

Abstract
Significance: Premature and sick neonates are often exposed to high concentrations of oxygen, which results in lung injury and long-term adverse consequences. Nevertheless, neonates are more tolerant to hyperoxia than are adults. This may be, in part, explained by the high lung content of heme oxygenase-1 (HO-1), the rate-limiting enzyme in the degradation of heme and an important stress protein. The abundance of HO-1 dictates its cytoprotective and deleterious effects. Interestingly, in response to hyperoxia, lung HO-1 mRNA is not further up-regulated in neonates, suggesting that lung HO-1 gene expression is tightly regulated so as to optimize cytoprotection when faced with an oxidative stress such as hyperoxia. Recent Advances: In addition to the lack of induction of HO-1 mRNA, neonatal lung HO-1 protein is observed in the nucleus in neonatal mice exposed to hyperoxia but not in adults, which is further evidence for the developmental regulation of HO-1. Nuclear HO-1 had unique properties independent of its enzymatic activity. In addition, there has been increasing evidence that nuclear HO-1 contributes to cellular proliferation and malignant transformation in several human cancers. Critical Issues: Since HO-1 has dual effects in cytoprotection and cellular proliferation, the titration of HO-1 effects is critical to ensure beneficial actions against oxidative stress. Future Directions: Much more has to be understood about the specific roles of HO-1 so as to manipulate its abundance and/or nuclear migration to maximize the therapeutic benefit of this pleiotropic protein in the neonatal lung. Antioxid. Redox Signal. 21, 1881–1892.
Introduction
Sick and premature newborns are often exposed to high concentrations of oxygen, which leads to arrested alveolar and vascular development as seen in bronchopulmonary dysplasia (BPD) (6, 44, 45, 102), This has long-term implications for lung function in adolescence and adulthood, and it also impacts neurodevelopmental outcomes (26, 27, 34, 111, 112). The lung injury observed in BPD results, in part, from reactive oxygen species (ROS), which damage DNA and other molecules (7, 12, 14). Fortunately, antioxidant responses have evolved and protect against ROS, including heme oxygenase (HO), a stress protein that degrades heme to biliverdin. There are two forms of HO that have different roles: different regulation and different post-translational modifications (85). HO-1 is highly inducible in inflammation and hyperoxia among other stressors and has multiple transcriptional factor binding sites that regulate its induction with oxidative stresses (3, 56). Although HO is an integral protein of the smooth endoplasm reticulum, it can localize to other compartments, including caveolae (43, 47, 50), mitochondria (9, 19), and the nucleus, where it can mediate signaling functions (61).
This review will focus on how HO-1, through its pleiotropic effects, modulates lung injury and repair, and will describe how the regulation and expression of HO-1 is unique in the neonatal lung.
Oxidative Stress and Neonatal Lung Injury
The lung continues to develop in complexity and size throughout the postnatal period and into early childhood. At birth, the transition to air breathing and away from the low oxygen tension provided by the placental circulation represents an oxidative stress. In preterm human neonates, exposure to hyperoxia, inflammation, and ventilation at this critical time in development results in disruption of the normal developmental process in the vasculature, mesenchyme, and alveolar structure of the lung. This leads to a disruption of angiogenesis, increased fibroblast proliferation, and arrested alveolar development, resulting in abnormal lung architecture with decreased ventilation/perfusion matching and impaired pulmonary compliance characteristic of BPD. In the mouse, high concentrations of oxygen alone can result in decreased cell proliferation, decreased pulmonary compliance, and altered architecture (109) as in BPD. ROS produced during hyperoxia cause DNA strand breaks and other chromosomal aberrations (7, 12, 14), which stimulate the expression of the genes involved in inhibiting cell cycle-progression (95). The majority of strand breaks occur in small airway epithelial (Type II) cells (84, 116). This can result in the simplification of alveoli as seen in BPD. In addition, the DNA strand breaks from hyperoxia lead to activation of the ataxia telangiectasia mutant (ATM)-related protein kinase-dependent p53 phosphorylation (53), which can result in either arrest or induction of transcription, induction of signal transduction pathways such as the serine threonine kinase AKT and phospho-Extracellular signal-regulated kinase (p-ERK), replication errors, and genomic instability, all of which are seen in carcinogenesis (1, 46) (Fig. 1).
FIG. 1.
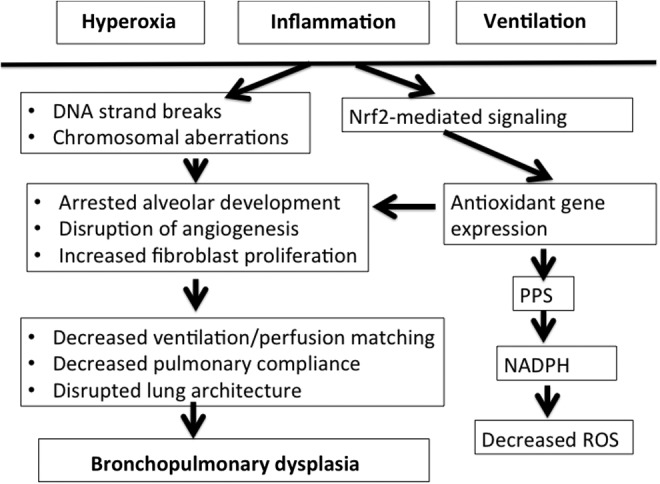
Pathophysiology of bronchopulmonary dysplasia (BPD). In the neonatal lung, hyperoxia, ventilation and inflammation contribute to changes in cellular function, leading to blunted repair and persistent distortion in lung architecture. In addition, oxidative-mediated signaling via NF-E2-related factor 2 (Nrf2) results in activation of the pentose phosphate shunt (PPS) with resultant conversion of NADP to NADPH. This provides reducing equivalents to detoxify reactive oxygen species (ROS).
Increased ROS also result in the activation of NF-E2-related factor 2 (Nrf2)-mediated pathways, which are a hallmark of the oxidative stress response, leading to the up-regulation of antioxidant defenses (16, 65, 83). This activation can also change metabolic signaling, resulting in the up-regulation of glucose-6-phosphate dehydrogenase (G6PDH) (103), the rate-limiting enzyme of the pentose phosphate shunt (PPS). With the latter, glucose is oxidized and nicotinamide adenine dinucleotide phosphate (NADPH) is produced, which provides reducing equivalents that detoxify ROS. In addition, the PPS facilitates the generation of ribose for the synthesis of macromolecules (Fig. 1). Although protective against oxidative stress, this response may enable the rapid proliferation of cancer cells even in adverse environments (10, 25).
Overall, oxidative stress in the lung affects the particularly vulnerable endothelial cells as well as alveolar type II cells, which are important in the recovery from lung injury. This leads to arrested alveolar development (6, 44, 45) as well as to the disruption of angiogenesis (102), which are characteristics of BPD. Protective responses against all aspects of this disease could mitigate this disease process. This article will explore the multiple roles of the antioxidant molecule HO in this process.
Important Aspects of HO-1 Regulation in Oxidative Stress
Induction of HO-1 is a generalized response to oxidative stress
There are two isoforms of HO. The constitutive form, HO-2 is found in abundance in the testes and brain and can be regulated by glucocorticoids via a glucocorticoid response element, but it is not readily inducible during oxidative stress (82). It plays a role in various signaling processes and neurotransmission. We have shown that HO-2 null mutant mice have increased evidence of oxidative stress after exposure to hyperoxia and that they accumulate reactive iron in the lung tissues, which exacerbates oxidative injury (22), proving that in neonatal animals, HO-2 also represents an important, albeit noninducible oxidative defense against hyperoxia.
The inducible isoform, HO-1, responds to the most oxidant stresses. This occurs via binding of the Nrf2/small Maf protein complex to the Maf recognition sites (multiple antioxidant response element [MARE]) (2, 3) (Fig. 2). Competitive binding between Nrf2 and heterodimer of BTB and CNC homology 1 (Bach1) at the MARE is important in down-regulating HO-1 expression (51, 98). Recently, transforming growth factor (TGF)-β was seen to suppress the transcriptional activation of HO-1 through Nrf2-independent mechanisms. In fact, TGF-β did not affect the stabilization or nuclear accumulation of Nrf2 but induced the expression of Maf K and Bach1, which suppresses HO-1 transcription despite the accumulation of Nrf2 in the nucleus. Knockdown of Maf-1 and Bach1 abolished the TGF-β-dependent suppression of HO-1 through the substitution of Nrf2 for Bach1 on the MARE of HO-1 (72). Other binding sites found on the proximal and distal enhancer regions of the HO-1 promoter include STAT and NF-KB, which also regulate HO-1 gene transcription (58).
FIG. 2.
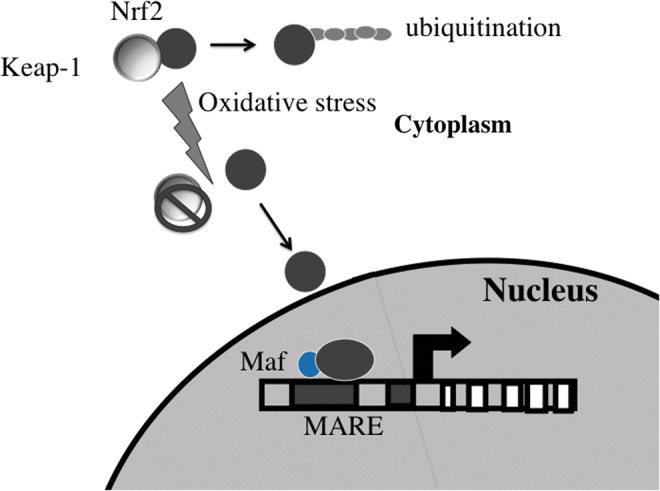
Activation of the Nrf2 pathway with oxidative stress. Nrf2 is sequestered in the cytoplasm with Keap-1, facilitating its ubiquination and subsequent degradation. With oxidative stress, Nrf2 is released and migrates to the nucleus, where it binds to multiple antioxidant response elements (MARE) on heme oxygenase (HO)-1 and other genes to mediate gene transcription. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
It is important to note that the Nrf2 signaling pathway not only results in HO-1 induction but also drives the expression of enzymes such as NAD(P)H:quinone reductase, an important electrophile-detoxifying enzyme, and G6PDH, the rate-limiting step in the PPS, thereby enhancing the generation of NADPH and reducing the generation of ROS (66). Nevertheless, the same genes downstream of Nrf2 may alter cellular metabolic fate and enable lung cancer cells to grow more rapidly (25). This will be discussed briefly in a later section.
How the enzymatic functions of HO-1 influence oxidative stress
HO enables the cleavage of heme specifically at the alpha-methene bridge of the molecule, in a multistep manner. Iron is then reduced to its ferric state through the action of cytochrome cP450 reductase. Carbon monoxide (CO) is released by elimination of the alpha methylene bridge of the porphyrin ring (Fig. 3). Each byproduct of HO is considered as having a significant signaling or cytoprotective function. Heme is a pro-oxidant molecule that can participate in the formation of oxidative radicals, leading to oxidative injury. Therefore, the sequestration of heme and the subsequent degradation by HO has antioxidant benefits. CO has important biological roles, including neurotransmission, vasodilation, and signal transduction. This product is unique to the HO reaction. Currently, the use of CO-releasing molecules (CORM) is being investigated as a cytoprotective strategy in several clinical models (37, 41, 52, 55). Biliverdin is an important antioxidant that can prevent lipid peroxidation (110). This compound does not accumulate endogenously and is rapidly converted to bilirubin by biliverdin reductase. Bilirubin is documented to decrease lipid peroxidation even better than vitamin E (97). Overall, by sequestering heme and forming antioxidant and bioreactive molecules, HO and bilirubin can lead to cytoprotection against oxidative injury. Despite the antioxidant benefits of HO-1 byproducts, there are examples where even an inactive form of HO-1 is cytoprotective (38). The exact mechanisms for this effect are not yet well characterized.
FIG. 3.
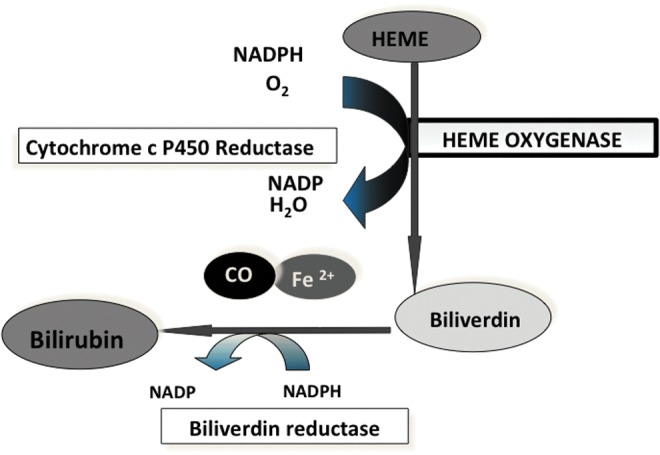
Catalytic reaction of HO. Heme is degraded in an energy requiring process to biliverdin. This is then converted to bilirubin by the nonrate limiting biliverdin reductase. Iron (Fe) and carbon monoxide (CO) are released in equimolar amounts. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Subcellular localization of HO-1 and implications for oxidative stress
Although HO-1 is predominantly found in the smooth endoplasmic reticulum, where it is anchored at its c-terminus, it has been identified in the nuclear compartment and the nuclear envelope (33, 61, 73, 86) as well as in the mitochondria (19, 93) and caveolae (43, 47, 50).
In the mitochondria, a 27 kD HO-1 immunoreactive fragment was increased in a model of hepatotoxicity. The induction of mitochondrial HO-1 improved respiration and prevented a further drop in ATP levels (70). In cultured A549 cells as well as in primary small airway and epithelial cell cultures, mitochondrial localization was observed. In addition, in vivo, after exposure to cigarette smoke, this phenomenon occurred. The over-expression of HO-1 inhibited cigarette smoke-induced cell death and preserved cellular ATP levels (93). Therefore, the compartmentalization of HO-1 in the mitochondria may help protect against cigarette smoke-induced cell death. This form of HO-1 increases mitochondrial heme turnover, preserves liver ATP levels and energy metabolism (19). The mechanism by which HO-1 migrates to the mitochondria is not known, and no mitochondrial targeting sequence has been found on the HO-1 protein. The process may involve deletion of the C-terminus (93).
When HO-1 was over-expressed, it could be recovered in a detergent-resistant fraction containing caveolin-1 and was found in plasma membrane, cytosol, and isolated caveolae. Caveolin-1 physically interacted with HO-1, and HO activity increased in cells expressing caveolin-1 antisense transcripts, suggesting a negative regulatory role for caveolin-1 in the expression of HO-1 (50). Others also confirmed the binding of HO-1 protein to caveolin-1 using immuno-precipitation experiments (43, 47). In addition, HO-1 activity can be inhibited by caveolin-1. This binding occurred in the caveolin scaffolding domain, which plays an essential role in caveolin-related protein–protein interactions. The inhibition of HO activity by caveolin was correlated inversely to hemin concentration, suggesting that caveolin and hemin share a common binding site on the HO-1 protein (100). The proper distribution of HO-1 to caveolae appears to be required for normal toll-like receptor (TLR) signaling in response to inflammation (117).
With exposure to hypoxia and to hemin, we observed a faster migrating HO-1 immunoreactive band, which was enriched in nuclear extracts, suggesting that HO-1 could be cleaved to enables nuclear entry (61). The absence of HO-1 immunoreactive signal with an antibody against the C-terminus confirmed this as did the absence of a C-terminal sequence by gas chromatography/mass spectrometry. Furthermore, nuclear entry could be prevented by preincubation with a cysteine protease inhibitor, demonstrating the necessity for protease-mediated C-terminal cleavage for the nuclear transport of HO-1 (61). Nuclear localization was also associated with a reduction of HO activity (61). We also demonstrated that nuclear HO-1 regulates the activation of various transcription factors, including AP-1, an important mediator of the antioxidant response (61). In preliminary work, we document the activation of Nrf2 by HO-1 protein and the lack of ARE activation in HO-1 null mutant mouse embryonic fibroblast (MEF) cells transfected with an ARE-driven luciferase reporter, further indicating that not only does Nrf2 regulate HO-1 but also HO-1, in turn, regulates Nrf2 (Biswas, unpublished observations, 2013). Interestingly, despite reduced activity, nuclear HO-1 protected cells against hydrogen peroxide-mediated toxicity and prevented oxidative DNA damage in HO-1 null mutant MEF cells stably infected with nuclear HO-1 that were exposed to hyperoxia (61). The effects of nuclear HO-1 appear to be cell specific. After exposure to hyperoxia, cultured tracheal smooth muscle cells that were recovered from aborted human fetuses showed nuclear distribution of HO-1 only when they were not proliferating (73) Our studies reveal that neonatal mice do not induce HO-1 but have increased nuclear HO-1 in response to hyperoxia in the acute phase of hyperoxic exposure, but this nuclear expression does not persist during room air recovery (Yang, unpublished observations). This pattern of over-expression may be beneficial, because adults that induce lung HO-1 and do not demonstrate nuclear localization are more susceptible to oxidative stress. Corroborating this, mouse HO-1 null mutant MEFs with stable over-expression of nuclear HO-1 show decreased cellular proliferation and are relatively tolerant to 24 h of hyperoxia compared with MEFs expressing cytoplasmic HO-1 or empty vector controls (Fernando, unpublished observations). Intriguingly, transgenic mice over-expressing nuclear HO-1 in type II cells initially showed improved alveolarization with (3 days) hyperoxic exposure but had increased oxidative DNA damage and abnormal lung histology and pulmonary function tests as adults (69), raising the possibility that the duration of nuclear HO-1 protein signaling is key to cytoprotective responses to injury and repair.
Overall, these observations suggest that the level, localization, and duration of expression of HO-1 may be extremely important in determining its cytoprotective and proliferative effects.
Physiologic Effects of HO-1 That Influence Oxidative Lung Injury
To mitigate neonatal lung injury and enhance repair, HO would need to prevent the key pathologic aspects of BPD, namely increased oxidative stress, increased inflammation, disrupted vascular development, and disrupted alveolarization. All of these roles have been documented for HO-1, in particular. In addition, maladaptive consequences of the proliferative actions of HO-1 have been observed
HO-1 abundance and localization alters cellular differentiation and proliferation
Although cell proliferation is disrupted by hyperoxia, others have suggested that decreased cell proliferation may be beneficial in acute hyperoxic injury (57, 71). Nevertheless, long-term suppression of cell proliferation could lead to arrested lung development. In vivo, disruption of HO-1 in the neonatal mice had little effect at 3 days or exposure (23), but when the animals were allowed to recover in air for 11 days, they had significant dysregulation of cell-cycle gene expression compared with similarly exposed wild-type (WT) (115). In vitro, tracheal smooth muscle cells from human fetuses exposed to hyperoxia showed nuclear distribution of HO-1 only when they were in a nonproliferative state (73). These results are in agreement with several other publications showing that HO-1 is anti-proliferative in smooth muscle cells (59, 75). In contrast, in epithelial cells, HO activity is associated with pro-proliferative effects (18) and, in endothelial cells, knockdown of HO-1 suppressed proliferation (118). HO-1 also influences naive T-cell homeostatic proliferation (13) and in the differentiation of induced pluripotent stem cells in response to oxidative stress (60), as well as Wnt signaling-mediated differentiation of preadipocytes to adipocytes. (106).
To maximize the cytoprotective effects HO-1, one should account for the specific effects of its subcellular localization and expression levels. To this effect, lung (Type II cell)-specific transgenic mice expressing high or low levels of full-length HO-1 (cytoplasmic) or C-terminally truncated HO-1 (nuclear) were generated (69). Mice were exposed to hyperoxia for 3 days as neonates and then allowed to recover in room air for approximately 8 weeks. During recovery from hyperoxia, the mice expressing low levels of full-length HO-1 had normal alveoli and minimal oxidative damage, whereas those expressing high levels of HO-1 had increased alveolar wall thickness with type II cell hyperproliferation, worsened pulmonary function, and evidence of abnormal lung cell hyperproliferation at 8 weeks of age. In the mice expressing C-terminally truncated HO-1 in the nucleus, increased lung DNA oxidative damage, increased poly (ADP-ribose) polymerase protein expression, and reduced poly (ADP-ribose) hydrolysis as well as reduced pulmonary function were observed during recovery from hyperoxia. This demonstrates that low cytoplasmic levels of HO-1 protect against hyperoxia-induced lung injury by attenuating oxidative stress, whereas high cytoplasmic levels worsen lung injury by increasing type II cell proliferation, alveolar wall thickness, thereby impeding gas exchange. Enhanced lung nuclear HO-1 impairs recovery by disabling poly (ADP-ribose)-dependent regulation of DNA repair (69). Interestingly, when HO-1 null mutant mice exposed to hyperoxia as neonates were evaluated after 11 days of recovery in room air, these mice exhibited significant changes in lung alveolarization and altered expression of genes which were important in cell proliferation and DNA damage (115). With regard to fibroblasts' myofibroblast proliferation, myocardial infarct was reduced with both in vivo and in vitro HO-1 over-expression (62) Overall, as a regulator of cell proliferation and differentiation, moderate levels of HO-1 could have a significant impact on lung injury and repair processes in hyperoxia. Perhaps in the neonatal lung exposed to hyperoxia, basal moderate over-expression of HO-1 in epithelial and endothelial cells promotes their proliferation; whereas it suppresses the over-proliferation of smooth muscle cells and fibroblasts, thereby maintaining lung vascularization and alveolarization while suppressing pulmonary hypertension and fibrosis during the repair phase. This would mitigate the phenotypic changes of BPD (Fig. 4).
FIG. 4.
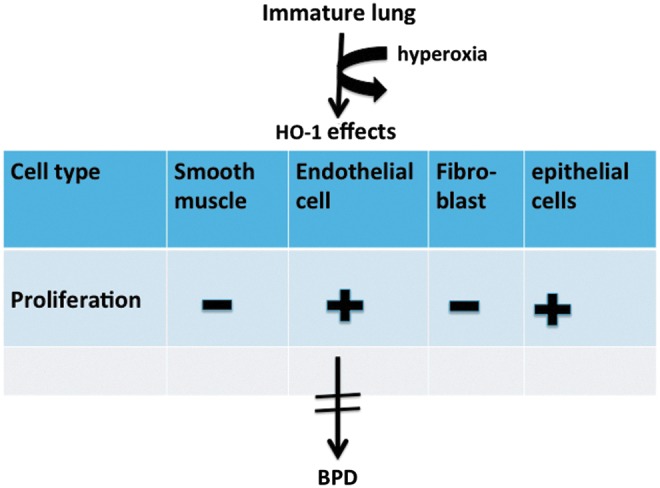
Effects of HO-1 on cell proliferation. Increased proliferation is shown by a+, decreased proliferation by a −, based on existing literature. In the developing lung exposed to hyperoxia, the net effect of HO-1 on the different cell lineages prevents the phenotypic changes of BPD. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
HO-1 reduces inflammation
Through the degradation of heme, HO may have cytoprotective effects against systemic infections (Fig. 5). The regulation of HO-1 expression in macrophages was strictly required for protection against mycobacterial infection in mice, and HO-1-deficient mice were more susceptible to intravenous mycobacterium avium infections, and failed to mount a protective granulomatous response in mice lacking mature B cells (91). In a mouse model of noneosinophilic asthma, HO-1 provided anti-inflammatory effects by inhibiting the p-STAT3-RORγt pathway (120). HO can also work along with the scaffold protein caveolin-1 and negatively regulate TLR-4 signaling. In LPS-challenged cystic fibrosis macrophages, HO-1 accumulated intracellularly. The over-expression of HO-1 or the stimulation of CO release with a CORM enhanced caveolin-1 expression in the macrophages and re-established HO-1 cell surface localization, which restored the normal TLR signaling pattern (119). Mice deficient in Irak-M, an important regulator of TLR-4, that were exposed to 95% oxygen had reduced mortality compared with WT mice, and this was associated with increased expression of HO-1 and Nrf2 (5). Treatment of the mice in vivo and incubation of cells in vitro with metalloporphyrins that suppress HO activity decreased survival and reduced the number of live cells after hypoxic exposure; this attenuated anti-inflammatrory cytokines, including interleukin-10, and up-regulated pro-inflammatory cytokines (4). In a premature lamb model, intra-amniotic endotoxin decreased lung caveolin-1 expression. This was associated with increased expression of HO-1 (54). It was not obvious whether this induction of HO-1 later resulted in improved lung histology or function. The HO-1 null mice developed by Poss and colleagues (77, 78) show evidence of oxidative damage and chronic inflammation. Surprisingly, the exposure of neonatal (<12 h old) HO-1 null mice to hyperoxia did not result in increased lung inflammation compared with WT littermates, suggesting developmental differences in the effects of HO-1 (23).
FIG. 5.
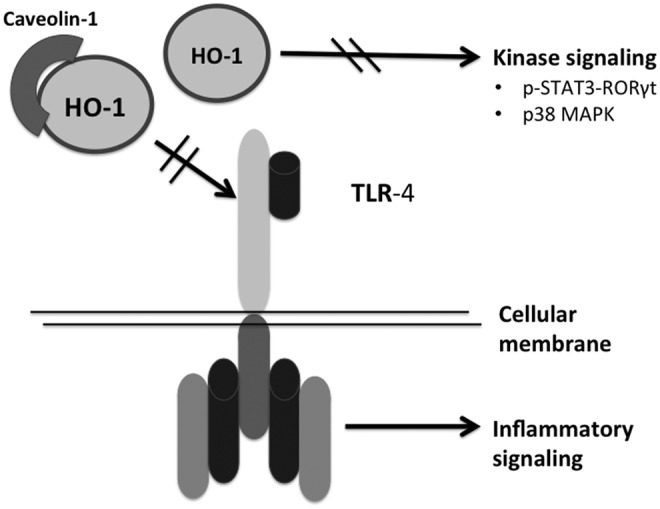
Mechanisms by which HO-1 influences inflammation. HO-1 can bind to caveolin to prevent toll-like receptor (TLR)-4 signaling. In addition, the activation of p-STAT3-RORγ and p38 MAP kinase signaling is reduced by HO-1, which also dampens inflammatory responses.
Since inflammation is an important component of BPD, abundance and caveolar localization of HO-1 may serve to mitigate this disease process.
Multiple roles of HO-1 in angiogenesis and vascular proliferation
There is significant evidence in both humans and animals that the disruption of lung vascular development disrupts alveolarization. In vitro, transfection with the human HO-1 gene increased blood vessel formation (24). In endothelial cells, hemodynamically relevant cyclic strain stimulated HO-1 gene expression and inhibited cell death (63). In vivo, lentiviral vectors with microRNA sequences controlled by vascular endothelium cadherin were used to study the role of lung endothelial HO-1 in mice exposed to hyperoxia (119). When HO-1 was knocked down by 55% in the lung endothelium, there was a twofold increase in apoptosis and ROS generation, and this had the same effect on lung injury and survival as silencing HO-1 in multiple lung cell types. Furthermore, HO-1 regulated caspase 3 activation and autophagy in the endothelium during hyperoxia (119). Pulmonary inflammation arterial remodeling and right ventricular hypertrophy were attenuated in transgenic mice over-expressing HO-1 in Type II cells exposed to hyperoxia. Type II cell-specific over-expression of HO-1 also reduced hyperoxia-mediated pulmonary edema, hemosiderosis and prevented the loss of blood vessels observed in similarly exposed WT animals (28). Interestingly, lung-specific HO-1 over-expression neither prevented alveolar simplification nor altered ferritin and lactoferrin levels in this model, suggesting that HO-1 over-expression primarily protects the vascular system through iron-independent antioxidant and anti-inflammatory pathways (28). In summary, HO-1 plays an important vasculoprotective role in the lung, and this could be beneficial for preventing BPD.
Clues of the role of HO-1 in lung cytoprotective defenses in humans
So far, there have been only two reported cases of HO-1 deficiency in humans. A 6-year-old boy with severe growth restriction was evaluated for persistent hemolytic anemia with a paradoxical absence of hyperbilirubinemia. He also suffered from severe endothelial damage as well as from iron deposition in his kidneys and liver. Sequence analysis revealed complete loss of exon 2 of the maternal allele and a two-nucleotide deletion within exon 3 of the paternal allele for HO-1 (113). Another case of human HO-1 deficiency was recently reported in a 12 year-old girl with congenital asplenia, who presented with severe hemolysis, inflammation, and nephritis, refractory to therapy. Mutation analysis showed homozygous missense mutations in exon 2 on chromosome 22q12, which would result in the absence of the functional HO-1 protein. Furthermore, the patient's kidneys were devoid of HO-1 immunostain (81). These two cases show common phenotypes involving inflammation, hemosiderosis, and oxidative stress. There are no reports of abnormal lung function in these patients. Perhaps a second insult would be needed to unmask the lung phenotype.
Several HO-1 promoter polymorphisms have been documented in lung diseases (30, 36, 90). Since GT dinucleotide repeats in the 5′′ flanking region of the human HO-1 gene can modulate its transcription, differences in GT repeat length could alter HO activity (Fig. 6). The frequency of longer repeat alleles was significantly higher in the smokers with chronic pulmonary emphysema than in smokers without it, suggesting that diminished HO-1 promoter activation was associated with increased susceptibility to emphysema (114). In vitro studies showed that hydrogen peroxide induced HO-1 only in the short and medium promoter repeats, suggesting that longer GT repeats prevent HO-1 induction. Other functional polymorphisms were tested as well. No association between the various single nucleotide polymorphisms of HO-1 and lung function decline could be found, nor was there any evidence that three promoter polymorphisms affected the regulation of HO-1 gene (101). In 44 asbestos-exposed subjects without mesothelioma and 78 asbestos-exposed subjects with mesothelioma, long GT repeats were significantly higher in the asbestos-exposed subjects with mesothelioma, suggesting that decreased induction of HO-1 is associated with a higher risk of malignant mesothelioma (68). In 749 French subjects aged 20–44, lung function was assessed and compared with the length of HO-1 promoter polymorphisms. The long allele carriers showed lower forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) than other noncarriers with steeper decline in FEV1 over time than noncarriers. In addition, HO-1 in the serum was lower in rapid decliners than in normal decliners, suggesting that HO-1 may be a predictor of lung function decline in these patients (87). Overall, lower inducibility of HO-1 is associated with worsened lung disease in humans. As evidence in children and neonates, a boy with marked elevation of serum bilirubin during autoimmune hemolytic anemia was seen to be a homozygous carrier of short GT dinucleotide-repeat promoter polymorphism (42), and short GT repeat alleles have been associated with prolonged neonatal jaundice (11). No information exists about neonatal lung disease and HO-1 promoter polymorphisms.
FIG. 6.
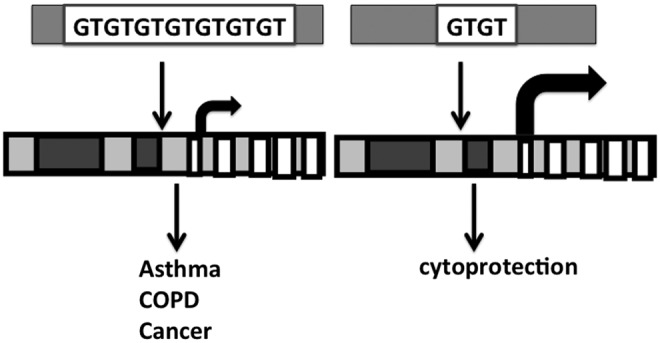
Proposed effects of promoter polymorphisms on HO-1 in lung disease. Long GT repeats on the HO-1 promoter (left) are associated with the development of several lung pathologic states. It remains to be determined whether short GT repeats are protective against lung disease.
Maturation alters the role and regulation of HO-1 in oxidative stress
Overall, HO-1 can exert pleiotropic effects in the lung depending on localization and abundance (Fig. 7). This is a very important consideration when devising therapeutic strategies using HO-1 to prevent BPD.
FIG. 7.
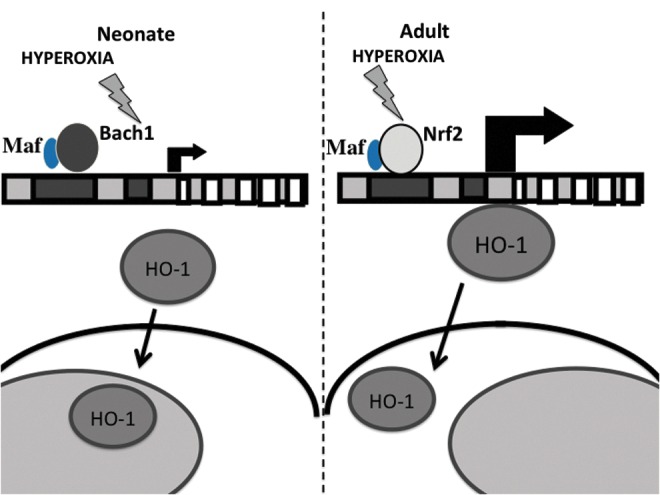
Maturational differences in HO-1 gene regulation and protein localization. In response to hyperoxia, neonates (left) do not up-regulate HO-1 mRNA but translocate HO-1 to the nucleus. In contrast, adults (right) induce HO-1 mRNA but do not translocate HO-1 to the nucleus. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Most studies on HO-1 have been done in adult animals. Neonatal rodents are more tolerant to hyperoxia and have a high expression of lung HO-1 than do adults (29, 96). This is not attributable to less inflammation (8, 32, 89). Theoretically, antioxidant defenses up-regulated at birth could protect the lung against oxidative injury (29). In fact, HO-1 is found at highest levels in the first postnatal days and then decreases to adult values by the second week of life (21). There may be a benefit to having high constitutive levels of this cytoprotective molecule at the time of delivery, where the neonatal animal transitions to the relatively hyperoxic ex-utero environment. At birth, with the transition from the placenta to air breathing, pulmonary vessels are exposed to oxidative stress and undergo remodeling. Although some argue that the lungs from HO-1-deficient mice develop normally after birth, which suggests that HO-1 induction plays no role in the development of the vasodilator response and remodeling which occurs at birth (94), we show evidence to the contrary. In fact, HO-1 null mutant mice showed mild alveolar simplification, disorganization, and reduced secondary crest formation. These defects were more pronounced when these mice were challenged with dexamethasone treatment. The latter further decreased levels of both endothelial and alveolar epithelial markers (121).
Despite the cytoprotective effects of moderate HO-1 over-expression, we have shown that at high levels, HO-1 may be deleterious (108), resulting in enhanced oxidative stress and apoptosis and decreased cell proliferation in vitro (99) and, when targeted to type II cells in vivo, this leads to a maladaptive over-proliferation of type II cells and perhaps a failure to differentiate to type I cells which are important for recapitulation of the normal alveolar epithelium. This then manifests as increased alveolar thickness and decreased lung function (69). In neonatal rats exposed to hyperoxia, no significant increase in lung HO-1 mRNA was seen in contrast to similarly exposed adults (21). In addition, with prolonged hyperoxic exposure, lung HO-1 mRNA only increased after 10 days in neonatal mice (107); whereas this occurred within 24 h in adult mice (74). Furthermore, neonatal lungs have enhanced expression of Bach1, suggesting a developmental cue to prevent further up-regulation of HO-1 in hyperoxia (48). These differences may explain the lack of up-regulation of neonatal lung HO-1 in hyperoxia, but other mechanisms could also be involved. The role of microRNAs in regulating HO-1 abundance is being explored (39, 40, 79, 80, 92, 119), but it is not known whether there are developmental differences in microRNAs that explain both the increased abundance and the relative lack of hyperoxic induction of lung HO-1 in neonatal mice.
Using a pig and mouse model, HO-1 expression was investigated during adaptation to extrauterine life. HO-2 expression was constitutive, whereas HO-1 protein was induced after birth in the blood vessels and airways, peaking at 14 days in the pig and at 4 days in the mouse. Inhibitors of HO-1 did not alter vasodilatory responses in the pigs (94), suggesting that these effects may not be related to the enzymatic activity of HO-1.
In the acute phase of hyperoxic exposure (3 days), neonatal mice have increased nuclear HO-1 compared with similarly exposed adults (115). This pattern of over-expression may be beneficial, because adults that induce lung HO-1 and do not demonstrate nuclear localization are more susceptible to oxidative stress. Corroborating this, HO-1 null mutant MEFs with stable over-expression of nuclear HO-1 show decreased cellular proliferation and are relatively tolerant to 24 h of hyperoxia compared with MEFs expressing cytoplasmic HO-1 or empty vector controls (Fernando, unpublished observations). Intriguingly, transgenic mice over-expressing nuclear HO-1 in type II cells initially showed improved alveolarization with (3 days) hyperoxic exposure but had increased oxidative DNA damage and abnormal lung histology and pulmonary function tests as adults (69), raising the possibility that the duration of nuclear HO-1 protein signaling is key to cytoprotective responses to injury and repair.
Overall, these observations suggest that the level, localization, and duration of expression of HO-1 may be extremely important in determining its cytoprotective and proliferative effects in the neonatal lung.
Maladaptive Consequences of HO-1 Overexpression and Cellular Localization in the Lung
Obviously, HO-1 plays a significant role in cellular proliferation. Important features of tumorigenesis are excessive proliferation and invasiveness (67, 105). Therefore, by enhancing cellular proliferation and mitigating oxidative stress, HO-1 could also promote tumor cell growth (Fig. 8). Many examples suggest that HO-1 abundance and localization are associated with tumor formation (20, 31). In nonsmall lung cells, cancer patients with a high HO-1 expression ratio in tumor tissue compared with normal tissue had a significantly poorer prognosis and a higher metastatic rate compared with those with a low HO-1 expression ratio (20). In vitro, the invasive and migratory abilities of A549 and H441 lung cancer cells significantly increased after high (20-fold increase) exogenous HO-1 over-expression and significantly decreased after HO-1 silencing. Furthermore, HO-1 over-expression positively correlated with the expression of metastasis-associated proteins (104). Adenocarcinoma is the most prevalent subtype of lung cancer, and it is often associated with mutations in the Kras oncogene (49). MAP kinase signal amplification characteristic of Kras lung adenocarcinomas drives toward the progression of malignancy (64). These result in constitutive signaling, which regulates proliferation, differentiation, and cell survival (15). Oncogenic ras alone results in a permanent G1 arrest, as in senescence, but can transform most immortal rodent cells to a tumorigenic state with the inactivation of tumor suppressors such as p53 (88). Growing tumors also preferentially utilize glycolysis over mitochondrial oxidative phosphorylation for glucose-dependent ATP production even in the presence of oxygen (35, 76). In addition to energy, glycolysis generates intermediates that are important to cell growth such as ribose-5-phosphate, a key intermediate in nucleotide biosynthesis (17) which supports the proliferation of tumor cells. In neonatal HO-1 null mutant mice and WT littermates exposed to hyperoxia for 3 days and allowed to recover in room air for 11 days, six DNA damage-response genes were down-regulated in the WT; whereas these were up-regulated many-fold in the knockout, suggesting that HO-1 disruption modifies DNA repair pathways which are important in tumorigenesis (115). We also show that transgenic mice with cytoplasmic over-expression of HO-1 in type II cells exposed to 3 days of hyperoxia as neonates had increased numbers of multinucleated hyper-proliferating type II cells and foamy macrophages but no evidence of fibrosis or inflammation (69). This correlated with lung lesions on MRI with enhanced p-ERK (which is seen in early tumorigenesis) and PCNA staining (69). Interestingly, adult nuclear HO-1 transgenics exposed to hyperoxia as newborns also had increased p-ERK (69). Furthermore, HO-1 null mutant MEF cells stably transfected with nuclear HO-1 showed the most migration toward EGF in an agarose assay, suggesting that nuclear HO-1 is a stronger stimulus for abnormal cell migration than cytoplasmic HO-1 (69). Lastly, G6PDH synthesis and activity was facilitated in nuclear HO-1-infected MEFs more so than in cytoplasmic HO-1-infected MEFs or empty vector controls (Biswas, unpublished observations). This suggests that nuclear HO-1 in conjunction with hyperoxia results in a metabolic switch that favors cancer cell survival. The precise mechanisms by which abundance and localization of HO-1 influence lung tumorigenesis in hyperoxia remain to be determined.
FIG. 8.
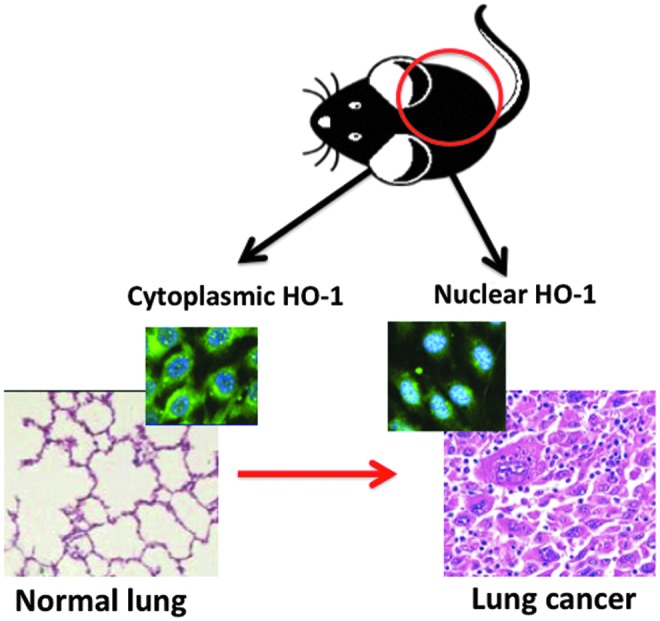
Association of nuclear HO-1 with lung cancer. In both rodents and humans, increased nuclear distribution of HO-1 (inset on the right where DAPI nuclear stain and HO-1 are co-localized as shown by the cyan color) is associated with abnormal lung histology (as shown in the hematoxylin and eosin-stained tissue on the right). With cytoplasmic localization of HO-1 (left inset), lung tissue histology is more likely to be normal (left). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Conclusions
HO is a complex and pleiotropic protein that has multiple roles depending on its abundance, intracellular localization, and duration of action. In the lung, many examples demonstrate its beneficial cytoprotective effects. Nevertheless, equal evidence exists which shows that abnormally high levels are detrimental. In the neonatal lung, HO-1 is found at high abundance but is not further inducible in response to hyperoxia, suggesting the importance of tight regulation of this protein. Furthermore, excessive induction of HO-1 may have abnormal consequences, including favoring a tumorigenic phenotype. In order to take advantage of this important protein, caution should be taken to enhance its cytoprotective properties while preventing adverse effects due to excessive expression, prolonged action, or subcellular localization.
Abbreviations Used
- Bach1
heterodimer of BTB and CNC homology 1
- BPD
bronchopulmonary dysplasia
- CO
carbon monoxide
- CORM
CO-releasing molecules
- G6PDH
glucose-6-phosphate dehydrogenase
- HO
heme oxygenase
- MARE
multiple antioxidant response element
- MEF
mouse embryonic fibroblast
- NADPH
nicotinamide adenine dinucleotide phosphate
- Nrf2
NF-E2-related factor 2
- p-ERK
phospho-Extracellular signal-regulated kinase
- PPS
pentose phosphate shunt
- ROS
reactive oxygen species
- TGF
transforming growth factor
- TLR
toll-like receptor
Acknowledgment
This work is funded by grants HL-58752 and HL-111907 from the National Institutes of Health.
References
- 1.Adamson IY, Young L, and Orr FW. Tumor metastasis after hyperoxic injury and repair of the pulmonary endothelium. Lab Invest 57: 71–77, 1987 [PubMed] [Google Scholar]
- 2.Alam J. and Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des 9: 2499–2511, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, and Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071–26078, 1999 [DOI] [PubMed] [Google Scholar]
- 4.An L, Liu CT, Qin XB, Liu QH, Liu Y, and Yu SY. Protective effects of hemin in an experimental model of ventilator-induced lung injury. Eur J Pharmacol 661: 102–108, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Ballinger MN, Newstead MW, Zeng X, Bhan U, Horowitz JC, Moore BB, Pinsky DJ, Flavell RA, and Standiford TJ. TLR signaling prevents hyperoxia-induced lung injury by protecting the alveolar epithelium from oxidant-mediated death. J Immunol 189: 356–364, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bancalari E. Changes in the pathogenesis and prevention of chronic lung disease of prematurity. Am J Perinatol 18: 1–9, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Barker GF, Manzo ND, Cotich KL, Shone RK, and Waxman AB. DNA damage induced by hyperoxia: quantitation and correlation with lung injury. Am J Respir Cell Mol Biol 35: 277–288, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med 15: 223–229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, and Bandyopadhyay U. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem 286: 39387–39402, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bokun R, Bakotin J, and Milasinovic D. Semiquantitative cytochemical estimation of glucose-6-phosphate dehydrogenase activity in benign diseases and carcinoma of the breast. Acta Cytol 31: 249–252, 1987 [PubMed] [Google Scholar]
- 11.Bozkaya OG, Kumral A, Yesilirmak DC, Ulgenalp A, Duman N, Ercal D, and Ozkan H. Prolonged unconjugated hyperbilirubinaemia associated with the haem oxygenase-1 gene promoter polymorphism. Acta Paediatr 99: 679–683, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Buckley S, Barsky L, Driscoll B, Weinberg K, Anderson KD, and Warburton D. Apoptosis and DNA damage in type 2 alveolar epithelial cells cultured from hyperoxic rats. Am J Physiol 274: L714–L720, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Burt TD, Seu L, Mold JE, Kappas A, and McCune JM. Naive human T cells are activated and proliferate in response to the heme oxygenase-1 inhibitor tin mesoporphyrin. J Immunol 185: 5279–5288, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cacciuttolo MA, Trinh L, Lumpkin JA, and Rao G. Hyperoxia induces DNA damage in mammalian cells. Free Radic Biol Med 14: 267–276, 1993 [DOI] [PubMed] [Google Scholar]
- 15.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, and Der CJ. Increasing complexity of Ras signaling. Oncogene 17: 1395–1413, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Cho HY, van Houten B, Wang X, Miller-DeGraff L, Fostel J, Gladwell W, Perrow L, Panduri V, Kobzik L, Yamamoto M, Bell DA, and Kleeberger SR. Targeted deletion of nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid Redox Signal 17: 1066–1082, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, and Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Clark JE, Green CJ, and Motterlini R. Involvement of the heme oxygenase-carbon monoxide pathway in keratinocyte proliferation. Biochem Biophys Res Commun 241: 215–220, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Converso DP, Taille C, Carreras MC, Jaitovich A, Poderoso JJ, and Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J 20: 1236–1238, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Degese MS, Mendizabal JE, Gandini NA, Gutkind JS, Molinolo A, Hewitt SM, Curino AC, Coso OA, and Facchinetti MM. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer 77: 168–175, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dennery PA, Lee CS, Ford BS, Weng YH, Yang G, and Rodgers PA. Developmental expression of heme oxygenase in the rat lung. Pediatr Res 53: 42–47, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Dennery PA, Spitz DR, Yang G, Tatarov A, Lee CS, Shegog ML, and Poss KD. Oxygen toxicity and iron accumulation in the lungs of mice lacking heme oxygenase-2. J Clin Invest 101: 1001–1011, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dennery PA, Visner G, Weng Y, Nguyen X, Lu F, Zander D, and Yang G. Resistance to hyperoxia with heme oxygenase-1 disruption: role of iron. Free Radic Biol Med 34: 124–133, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Deramaudt BM, Braunstein S, Remy P, and Abraham NG. Gene transfer of human heme oxygenase into coronary endothelial cells potentially promotes angiogenesis. J Cell Biochem 68: 121–127, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Dessi S, Batetta B, Cherchi R, Onnis R, Pisano M, and Pani P. Hexose monophosphate shunt enzymes in lung tumors from normal and glucose-6-phosphate-dehydrogenase-deficient subjects. Oncology 45: 287–291, 1988 [DOI] [PubMed] [Google Scholar]
- 26.Doyle LW. Growth and respiratory health in adolescence of the extremely low-birth weight survivor. Clin Perinatol 27: 421–432, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Doyle LW. and Anderson PJ. Long-term outcomes of bronchopulmonary dysplasia. Semin Fetal Neonatal Med 14: 391–395, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Fernandez-Gonzalez A, Alex Mitsialis S, Liu X, and Kourembanas S. Vasculoprotective effects of heme oxygenase-1 in a murine model of hyperoxia-induced bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 302: L775–L784, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank L. Developmental aspects of experimental pulmonary oxygen toxicity. Free Radic Biol Med 11: 463–494, 1991 [DOI] [PubMed] [Google Scholar]
- 30.Fredenburgh LE, Perrella MA, and Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol 36: 158–165, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gandini NA, Fermento ME, Salomon DG, Blasco J, Patel V, Gutkind JS, Molinolo AA, Facchinetti MM, and Curino AC. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp Mol Pathol 93: 237–245, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Gerik SM, Keeney SE, Dallas DV, Palkowetz KH, and Schmalstieg FC. Neutrophil adhesion molecule expression in the developing neonatal rat exposed to hyperoxia. Am J Respir Cell Mol Biol 29: 506–512, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Giordano A, Nisoli E, Tonello C, Cancello R, Carruba MO, and Cinti S. Expression and distribution of heme oxygenase-1 and −2 in rat brown adipose tissue: the modulatory role of the noradrenergic system. FEBS Lett 487: 171–175, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Gough A, Linden M, Spence D, Patterson CC, Halliday HL, and McGarvey LP. Impaired lung function and health status in adult survivors of bronchopulmonary dysplasia. Eur Respir J 2013[Epub ahead of print]; DOI: 10.1183/09031936.00039513 [DOI] [PubMed] [Google Scholar]
- 35.Griguer CE, Oliva CR, and Gillespie GY. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J Neurooncol 74: 123–133, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Guenegou A, Leynaert B, Benessiano J, Pin I, Demoly P, Neukirch F, Boczkowski J, and Aubier M. Association of lung function decline with the heme oxygenase-1 gene promoter microsatellite polymorphism in a general population sample. Results from the European Community Respiratory Health Survey (ECRHS), France. J Med Genet 43: e43, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hervera A, Leanez S, Motterlini R, and Pol O. Treatment with carbon monoxide-releasing molecules and an HO-1 inducer enhances the effects and expression of micro-opioid receptors during neuropathic pain. Anesthesiology 118: 1180–1197, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Hori R, Kashiba M, Toma T, Yachie A, Goda N, Makino N, Soejima A, Nagasawa T, Nakabayashi K, and Suematsu M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide-induced cytotoxicity. J Biol Chem 277: 10712–10718, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Hou W, Tian Q, Steuerwald NM, Schrum LW, and Bonkovsky HL. The let-7 microRNA enhances heme oxygenase-1 by suppressing Bach1 and attenuates oxidant injury in human hepatocytes. Biochim Biophys Acta 1819: 1113–1122, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou W, Tian Q, Zheng J, and Bonkovsky HL. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 51: 1494–1504, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibanez L, Alcaraz MJ, Maicas N, Guede D, Caeiro JR, Motterlini R, and Ferrandiz ML. Downregulation of the inflammatory response by CORM-3 results in protective effects in a model of postmenopausal arthritis. Calcif Tissue Int 91: 69–80, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Immenschuh S, Shan Y, Kroll H, Santoso S, Wossmann W, Bein G, and Bonkovsky HL. Marked hyperbilirubinemia associated with the heme oxygenase-1 gene promoter microsatellite polymorphism in a boy with autoimmune hemolytic anemia. Pediatrics 119: e764–e767, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Jin Y, Kim HP, Chi M, Ifedigbo E, Ryter SW, and Choi AM. Deletion of caveolin-1 protects against oxidative lung injury via up-regulation of heme oxygenase-1. Am J Respir Cell Mol Biol 39: 171–179, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr 23: 167–172, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res 46: 641–643, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Joenje H. Genetic toxicology of oxygen. Mutat Res 219: 193–208, 1989 [DOI] [PubMed] [Google Scholar]
- 47.Jung NH, Kim HP, Kim BR, Cha SH, Kim GA, Ha H, Na YE, and Cha YN. Evidence for heme oxygenase-1 association with caveolin-1 and −2 in mouse mesangial cells. IUBMB Life 55: 525–532, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Kassovska-Bratinova S, Yang G, Igarashi K, and Dennery PA. Bach1 modulates heme oxygenase-1 expression in the neonatal mouse lung. Pediatr Res 65: 145–149, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kerr KM. Pulmonary preinvasive neoplasia. J Clin Pathol 54: 257–271, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HP, Wang X, Galbiati F, Ryter SW, and Choi AM. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J 18: 1080–1089, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Kitamuro T, Takahashi K, Ogawa K, Udono-Fujimori R, Takeda K, Furuyama K, Nakayama M, Sun J, Fujita H, Hida W, Hattori T, Shirato K, Igarashi K, and Shibahara S. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J Biol Chem 278: 9125–9133, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Kramkowski K, Leszczynska A, Mogielnicki A, Chlopicki S, Fedorowicz A, Grochal E, Mann B, Brzoska T, Urano T, Motterlini R, and Buczko W. Antithrombotic properties of water-soluble carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol 32: 2149–2157, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Kulkarni A. and Das KC. Differential roles of ATR and ATM in p53, Chk1, and histone H2AX phosphorylation in response to hyperoxia: ATR-dependent ATM activation. Am J Physiol Lung Cell Mol Physiol 294: L998–L1006, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kunzmann S, Collins JJ, Yang Y, Uhlig S, Kallapur SG, Speer CP, Jobe AH, and Kramer BW. Antenatal inflammation reduces expression of caveolin-1 and influences multiple signaling pathways in preterm fetal lungs. Am J Respir Cell Mol Biol 45: 969–976, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lancel S, Montaigne D, Marechal X, Marciniak C, Hassoun SM, Decoster B, Ballot C, Blazejewski C, Corseaux D, Lescure B, Motterlini R, and Neviere R. Carbon monoxide improves cardiac function and mitochondrial population quality in a mouse model of metabolic syndrome. PLoS One 7: e41836, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee PJ, Alam J, Sylvester SL, Inamdar N, Otterbein L, and Choi AM. Regulation of heme oxygenase-1 expression in vivo and in vitro in hyperoxic lung injury. Am J Respir Cell Mol Biol 14: 556–568, 1996 [DOI] [PubMed] [Google Scholar]
- 57.Lee PJ, Alam J, Wiegand GW, and Choi AM. Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci U S A 93: 10393–10398, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee PJ, Camhi SL, Chin BY, Alam J, and Choi AM. AP-1 and STAT mediate hyperoxia-induced gene transcription of heme oxygenase-1. Am J Physiol Lung Cell Mol Physiol 279: L175–L182, 2000 [DOI] [PubMed] [Google Scholar]
- 59.Li M, Liu Y, Shi H, Zhang Y, Wang G, Xu J, Lu J, Zhang D, Xie X, Han D, Wu Y, and Li S. Statins inhibit pulmonary artery smooth muscle cell proliferation by upregulation of HO-1 and p21WAF1. Naunyn Schmiedebergs Arch Pharmacol 385: 961–968, 2012 [DOI] [PubMed] [Google Scholar]
- 60.Lin CY, Peng CY, Huang TT, Wu ML, Lai YL, Peng DH, Chen PF, Chen HF, Yen BL, Wu KK, and Yet SF. Exacerbation of oxidative stress-induced cell death and differentiation in induced pluripotent stem cells lacking heme oxygenase-1. Stem Cells Dev 21: 1675–1687, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, and Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 282: 20621–20633, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Liu X, Pachori AS, Ward CA, Davis JP, Gnecchi M, Kong D, Zhang L, Murduck J, Yet SF, Perrella MA, Pratt RE, Dzau VJ, and Melo LG. Heme oxygenase-1 (HO-1) inhibits postmyocardial infarct remodeling and restores ventricular function. FASEB J 20: 207–216, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Liu XM, Peyton KJ, and Durante W. Physiological cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am J Physiol Heart Circ Physiol 304: H1634–H1643, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malkinson AM. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp Lung Res 24: 541–555, 1998 [DOI] [PubMed] [Google Scholar]
- 65.McGrath-Morrow S, Lauer T, Yee M, Neptune E, Podowski M, Thimmulappa RK, O'Reilly M, and Biswal S. Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol 296: L565–L573, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller CJ, Gounder SS, Kannan S, Goutam K, Muthusamy VR, Firpo MA, Symons JD, Paine R, 3rd, Hoidal JR, and Rajasekaran NS. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim Biophys Acta 1822: 1038–1050, 2012 [DOI] [PubMed] [Google Scholar]
- 67.Mori I, Yasuhara K, Hayashi SM, Nonoyama T, Nomura T, Yanai T, Masegi T, and Mitsumori K. Aberrant expression of cyclin D1 in pulmonary proliferative lesions induced by high doses of urethane in transgenic mice carrying the human prototype c-H-ras gene. J Vet Med Sci 63: 261–268, 2001 [DOI] [PubMed] [Google Scholar]
- 68.Murakami A, Fujimori Y, Yoshikawa Y, Yamada S, Tamura K, Hirayama N, Terada T, Kuribayashi K, Tabata C, Fukuoka K, Tamaoki T, and Nakano T. Heme oxygenase-1 promoter polymorphism is associated with risk of malignant mesothelioma. Lung 190: 333–337, 2012 [DOI] [PubMed] [Google Scholar]
- 69.Namba F, Go H, Murphy JA, La P, Yang G, Sengupta S, Fernando AP, Yohannes M, Biswas C, Wehrli S, and Dennery PA. Expression level and subcellular localization of heme oxygenase-1 modulates its cytoprotective properties in response to lung injury: a mouse model. PLoS One, [In Press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nikam A, Patankar JV, Lackner C, Schock E, Kratky D, Zatloukal K, and Abuja PM. Transition between acute and chronic hepatotoxicity in mice is associated with impaired energy metabolism and induction of mitochondrial heme oxygenase-1. PLoS One 8: e66094, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Reilly MA, Staversky RJ, Finkelstein JN, and Keng PC. Activation of the G2 cell cycle checkpoint enhances survival of epithelial cells exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol 284: L368–L375, 2003 [DOI] [PubMed] [Google Scholar]
- 72.Okita Y, Kamoshida A, Suzuki H, Itoh K, Motohashi H, Igarashi K, Yamamoto M, Ogami T, Koinuma D, and Kato M. Transforming growth factor-beta induces transcription factors MafK and Bach1 to suppress expression of the heme oxygenase-1 gene. J Biol Chem 288: 20658–20667, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pandya HC, Snetkov VA, Twort CH, Ward JP, and Hirst SJ. Oxygen regulates mitogen-stimulated proliferation of fetal human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 283: L1220–L1230, 2002 [DOI] [PubMed] [Google Scholar]
- 74.Perkowski S, Sun J, Singhal S, Santiago J, Leikauf GD, and Albelda SM. Gene expression profiling of the early pulmonary response to hyperoxia in mice. Am J Respir Cell Mol Biol 28: 682–696, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Peyton KJ, Shebib AR, Azam MA, Liu XM, Tulis DA, and Durante W. Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front Pharmacol 3: 48, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Portais JC, Schuster R, Merle M, and Canioni P. Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon [1-13C]glucose incubation. Eur J Biochem 217: 457–468, 1993 [DOI] [PubMed] [Google Scholar]
- 77.Poss KD. and Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A 94: 10919–10924, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poss KD. and Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A 94: 10925–10930, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pulkkinen KH, Yla-Herttuala S, and Levonen AL. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med 51: 2124–2131, 2011 [DOI] [PubMed] [Google Scholar]
- 80.Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, Chen L, Chen Y, Duan Z, and Meng S. miR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun 398: 771–777, 2010 [DOI] [PubMed] [Google Scholar]
- 81.Radhakrishnan N, Yadav SP, Sachdeva A, Pruthi PK, Sawhney S, Piplani T, Wada T, and Yachie A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol 33: 74–78, 2011 [DOI] [PubMed] [Google Scholar]
- 82.Raju VS, McCoubrey WK, Jr., and Maines MD. Regulation of heme oxygenase-2 by glucocorticoids in neonatal rat brain: characterization of a functional glucocorticoid response element. Biochim Biophys Acta 1351: 89–104, 1997 [DOI] [PubMed] [Google Scholar]
- 83.Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, and Reddy SP. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J Immunol 182: 7264–7271, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roper JM, Mazzatti DJ, Watkins RH, Maniscalco WM, Keng PC, and O'Reilly MA. In vivo exposure to hyperoxia induces DNA damage in a population of alveolar type II epithelial cells. Am J Physiol Lung Cell Mol Physiol 286: L1045–L1054, 2004 [DOI] [PubMed] [Google Scholar]
- 85.Rotenberg MO. and Maines MD. Isolation, characterization, and expression in Escherichia coli of a cDNA encoding rat heme oxygenase-2. J Biol Chem 265: 7501–7506, 1990 [PubMed] [Google Scholar]
- 86.Sacca P, Meiss R, Casas G, Mazza O, Calvo JC, Navone N, and Vazquez E. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br J Cancer 97: 1683–1689, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sato T, Saito Y, Inoue S, Shimosato T, Takagi S, Kaneko T, and Ishigatsubo Y. Serum heme oxygenase-1 as a marker of lung function decline in patients with chronic silicosis. J Occup Environ Med 54: 1461–1466, 2012 [DOI] [PubMed] [Google Scholar]
- 88.Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602, 1997 [DOI] [PubMed] [Google Scholar]
- 89.Sherman MP, Evans MJ, and Campbell LA. Prevention of pulmonary alveolar macrophage proliferation in newborn rabbits by hyperoxia. J Pediatr 112: 782–786, 1988 [DOI] [PubMed] [Google Scholar]
- 90.Sheu CC, Zhai R, Wang Z, Gong MN, Tejera P, Chen F, Su L, Thompson BT, and Christiani DC. Heme oxygenase-1 microsatellite polymorphism and haplotypes are associated with the development of acute respiratory distress syndrome. Intensive Care Med 35: 1343–1351, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Silva-Gomes S, Appelberg R, Larsen R, Soares MP, and Gomes MS. Heme catabolism by heme oxygenase-1 confers host resistance to Mycobacterium infection. Infect Immun 81: 2536–2545, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skrzypek K, Tertil M, Golda S, Ciesla M, Weglarczyk K, Collet G, Guichard A, Kozakowska M, Boczkowski J, Was H, Gil T, Kuzdzal J, Muchova L, Vitek L, Loboda A, Jozkowicz A, Kieda C, and Dulak J. Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis. Antioxid Redox Signal 19: 644–660, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, Lee JS, Postma DS, Kauffman HF, and Choi AM. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol 36: 409–417, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Stanford SJ, Hislop AA, Oltmanns U, Nabel EG, Sang H, Haworth SG, and Mitchell JA. Transition from placental to air breathing stimulates haem-oxygenase-1 expression without functional consequence for pulmonary vascular adaptation in pigs and mice. Br J Pharmacol 144: 467–476, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Staversky RJ, Watkins RH, Wright TW, Hernady E, LoMonaco MB, D'Angio CT, Williams JP, Maniscalco WM, and O'Reilly MA. Normal remodeling of the oxygen-injured lung requires the cyclin-dependent kinase inhibitor p21(Cip1/WAF1/Sdi1). Am J Pathol 161: 1383–1393, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stevens JB. and Autor AP. Proposed mechanism for neonatal rat tolerance to normobaric hyperoxia. Fed Proc 39: 3138–3143, 1980 [PubMed] [Google Scholar]
- 97.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, and Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science 235: 1043–1046, 1987 [DOI] [PubMed] [Google Scholar]
- 98.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, and Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A 101: 1461–1466, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suttner DM. and Dennery PA. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J 13: 1800–1809, 1999 [DOI] [PubMed] [Google Scholar]
- 100.Taira J, Sugishima M, Kida Y, Oda E, Noguchi M, and Higashimoto Y. Caveolin-1 is a competitive inhibitor of heme oxygenase-1 (HO-1) with heme: identification of a minimum sequence in caveolin-1 for binding to HO-1. Biochemistry 50: 6824–6831, 2011 [DOI] [PubMed] [Google Scholar]
- 101.Tanaka K, Kanno T, Yanagisawa Y, Yasutake K, Hadano S, Yoshii F, and Ikeda JE. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp Neurol 232: 41–52, 2011 [DOI] [PubMed] [Google Scholar]
- 102.Thebaud B. Angiogenesis in lung development, injury and repair: implications for chronic lung disease of prematurity. Neonatology 91: 291–297, 2007 [DOI] [PubMed] [Google Scholar]
- 103.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, and Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196–5203, 2002 [PubMed] [Google Scholar]
- 104.Tsai JR, Wang HM, Liu PL, Chen YH, Yang MC, Chou SH, Cheng YJ, Yin WH, Hwang JJ, and Chong IW. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol (Dordr) 35: 461–471, 2012 [DOI] [PubMed] [Google Scholar]
- 105.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, and Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387, 2004 [DOI] [PubMed] [Google Scholar]
- 106.Vanella L, Sodhi K, Kim DH, Puri N, Maheshwari M, Hinds TD, Jr., Bellner L, Goldstein D, Peterson SJ, Shapiro JI, and Abraham NG. Increased heme-oxygenase 1 expression in mesenchymal stem cell-derived adipocytes decreases differentiation and lipid accumulation via upregulation of the canonical Wnt signaling cascade. Stem Cell Res Ther 4: 28, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJ, and Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med 36: 782–801, 2004 [DOI] [PubMed] [Google Scholar]
- 108.Wang J. and Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 130: 1643–1652, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Warner BB, Stuart LA, Papes RA, and Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol 275: L110–L117, 1998 [DOI] [PubMed] [Google Scholar]
- 110.Wolfram I, Vegh M, and Horvath I. Bile pigments inhibit microsomal lipid peroxidation. Acta Biochim Biophys Hung 21: 307–311, 1986 [PubMed] [Google Scholar]
- 111.Wong P, Murray C, Louw J, French N, and Chambers D. Adult bronchopulmonary dysplasia: computed tomography pulmonary findings. J Med Imaging Radiat Oncol 55: 373–378, 2011 [DOI] [PubMed] [Google Scholar]
- 112.Wong PM, Lees AN, Louw J, Lee FY, French N, Gain K, Murray CP, Wilson A, and Chambers DC. Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J 32: 321–328, 2008 [DOI] [PubMed] [Google Scholar]
- 113.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, and Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 103: 129–135, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, and Sasaki H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet 66: 187–195, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang G, Biswas C, Lin QS, La P, Namba F, Zhuang T, Muthu M, and Dennery PA. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: role of β-catenin/hnRNPK signaling. Redox Biol 1: 234–243, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yee M, Vitiello PF, Roper JM, Staversky RJ, Wright TW, McGrath-Morrow SA, Maniscalco WM, Finkelstein JN, and O'Reilly MA. Type II epithelial cells are critical target for hyperoxia-mediated impairment of postnatal lung development. Am J Physiol Lung Cell Mol Physiol 291: L1101–L1111, 2006 [DOI] [PubMed] [Google Scholar]
- 117.Zhang PX, Murray TS, Villella VR, Ferrari E, Esposito S, D'Souza A, Raia V, Maiuri L, Krause DS, Egan ME, and Bruscia EM. Reduced caveolin-1 promotes hyperinflammation due to abnormal heme oxygenase-1 localization in lipopolysaccharide-challenged macrophages with dysfunctional cystic fibrosis transmembrane conductance regulator. J Immunol 190: 5196–5206, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang W, Zhang X, Lu H, Matsukura M, Zhao J, and Shinohara M. Silencing heme oxygenase-1 gene expression in retinal pigment epithelial cells inhibits proliferation, migration and tube formation of cocultured endothelial cells. Biochem Biophys Res Commun 434: 492–497, 2013 [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Jiang G, Sauler M, and Lee PJ. Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J 27: 4041–4058, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Y, Zhang L, Wu J, Di C, and Xia Z. Heme oxygenase-1 exerts a protective role in ovalbumin-induced neutrophilic airway inflammation by inhibiting response in the immune cells Th17. J Biol Chem 2013[Epub ahead of print]; DOI: 10.1074/jbc.M113.494369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhuang T, Zhang M, Zhang H, Dennery PA, and Lin QS. Disrupted postnatal lung development in heme oxygenase-1 deficient mice. Respir Res 11: 142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]