials have been carried out to address this issue. One phase I trial in patients with SCCHN established that 7.5 mg/kg per day of lovastatin for 2 weeks (the dose for dyslipidemia is 1 mg/kg per day) followed by a 1-week break was a well-tolerated scheme (provided that creatinine clearance is > 70 ml/min) [38]. Nevertheless, lovastatin doses can be safely escalated (35 mg/kg per day) as long as ubiquinone (co-enzyme Q) is given concomitantly [37], [39]. The existing uncertainties about the effective dose of statins in cancer therapy are aggravated by the fact that lovastatin and simvastatin are administered as inactive prodrugs and need to be enzymatically activated to β-hydroxy acid by esterase and paraoxonase-mediated hydrolysis [40]. To our knowledge, no published studies have measured the actual active acid form of simvastatin or lovastatin in cell cultures and/or in mice—in which liver statins undergo active transformation—to properly infer the statin dose that should be used in clinical cancer trials. Although clinical and epidemiological data suggest that relative low plasma concentrations of statins could be sufficient to achieve an antitumor effect, reasonably, new phase I trials with pharmacokinetic and pharmacodynamic studies are warranted.
In conclusion, we have presented a proof-of-concept study that demonstrates that simvastatin may enhance antitumor response of concomitant XRT and C225. In this preclinical work, we have provided evidence that supports further basic and clinical investigation of simvastatin in SCCHN disease.
Acknowledgments
We are grateful to Bradley Londres for his excellent assistance in improving the English of the manuscript. Disclosures: L.I.d.L. and M.B. are the recipients of laboratory research awards from Merck KGaA. R.M. receives lecture fees and grant support from Merck and serves on a paid advisory board. J.B. is the principal investigator of this study and received financial support from Merck KGaA. The study sponsors had no involvement in the study design, in the collection, analysis, and interpretation of data, in the writing of the manuscript, and in the decision to submit the manuscript for publication. None of the authors hold stock options in the company.
Footnotes
This work received financial support from Merck KGaA, Spanish Association Against Cancer, Barcelona Committee, and Agència de Gestió d'Ajuts Universitaris i de Recerca (SGR681) from the Catalan Government (Spain).
References
- 1.Bonner J.A., Harari P.M., Giralt J., Azarnia N., Shin D.M., Cohen R.B., Jones C.U., Sur R., Raben D., Jassem J. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 2.Cetuximab approved by FDA for treatment of head and neck squamous cell cancer. Cancer Biol Ther. 2006;5:340–342. [PubMed] [Google Scholar]
- 3.Benavente S., Huang S., Armstrong E.A., Chi A., Hsu K.T., Wheeler D.L., Harari P.M. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin Cancer Res. 2009;15:1585–1592. doi: 10.1158/1078-0432.CCR-08-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prior I.A., Muncke C., Parton R.G., Hancock J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reuter C.W., Morgan M.A., Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000;96:1655–1669. [PubMed] [Google Scholar]
- 6.Simons K., Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 7.Freed-Pastor W.A., Mizuno H., Zhao X., Langerod A., Moon S.H., Rodriguez-Barrueco R., Barsotti A., Chicas A., Li W., Polotskaia A. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2011;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song J.Y., Lee S.W., Hong J.P., Chang S.E., Choe H., Choi J. Epidermal growth factor competes with EGF receptor inhibitors to induce cell death in EGFR-overexpressing tumor cells. Cancer Lett. 2009;283:135–142. doi: 10.1016/j.canlet.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 9.Baro M., de Llobet L.I., Modolell I., Guedea F., Visa J., Balart J. Development and refinement of a technique using a medical radiation therapy facility to irradiate immunodeficient mice bearing xenografted human tumours. Lab Anim. 2012;46:345–348. doi: 10.1258/la.2012.011147. [DOI] [PubMed] [Google Scholar]
- 10.Johnson-Anuna L.N., Eckert G.P., Keller J.H., Igbavboa U., Franke C., Fechner T., Schubert-Zsilavecz M., Karas M., Müller W.E., Wood W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005;312:786–793. doi: 10.1124/jpet.104.075028. [DOI] [PubMed] [Google Scholar]
- 11.Wu H., Jiang H., Lu D., Xiong Y., Qu C., Zhou D., Mahmood A., Chopp M. Effect of simvastatin on glioma cell proliferation, migration, and apoptosis. Neurosurgery. 2009;65:1087–1096. doi: 10.1227/01.NEU.0000360130.52812.1D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J., Lee I., Han B., Park J.O., Jang J., Park C., Kang W.K. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst. 2011;103:674–688. doi: 10.1093/jnci/djr070. [DOI] [PubMed] [Google Scholar]
- 13.Pueyo G., Mesia R., Figueras A., Lozano A., Baro M., Vazquez S., Capella G., Balart J. Cetuximab may inhibit tumor growth and angiogenesis induced by ionizing radiation: a preclinical rationale for maintenance treatment after radiotherapy. Oncologist. 2010;15:976–986. doi: 10.1634/theoncologist.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanli T., Liu C., Rashid A., Hopmans S.N., Tsiani E., Schultz C., Farrell T., Singh G., Wright J., Tsakiridis T. Lovastatin sensitizes lung cancer cells to ionizing radiation: modulation of molecular pathways of radioresistance and tumor suppression. J Thorac Oncol. 2011;6:439–450. doi: 10.1097/JTO.0b013e3182049d8b. [DOI] [PubMed] [Google Scholar]
- 15.de Llobet L.I., Baro M., Figueras A., Modolell I., Da Silva M.V., Muñoz P., Navarro A., Mesia R., Balart J. Development and characterization of an isogenic cell line with a radioresistant phenotype. Clin Transl Oncol. 2013;15:189–197. doi: 10.1007/s12094-012-0898-8. [DOI] [PubMed] [Google Scholar]
- 16.Viñals F., Chambard J.C., Pouysségur J. p70 S6 Kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem. 1999;274:26776–26782. doi: 10.1074/jbc.274.38.26776. [DOI] [PubMed] [Google Scholar]
- 17.Sleijfer S., van der Gaast A., Planting A.S., Stoter G., Verweij J. The potential of statins as part of anti-cancer treatment. Eur J Cancer. 2005;41:516–522. doi: 10.1016/j.ejca.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Huang S.M., Bock J.M., Harari P.M. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–1940. [PubMed] [Google Scholar]
- 19.Schmidt-Ullrich R.K., Mikkelsen R.B., Dent P., Todd D.G., Valerie K., Kavanagh B.D., Contessa J.N., Rorrer W.K., Chen P.B. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15:1191–1197. doi: 10.1038/sj.onc.1201275. [DOI] [PubMed] [Google Scholar]
- 20.Chan K.K., Oza A.M., Siu L.L. The statins as anticancer agents. Clin Cancer Res. 2003;9:10–19. [PubMed] [Google Scholar]
- 21.Hindler K., Cleeland C.S., Rivera E., Collard C.D. The role of statins in cancer therapy. Oncologist. 2006;11:306–315. doi: 10.1634/theoncologist.11-3-306. [DOI] [PubMed] [Google Scholar]
- 22.Gauthaman K., Fong C.Y., Bongso A. Statins, stem cells, and cancer. J Cell Biochem. 2009;106:975–983. doi: 10.1002/jcb.22092. [DOI] [PubMed] [Google Scholar]
- 23.Schartl M., Bocksch W., Koschyk D.H., Voelker W., Karsch K.R., Kreuzer J., Hausmann D., Beckmann S., Gross M. Use of intravascular ultrasound to compare effects of different strategies of lipid-lowering therapy on plaque volume and composition in patients with coronary artery disease. Circulation. 2001;104:387–392. doi: 10.1161/hc2901.093188. [DOI] [PubMed] [Google Scholar]
- 24.Danesh F.R., Sadeghi M.M., Amro N., Philips C., Zeng L., Lin S., Sahai A., Kanwar Y.S. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/p21 signaling pathway: Implications for diabetic nephropathy. Proc Natl Acad Sci U S A. 2002;99:8301–8305. doi: 10.1073/pnas.122228799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soma M.R., Pagliarini P., Butti G., Paoletti R., Paoletti P., Fumagalli R. Simvastatin, an inhibitor of cholesterol biosynthesis, shows a synergistic effect with N,N′-bis(2-chloroethyl)-N-nitrosourea and β-interferon on human glioma cells. Cancer Res. 1992;52:4348–4355. [PubMed] [Google Scholar]
- 26.Dimitroulakos J., Ye L.Y., Benzaquen M., Moore M.J., Kamel-Reid S., Freedman M.H., Yeger H., Penn L.Z. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res. 2001;7:158–167. [PubMed] [Google Scholar]
- 27.Khanzada U.K., Pardo O.E., Meier C., Downward J., Seckl M.J., Arcaro A. Potent inhibition of small-cell lung cancer cell growth by simvastatin reveals selective functions of Ras isoforms in growth factor signalling. Oncogene. 2006;25:877–887. doi: 10.1038/sj.onc.1209117. [DOI] [PubMed] [Google Scholar]
- 28.Wang W., Collie-Duguid E., Cassidy J. Cerivastatin enhances the cytotoxicity of 5-fluorouracil on chemosensitive and resistant colorectal cancer cell lines. FEBS Lett. 2002;531:415–420. doi: 10.1016/s0014-5793(02)03575-5. [DOI] [PubMed] [Google Scholar]
- 29.Werner M., Sacher J., Hohenegger M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br J Pharmacol. 2004;143:715–724. doi: 10.1038/sj.bjp.0705928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller A.C., Kariko K., Myers C.E., Clark E.P., Samid D. Increased radioresistance of EJras-transformed human osteosarcoma cells and its modulation by lovastatin, an inhibitor of p21ras isoprenylation. Int J Cancer. 1993;53:302–307. doi: 10.1002/ijc.2910530222. [DOI] [PubMed] [Google Scholar]
- 31.Graaf M.R., Beiderbeck A.B., Egberts A.C., Richel D.J., Guchelaar H.J. The risk of cancer in users of statins. J Clin Oncol. 2004;22:2388–2394. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 32.Boudreau D.M., Gardner J.S., Malone K.E., Heckbert S.R., Blough D.K., Daling J.R. The association between 3-hydroxy-3-methylglutaryl coenzyme A inhibitor use and breast carcinoma risk among postmenopausal women: a case-control study. Cancer. 2004;100:2308–2316. doi: 10.1002/cncr.20271. [DOI] [PubMed] [Google Scholar]
- 33.Gutt R., Tonlaar N., Kunnavakkam R., Karrison T., Weichselbaum R.R., Liauw S.L. Statin use and risk of prostate cancer recurrence in men treated with radiation therapy. J Clin Oncol. 2010;28:2653–2659. doi: 10.1200/JCO.2009.27.3003. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen S.F., Nordestgaard B.G., Bojesen S.E. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367:1792–1802. doi: 10.1056/NEJMoa1201735. [DOI] [PubMed] [Google Scholar]
- 35.Kawata S., Yamasaki E., Nagase T., Inui Y., Ito N., Matsuda Y., Inada M., Tamura S., Noda S., Imai Y. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br J Cancer. 2001;84:886–891. doi: 10.1054/bjoc.2000.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang K., Ang K.K., Milas L., Hunter N., Fan Z. The epidermal growth factor receptor mediates radioresistance. Int J Radiat Oncol Biol Phys. 2003;57:246–254. doi: 10.1016/s0360-3016(03)00511-x. [DOI] [PubMed] [Google Scholar]
- 37.Thibault A., Samid D., Tompkins A.C., Figg W.D., Cooper M.R., Hohl R.J., Trepel J., Liang B., Patronas N., Venzon D.J. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2:483–491. [PubMed] [Google Scholar]
- 38.Knox J.J., Siu L.L., Chen E., Dimitroulakos J., Kamel-Reid S., Moore M.J., Chin S., Irish J., LaFramboise S., Oza A.M. A phase I trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur J Cancer. 2005;41:523–530. doi: 10.1016/j.ejca.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 39.Kim W.S., Kim M.M., Choi H.J., Yoon S.S., Lee M.H., Park K., Park C.H., Kang W.K. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Invest New Drugs. 2001;19:81–83. doi: 10.1023/a:1006481423298. [DOI] [PubMed] [Google Scholar]
- 40.Billecke S., Draganov D., Counsell R., Stetson P., Watson C., Hsu C., La Du B.N. Human serum paraoxonase (PON1) isozymes Q and R hydrolyze lactones and cyclic carbonate esters. Drug Metab Dispos. 2000;28:1335–1342. [PubMed] [Google Scholar]