

Abstract
Protein tyrosine phosphatase receptor U (PTPRU) has been shown to be a tumor suppressor in colon cancer by dephosphorylating β-catenin and reducing the activation of β-catenin signaling. Here, we investigate the expression of PTPRU protein in gastric cancer cell lines, gastric cancer tissues and respective adjacent non-cancer tissues and find that the 130kDa nuclear-localized PTPRU fragment is the main PTPRU isoform in gastric cancer cells, whereas the full-length PTPRU is relatively lowly expressed. The level of the 130kDa PTPRU is higher in gastric cancer tissues than in adjacent non-cancer tissues. Knockdown of endogenous PTPRU in gastric cancer cells using lentivirus-delivered specific shRNA results in the attenuation of cell growth, migration, invasion and adhesion. Knockdown of PTPRU also inhibits tyrosine phosphorylation and transcriptional activity of β-catenin as well as levels of focal adhesion proteins and lysine methylation of histone H3. These results indicate that PTPRU is required for gastric cancer progression and may serve as a potential therapeutic target.
Keywords: PTPRU, gastric cancer, proliferation, motility, β-catenin
Introduction
Gastric cancer is the second most common cancer type and one of the deadliest epithelial malignancies, with a 5-year survival rate of approximate 24% worldwide and high frequency of occurrence in Eastern Asia [1,2]. Currently, surgical resection remains the most important way to prolong the survival time of gastric cancer patients, but nevertheless with a relapse rate of more than 50%, especially in patients with lymph node metastasis [2]. Compared with other more intensively investigated cancers, such as breast cancer, colorectal cancer and glioma, the molecular mechanisms underlying gastric cancer progression are largely unclear so far. Therefore, developing new strategies to complement the shortcoming of the current therapies is warranted.
Tyrosine phosphorylation as a molecular switch, is dynamically regulated by protein tyrosine phosphatase (PTPs) and kinases (PTKs). It plays an important role in various cellular signalings that affect growth, differentiation, adhesion, apoptosis, migration, and invasion [3-5]. The role of PTKs in cancers is well characterized and their inhibitors have been utilized as anticancer drugs [6]. Although the role of PTPs in cancers is not as well studied as that of PTKs, growing evidence suggests that functional aberration of PTPs is associated with the progression of multiple types of human cancers [7-12]. The PTP superfamily can be divided into several subfamilies based on the structural diversity. Among them, the R2B RPTP subfamily comprises four members, PTPRU, PTPRM, PTPRT and PTPRK. A myriad of reports indicate that R2B RPTPs are negative regulators in tumorigenesis. For example, PTPRK is mutant in sporadic endocrine pancreatic tumors, glioma, and primary central nervous system lymphomas [13-15]. It also negatively regulates the proliferation and motility of melanoma and breast cancer cells [12,16]. The aberrant function of PTPRM is associated with the progression of ovarian carcinoma, prostate cancer, breast cancer and glioma [17-20]. PTPRT is the most frequently mutated phosphatase gene in cancers including colon, lung, skin and stomach [21,22]. Similar to other members of the R2B RPTP subfamily, PTPRU negatively regulates the growth and migration of colon cancer cell SW480 [23,24]. In contrast to the tumor suppressive role of full-length and mutant R2B RPTPs, some of their proteolytic fragments contribute to tumorigenesis [20,25,26]. Intracellular fragments of PTPRK and PTPRM generated by furin-initiated proteolytic cleavage can translocate to the nucleus and activate the oncogenic signaling [25,26]. PTPRM fragments positively regulate the growth and motility of glioma cells probably in this kind of way [20,26]. Together, these findings suggest a pro-oncogenic role of the proteolytic fragments of R2B RPTPs.
The expression, function and mechanism of PTPRU in cancers are not as well characterized as other R2B RPTPs. In the present study, we found that the expression of a 130KDa PTPRU isoform is higher in gastric cancer tissues than respective adjacent non-cancer tissue. Knockdown of PTPRU suppresses growth and motility and β-catenin transcriptional activity of gastric cancer cells. We conclude that endogenous PTPRU play oncogenic roles in gastric cancer and may be of clinical value.
Materials and methods
Tissue samples
A total of 24 pairs of gastric cancer (GC) and their adjacent non-cancer tissues were collected immediately after surgery from Department of Gastrointestinal Surgery, Ningbo First Hospital after surgery and stored at -80°C until being used. This work was approved by the Ethics Committee of Ningbo First Hospital.
Cell culture
The human gastric cancer cell lines AGS, SGC7901, MKN45 and MGC803 were cultured in RPMI 1640 medium (Corning, Williamsburg, USA) supplemented with 10% fetal bovine serum (Life technologies, Grand Island, USA), 100U/ml penicillin, 0.1 mg/ml streptomycin at 37°C in a 5% CO2 incubator. These cell lines were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China).
Western blot
Cells and tissues were collected and lysed on ice in lysis buffer (20 mM Tris-HCl pH 7.5, 2 mM EDTA, 1% NP-40, 150 mM NaCl, 1 mg/ml SDS and 0.25 mg/ml sodium deoxycholate) supplemented with PMSF (Beyotime, Nantong, China), phosphatase inhibitor (Sangon, Shanghai, China) and protease inhibitor cocktail (Thermo Scientific, Waltham, USA), then mixed with loading buffer and boiled. Nuclear and cytoplasmic protein fractions were prepared using the nuclear and cytoplasmic protein extraction kit (Beyotime). Equal amount of proteins were separated by SDS-PAGE and transferred onto polyvinylidenedifluoride membranes (Millipore, Billerica, USA). Membranes were blocked in 5% nonfat dry milk or bovine serum albumin in TBST (Tris-buffer saline with 0.05% Tween-20) for 1 h, then incubated overnight at 4°C with antibodies against PTPRU (Sigma-Aldrich, St. Louis, USA), PTPλ (Abcam, Cambridge, UK), β-catenin, EGFR, FAK (BD Biosciences, San Jose, USA), c-Myc, Dnmt1, p-ERK1/2 (T202/Y204), H3K27me2, Histone H3, p-Tyr-102, TCF4 (Cell Signaling Technology, Beverly, USA), ERK1/2, H3K9me3, paxillin, PYK2 (Bioworld Technology, Nanjing, China), cyclin B1, cyclin D1, VE-cadherin (Boster, Wuhan, China) and GAPDH (Kangchen, Shanghai, China). Corresponding horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (Cell Signaling Technology, Beverly, USA) were used. Protein bands were visualized by photo development using Kodak film. To analyze the relative levels of PTPRU, band grayscales of PTPRU and GAPDH were quantified using the Adobe Photoshop software and the ratio of PTPRU to GAPDH was calculated.
Lentivirus-mediated shRNA knockdown
Lentivirus encoding shRNA plasmids (on vector pLKD-CMV-GFP-U6-shRNA, Neuron Biotech, Shanghai, China) specifically targeting human PTPRU and a scrambled shRNA plasmid were packed by Neuron Biotech. Western blot and real time PCR were used to validate the knockdown efficacy and found that the most efficient shRNA sequence was: CCGGGGAGATGATCCGCATTGATCCCTCAGGGATCAATGCGGATCATCTCCTTTTTTG. The sequence of the scrabled shRNA was: CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTG.
Immunofluorescence
Cell suspension was plated onto sterile glass coverslips in 6-well plates for 24 h, then fixed with 4% paraformaldehyde at 4°C for 15 min. Cells were incubated in turn with blocking buffer (3% bovine serum albumin and 0.1% Triton in phosphate-buffered saline) for 1 h, with PTPRU or PTPλ antibody at 4°C overnight, with biotin labeled secondary antibody (Life technologies) for 2 h, and with Cy3 labeled avidin (Life technologies) and Hoechst 33342 (Beyotime) for 2 h. Fluorescent images were taken using a Nikon eclipse Ti-S inverted microscope.
Real-time quantitative PCR
Total RNA was extracted using Trizol (Life technologies), and reverse transcribed using RT M-MLV (Promega, Beijing, China) with oligo dT. Quantitative real-time PCR with SYBR Premix Ex Taq (Takara, Dalian, China) was performed according to the manufacturer’s protocol. PCR primers were used as follows: PTPRU, forward, 5’-GAGTGAAACTGCACCCGATG-3’, reverse, 5’-TACTCTGAGGGTCAATGCGGA-3’; GAPDH, forward, 5’-AAGGTGAAGGTCGGAGTCAAC-3’, reverse, 5’-GGGGTCATTGATGGCAACAATA-3’; Relative quantification of PTPRU expression was determined by the 2-ΔΔCt method with GAPDH as the endogenous control and normalized to the expression of the Controls [27].
MTT assay
Cells were trypsinized, counted and plated onto the 96-well plates in 150 μl culture medium at a density of 1,500 cells/well in quintuplicate wells. Each day for the next five days, 15 μl MTT (5 mg/ml) was added to the corresponding well. After incubating for an additional 4 h at 37°C, culture medium was replaced with 150 μl DMSO and optical density was measured at 490 nm in the BioTek synergy 2 microplate reader.
Colony formation assay
Cells were trypsinized, counted and seeded onto 6-well plates at a density of 500 cells per well in triplicate wells. Colonies were fixed with methanol and stained with 0.5% crystal violet after two weeks. Colonies were photographed and counted.
Flow cytometry
Flow cytometry was used for cell cycle distribution analysis. Cells in the log phase of growth were harvested, fixed with 70% (v/v) ethanol overnight at -20°C, incubated with 50 μg/ml RNase A (Thermo Scientific) and 50 μg/ml propidium iodide (Sigma-Aldrich) in PBS for 30 min at 37°C in the dark and filtered by sieves to get single-cell suspension. Stained nuclei were analyzed by a FACSCalibur Flow Cytometry with 10,000 gated events/determination. DNA ploidy was analyzed by FlowJo software.
Wound-healing assay
Cells were seeded onto a 6-well plate and scraped with a 10 μl pipet tip when they grew into confluent cell monolayer. Culture medium was replaced with serum free RPMI 1640 to exclude the factor of proliferation difference. Photographs were taken right after wounding and 24 h later. Cell migration distances in 24 h were measured using Image J software.
Transwell migration and invasion assay
Transwell migration and invasion assay were performed using the 24-well cell culture inserts without matrigel and matirgel invasion chambers (8 μm pore, BD Bioscience), respectively. Briefly, 5×104 cells were resuspended in 250 μl serum-free RPMI 1640 and added into the inserts. 500 μl RPMI 1640 with 10% FBS was added to the lower chamber. After allowing cells to migrate for 4 h or invasion for 22 h, cells on the upper surface of the membrane were removed using a cotton swab, and the membranes were fixed with methanol and stained with crystal violet. The number of migrating or invading cells was determined by averaging cell counts from nine random selected 100× fields.
Cell-matrix adhesion assay
To determine the matrix-dependent cell adhesion, 24-well plates was coated with 200 μl of 50 μg/ml matrigel (BD Biosciences) overnight at 4°C as previously described [28]. Briefly, 500 μl cell suspension (1×105 cells/ml) was added to the corresponding well and incubated for 30 min at 37°C. Non-adherent cells were removed by washing the wells for three times with D’ Hanks. Adherent cells were fixed with methanol and stained with crystal violet. The number of adherent cells was determined by averaging cell counts from three random selected 100× fields.
Immunoprecipitation
Cells were lysed on ice in lysis buffer for Western and IP (Beyotime). Lysates were adjusted for equal protein concentrations and incubated with β-catenin antibody overnight at 4°C, and with additional protein G agarose (Beyotime) for 3 h at 4°C. Precipitates were washed four times with lysis buffer before SDS-PAGE loading buffer was added and boiling.
Luciferase reporter assay
Cells were co-transfected with the TOP-FLASH firefly luciferase reporter plasmid (Genomeditech, Shanghai, China) containing multiple TCF/LEF1 binding sites and the Renilla luciferase plasmid pRL-CMV (Promega) using Lipofectamine 2000 (Life technologies). A dual luciferase reporter assay was carried out according to the manufacturers’ protocol (Promega). Luciferase activities were measured using the BioTek synergy 2 microplate reader. Firefly luciferase activities were normalized to Renilla luciferase activities. All experiments were performed in triplicate.
Statistical analysis
Results were expressed as mean ± standard deviation of the mean (SD). Statistical significance between groups was measured by Student’s t test, with statistically significant defined as *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
PTPRU expression in gastric cancer cells and tissues
We first examined the expression and subcellular localization of PTPRU protein in four human gastric cancer cell lines MKN45, SGC7901, MGC803 and AGS. As identified by an antibody raised against the N-terminal of PTPRU (residues 28-111), gastric cancer cells mainly expressed two PTPRU isoforms, a 200kDa isoform (PTPRU-FL) that corresponded to the full-length PTPRU in molecular weight and a 130kDa isoform (PTPRU130) (Figure 1A). PTPRU-FL localized to the cytoplasm and membrane while the PTPRU130 localized to the nucleus of gastric cancer cells (Figure 1B). Thus, we concentrated on the expression of PTPRU130 in gastric cancer and adjacent non-cancer tissues since PTPRU130 was the predominant PTPRU isoform. Level of PTPRU130 was higher in 18 of 24 pairs of gastric cancer tissues than their adjacent non-cancer tissues and was lower in 6 of 24 pairs (Figure 1C, 1D). These results suggest that PTPRU130 may be involved in gastric cancer progression.
Figure 1.
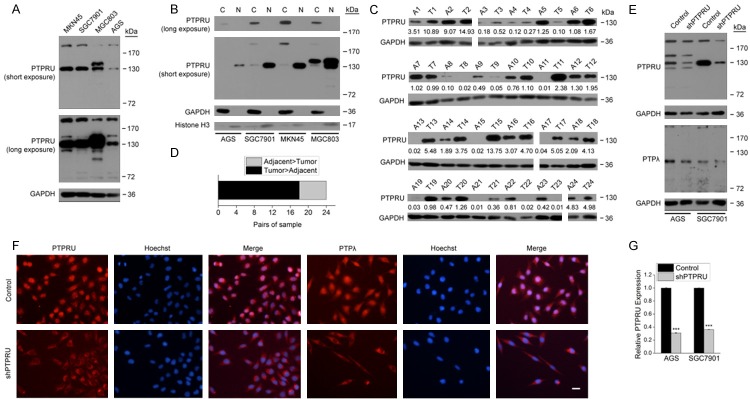
PTPRU protein expression in gastric cancer tissues and cell lines. (A) Western blot analysis of PTPRU protein expression in four human gastric cancer cell lines. (B) Distribution of PTPRU isoforms in the cytoplasm and nucleus of four gastric cancer cell lines. Abbreviations: C, cytoplasm; N, nucleus. (C, D) PTPRU130 levels in gastric cancer tissues and respective adjacent non-cancer tissues. Relative grayscales of PTPRU130 are indicated below the blots. Abbreviations: A, adjacent non-cancer tissues; T, gastric cancer tissues. The number of tissue pairs in which tumors express higher levels or lower levels of PTPRU130 than adjacent tissues are shown in (D). (E, F) AGS and SGC7901 were infected by lentiviruses encoding a scrambled shRNA (Control) or shRNA targeting human PTPRU (shPTPRU). PTPRU protein expression was analyzed by western blot (E) and immunofluorescence (F) using two antibodies against PTPRU (PTPRU and PTPλ). Magnification, ×200; scale bar, 50 μm. (G) PTPRU mRNA expression in AGS and SGC7901 cells treated as indicated was analyzed by real-time quantitative PCR. ***P < 0.001. GAPDH (A, C, E and cytoplasmic in B) and Histone H3 (nuclear in B) were used as loading controls. Molecular weights in kDa are indicated on the right-hand side of each blot.
To determine whether PTPRU130 and other bands detected by the PTPRU antibody are PTPRU-specific and to carry out functional study of PTPRU in gastric cancer cells, we knocked down PTPRU expression in AGS and SGC7901 cells using a lentivirus-delivered shRNA specifically targeting human PTPRU (shPTPRU), whose knockdown efficacy was verified in previous study [29]. PTPRU130 and some other bands were downregulated upon PTPRU knockdown, as revealed by western blot using the PTPRU antibody. Another PTPRU antibody called PTP λ, which is raised against residues 850-950 of human PTPRU, detected PTPRU-FL and a 120kDa isoform (Figure 1E). PTPRU immunofluorescence is mainly localized to the nucleus of AGS and SGC7901 cells, which is consistent with the results of western blot, and its intensity is decreased upon PTPRU knockdown (Figure 1F). Real-time quantitative PCR also showed that PTPRU mRNA was reduced following PTPRU knockdown (Figure 1G). These results provide compelling evidence for the effectiveness of the shPTPRU plasmid and PTPRU antibodies used in this study.
Knockdown of PTPRU inhibits growth of gastric cancer cells
Knockdown of endogenous PTPRU impeded the proliferation and survival of AGS and SGC7901 cells, as revealed by MTT assay and colony formation assay (Figure 2A, 2B). To investigate whether cell cycle arrest contributed to the growth inhibition, we analyzed the cell cycle distribution in AGS and SGC7901 cells using flow cytometry. As expected, knockdown of PTPRU arrested the cell cycle in G0/G1 phase in AGS and SGC7901 cells and accordingly decreased the cell number in S phase (Figure 2C). Consistently, protein levels of positive regulators of cell cycle cyclin D1, cyclin B1 were downregulated in shPTPRU AGS and SGC7901 cells (Figure 2D). Surprisingly, the level of p-ERK1/2 was upregulated and levels of H3K9me3 and H3K4me2 were downregulated in shPTPRU AGS and SGC7901 cells (Figure 2E, 2F). These results suggest that knockdown of PTPRU inhibits growth of gastric cancer cells, which may be regulated by multiple mechanisms.
Figure 2.

Kncokdown of PTPRU inhibits growth of gastric cancer cells. (A) AGS and SGC7901 cells expressing scrambled shRNA (Control) or PTPRU shRNA (shPTPRU) were seeded onto 96-well plates in quintuplicate, and proliferation rates were measured by MTT assay. (B) The colony formation ability of AGS and SGC7901 cells from each treatment group was measured by colony formation assay. Quantification of colonies in each group is shown on the right panel. (C) The cell cycle distribution of AGS and SGC7901 cells from each treatment group was analyzed by flow cytometry. The number of cells in G0/G1, S and G2/M phases are shown (n = 3). (D-F) Levels of various proteins in AGS and SGC7901 cells from each treatment group were assessed by western blot. GAPDH (D, E) and Histone H3 (F) were used as the loading control. *P < 0.05, **P < 0.01, ***P < 0.001.
Knockdown of PTPRU inhibits motility of gastric cancer cells
We further investigated the effect of PTPRU knockdown on gastric cancer cell motility. ShPTPRU AGS and SGC7901 cells showed attenuated migration abilities not only in wound-healing assay but also in chemoataxis cell migration assay (Figure 3A-D). PTPRU knockdown also reduced abilities of cell invasion and adhesion to the extracelluar matrix as revealed respectively by transwell invasion assay and cell-matrix adhesion assay (Figure 3E-H). Consistent with these phenotypic changes, levels of cell motility-associated proteins focal adhesion kinase (FAK), paxillin, proline-rich tyrosine kinase 2 (PYK2) and VE-cadherin were downregulated (Figure 3I). These results indicate that PTPRU knockdown inhibits motility of gastric cancer cells.
Figure 3.
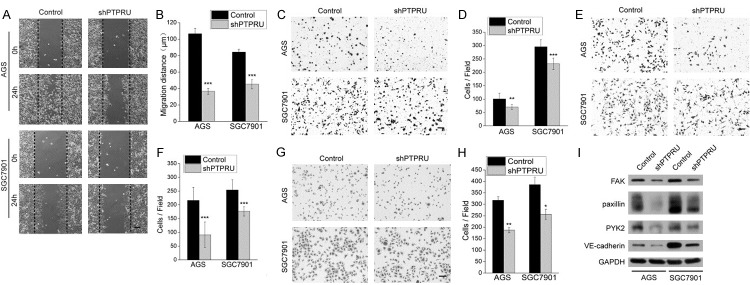
Knockdown of PTPRU inhibits migration, invasion and adhesion of gastric cancer cells. (A, B) Confluent monolayers of AGS and SGC7901 cells were wounded and edges of each wound were imaged right after wounding (0 h) and 24 h later. The black dotted line represents the initial wound edge. Cell migration distances in 24 h were measured using Image J software and shown in (B). (C, D) Transwell chambers without matrigel were used to assess the migratory ability of AGS and SGC7901 cells. The number of migrating cells, averaged over nine randomly selected fields of view, is quantified in (D). (E, F) Matrigel invasion chambers were used to assess the invasiveness of AGS and SGC7901 cells. The number of invading cells was counted and averaged over nine randomly selected fields, as showed in (F). (G, H) Cell-matrix adhesion assay was used to assess the adhesion ability of AGS and SGC7901 cells. The number of adherent cells, averaged over three random selected fields, is showed in (H). (I) Levels of focal adhesion proteins and VE-cadherin were detected by western blot. GAPDH was used as the loading control. Magnification, ×100 for all pictures; scale bar, 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
PTPRU regulates tyrosine phosphorylation and transcriptional activity of β-catenin in gastric cancer cells
Previous reports indicate that PTPRU-FL colocalized with β-catenin at cell junctions, which cause a significant reduction in the tyrosine phosphorylation (pY) level and transcriptional activity of β-catenin [23,24]. To determine the effect of PTPRU knockdown on β-catenin signaling, we carried out immunoprecipitation experiments to detect pY level of β-catenin and the interaction of β-catenin with one of its downstream transcription factor TCF4 in AGS and SGC7901 cells. Both pY level of β-catenin and β-catenin/TCF4 interaction were diminished in shPTPRU cells (Figure 4A, 4B). Consequently, β-catenin/TCF4 interaction-mediated transcriptional activity of TCF/LEF1 complex was downregulated, which finally resulted in the expression inhibition of several target genes of TCF/LEF1 complex, such as cyclin D1, c-Myc, Dnmt1 and EGFR (Figures 2D, 4C, 4D). These results suggest that knockdown of PTPRU suppresses β-catenin signaling in gastric cancer cells.
Figure 4.
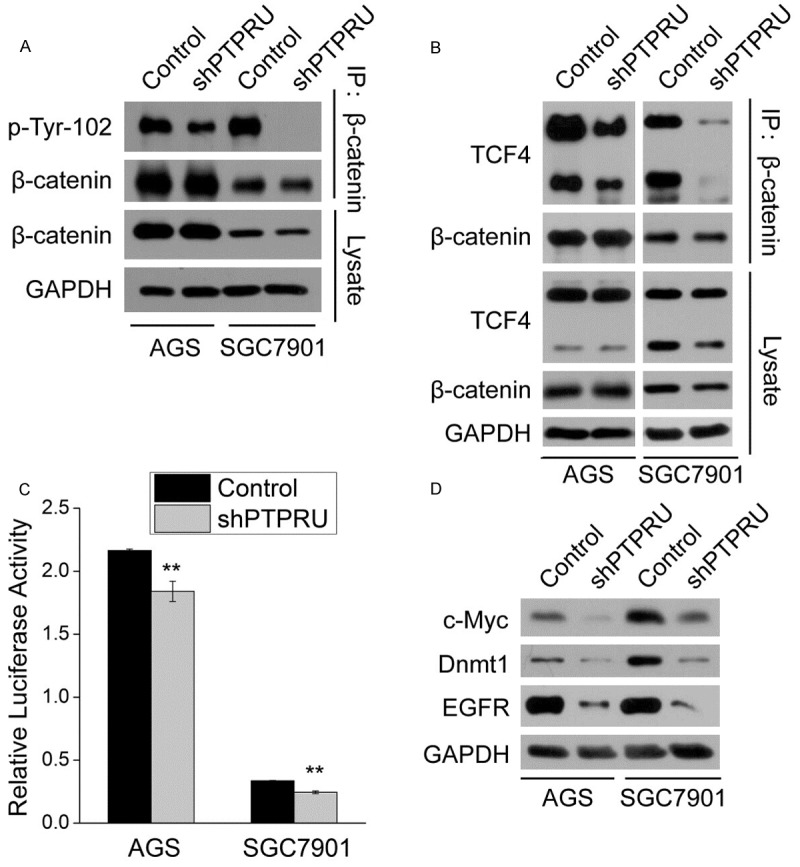
PTPRU regulates tyrosine phosphorylation and transcriptional activity of β-catenin in gastric cancer cells. A, B. β-catenin was immunoprecipitated (IP) from AGS and SGC7901 cells treated as indicated, and levels of β-catenin, pY β-catenin and TCF4 were detected by western blot. GAPDH was used as the loading control. C. AGS and SGC7901 cells treated as indicated were co-transfected with TOP-FLASH and pRL-CMV plasmids, and luciferase activity was measured 24 h after transfection (n = 3). Firefly luciferase activity was normalized to renilla luciferase activity. **P < 0.01. D. Protein levels of downstream target genes of β-catenin signaling in AGS and SGC7901 cells were detected by western blot. GAPDH was used as the loading control.
Discussion
Numerous members of RPTPs family are identified as tumor suppressors in many cancer types, and gene mutation, expression downregulation and protein modification that impede their tumor suppressing function are commonly seem in cancer tissues [11-16]. In this study, we find that PTPRU130, a non-full-length PTPRU isoform, is highly expressed in gastric cancer cells and tissues. Endogenous PTPRU is required for gastric cancer cell growth and motility.
β-catenin is a dual-functional protein that play roles in cell-cell adhesion and transcriptional regulation. pY of β-catenin is related to its dissociation from the adherens junctions, nuclear accumulation and transcriptional activity [30-33]. β-catenin internalization is associated with aggressive phenotypes of gastric cancers, like lymphatic vessel invasion and lymph node metastases [34]. Previous reports indicated that PTPRU-FL directly interacts with β-catenin and dephosphorylates it, thus improving the formation of β-catenin/E-cadherin complex at plasma membrane [23,24]. Contrary to PTPRU-FL alone, we find that endogenous PTPRU positively regulate pY and transcriptional activity of β-catenin in gastric cancer cells. Posttranslational modification of PTPRU, probably proteolytic cleavage, may contribute to opposite effects of PTPRU-FL and intracellular PTPRU on β-catenin signaling. All other members of the R2B RPTP subfamily can be cleaved by proteases and release intracellular domains to the nucleus. Nuclear import of intracellular PTPRT helps it better dephosphorylate STAT3 [35]. Intracellular PTPRK promotes transcriptional activity of β-catenin though it still dephosphorylates β-catenin like full-length PTPRK [25]. Therefore, intracellular PTPRK may take advantage of other mechanism, such as regulating cofactors of β-catenin transcriptional complex, to accomplish this. Although the function of intracellular PTPRM has not been characterized, the fact that PTPRM is highly cleaved in gliomas and its downregulation inhibits glioma cell growth and migration indicates that intracellular PTPRM is important for tumor progression [20,26]. Hence, it is reasonable that endogenous PTPRU promote growth and motility of gastirc cancer cells through β-catenin signaling. We postulate that endogenous PTPRU may indirectly regulate pY of β-catenin through protein modification or expression alteration of tyrosine kinases of β-catenin, such as Src, PYK2 or EGFR [33,36,37], which requires further investigation.
PTPRU knockdown also results in the upregulation of p-ERK1/2 and downregulation of lysine methylation of histone H3 and several focal adhesion proteins. The role of ERK signaling in growth promotion or inhibition depends on whether activation is acute or chronic. Acute activation of ERK stimulates DNA synthesis and promotes proliferation, whereas chronic activation causes cell cycle arrest followed by cellular differentiation [38,39]. PTPRM knockdown upregulates pY of ERK and promotes cancer cell growth [11]. In this study, however, PTPRU knockdown-induced p-ERK1/2 may negatively regulate of cell growth. Aberrant histone modification is linked to cancers through the activation of oncogene expression or inactivation of tumor suppressing gene expression [40-42]. As repressive histone modifications, demethylation of H3K9 and H3K27 upon PTPRU knockdown may lead to reexpression of tumor suppressing genes and mitigate the aggressiveness of gastric cancer cells. Assembly and turnover of focal adhesions is crucial for cancer cell motility [43]. Thus, inhibition of focal adhesion proteins, such as FAK, paxillin and PYK2, impedes cell motility [44-46]. These findings implicate that PTPRU-promoted aggressive behaviors of gastric cancers is mediated by multiple potential mechanisms.
In conclusion, we show here that PTPRU130 is highly expressed in gastric cancer cells and tissues, whereas the previous demonstrated tumor suppressor PTPRU-FL is lowly expressed compared to PTPRU fragments. Most of the gastric cancer tissues express higher levels of PTPRU130 than their adjacent non-cancer tissues. Loss-of-function experiments verified that endogenous PTPRU is required for growth and motility of gastirc cancer cells. PTPRU participate in the regulation of β-catenin signaling, ERK signaling and histone modification. Further studies are required to clarify the formation mechanism of non-full-length PTPRU isoforms and fully understand the mechanisms underlying the oncogenic role of PTPRU.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 31300904) and the Ningbo Natural Science Foundation (No. 2012A610209). The authors appreciate the International Postdoctoral Exchange Fellowship Program China (20130018) for the financial support of this work.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Van Cutsem E, Dicato M, Arber N, Benson A, Cunningham D, Diaz-Rubio E, Glimelius B, Goldberg R, Haller D, Haustermans K, Koo-Kang Y, Labianca R, Lang I, Minsky B, Nordlinger B, Roth A, Rougier P, Schmoll HJ, Sobrero A, Tabernero J, Szawlowski A, van de Velde C. The neo-adjuvant, surgical and adjuvant treatment of gastric adenocarcinoma. Current expert opinion derived from the Seventh World Congress on Gastrointestinal Cancer, Barcelona, 2005. Ann Oncol. 2006;17(Suppl 6):vi13–8. doi: 10.1093/annonc/mdl976. [DOI] [PubMed] [Google Scholar]
- 3.Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 4.Tonks NK, Neel BG. From form to function: signaling by protein tyrosine phosphatases. Cell. 1996;87:365–368. doi: 10.1016/s0092-8674(00)81357-4. [DOI] [PubMed] [Google Scholar]
- 5.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 7.Ostman A, Hellberg C, Böhmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 8.Nikolaienko RM, Agyekum B, Bouyain S. Receptor protein tyrosine phosphatases and cancer: new insights from structural biology. Cell Adh Migr. 2012;6:356–364. doi: 10.4161/cam.21242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laczmanska I, Sasiadek MM. Tyrosine phosphatases as a superfamily of tumor suppressors in colorectal cancer. Acta Biochim Pol. 2011;58:467–470. [PubMed] [Google Scholar]
- 10.Lui VW, Peyser ND, Ng PK, Hritz J, Zeng Y, Lu Y, Li H, Wang L, Gilbert BR, General IJ, Bahar I, Ju Z, Wang Z, Pendleton KP, Xiao X, Du Y, Vries JK, Hammerman PS, Garraway LA, Mills GB, Johnson DE, Grandis JR. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111:1114–1119. doi: 10.1073/pnas.1319551111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun PH, Ye L, Mason MD, Jiang WG. Protein tyrosine phosphatase μ (PTP μ or PTPRM), a negative regulator of proliferation and invasion of breast cancer cells, is associated with disease prognosis. PLoS One. 2012;7:e50183. doi: 10.1371/journal.pone.0050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novellino L, De Filippo A, Deho P, Perrone F, Pilotti S, Parmiani G, Castelli C. PTPRK negatively regulates transcriptional activity of wild type and mutated oncogenic beta-catenin and affects membrane distribution of beta-catenin/E-cadherin complexes in cancer cells. Cell Signal. 2008;20:872–883. doi: 10.1016/j.cellsig.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 13.Barghorn A, Speel EJ, Farspour B, Saremaslani P, Schmid S, Perren A, Roth J, Heitz PU, Komminoth P. Putative tumor suppressor loci at 6q22 and 6q23-q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol. 2001;158:1903–1911. doi: 10.1016/S0002-9440(10)64658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura M, Kishi M, Sakaki T, Hashimoto H, Nakase H, Shimada K, Ishida E, Konishi N. Novel tumor suppressor loci on 6q22-23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737–741. [PubMed] [Google Scholar]
- 15.Agarwal S, Al-Keilani MS, Alqudah MA, Sibenaller ZA, Ryken TC, Assem M. Tumor derived mutations of protein tyrosine phosphatase receptor type k affect its function and alter sensitivity to chemotherapeutics in glioma. PLoS One. 2013;8:e62852. doi: 10.1371/journal.pone.0062852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun PH, Ye L, Mason MD, Jiang WG. Protein tyrosine phosphatase kappa (PTPRK) is a negative regulator of adhesion and invasion of breast cancer cells, and associates with poor prognosis of breast cancer. J Cancer Res Clin Oncol. 2013;139:1129–1139. doi: 10.1007/s00432-013-1421-5. [DOI] [PubMed] [Google Scholar]
- 17.Györffy B, Dietel M, Fekete T, Lage H. A snapshot of microarray-generated gene expression signatures associated with ovarian carcinoma. Int J Gynecol Cancer. 2008;18:1215–1233. doi: 10.1111/j.1525-1438.2007.01169.x. [DOI] [PubMed] [Google Scholar]
- 18.Hellberg CB, Burden-Gulley SM, Pietz GE, Brady-Kalnay SM. Expression of the receptor protein-tyrosine phosphatase, PTPmu, restores E-cadherin-dependent adhesion in human prostate carcinoma cells. J Biol Chem. 2002;277:11165–11173. doi: 10.1074/jbc.M112157200. [DOI] [PubMed] [Google Scholar]
- 19.Burgoyne AM, Palomo JM, Phillips-Mason PJ, Burden-Gulley SM, Major DL, Zaremba A, Robinson S, Sloan AE, Vogelbaum MA, Miller RH, Brady-Kalnay SM. PTPμ suppresses glioma cell migration and dispersal. Neuro Oncol. 2009;11:767–778. doi: 10.1215/15228517-2009-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur H, Burden-Gulley SM, Phillips-Mason PJ, Basilion JP, Sloan AE, Brady-Kalnay SM. Protein tyrosine phosphatase mu regulates glioblastoma cell growth and survival in vivo. Neuro Oncol. 2012;14:561–573. doi: 10.1093/neuonc/nos066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 22.Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr Protoc Hum Genet. 2008 doi: 10.1002/0471142905.hg1011s57. Chapter 10: Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan HX, He YQ, Dong H, Zhang P, Zeng JZ, Cao HF, Wu MC, Wang HY. Physical and functional interaction between receptor-like protein tyrosine phosphatase PCP-2 and beta-catenin. Biochemistry. 2002;41:15854–15860. doi: 10.1021/bi026095u. [DOI] [PubMed] [Google Scholar]
- 24.Yan HX, Yang W, Zhang R, Chen L, Tang L, Zhai B, Liu SQ, Cao HF, Man XB, Wu HP, Wu MC, Wang HY. Protein-tyrosine phosphatase PCP-2 inhibits beta-catenin signaling and increases E-cadherin-dependent cell adhesion. J Biol Chem. 2006;281:15423–15433. doi: 10.1074/jbc.M602607200. [DOI] [PubMed] [Google Scholar]
- 25.Anders L, Mertins P, Lammich S, Murgia M, Hartmann D, Saftig P, Haass C, Ullrich A. Furin-, ADAM 10-, and gamma-secretase-mediated cleavage of a receptor tyrosine phosphatase and regulation of beta-catenin’s transcriptional activity. Mol Cell Biol. 2006;26:3917–3934. doi: 10.1128/MCB.26.10.3917-3934.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, Robinson S, Sloan AE, Miller RH, Brady-Kalnay SM. Proteolytic cleavage of protein tyrosine phosphatase mu regulates glioblastoma cell migration. Cancer Res. 2009;69:6960–6968. doi: 10.1158/0008-5472.CAN-09-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arocho A, Chen B, Ladanyi M, Pan Q. Validation of the 2-DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn Mol Pathol. 2006;15:56–61. doi: 10.1097/00019606-200603000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Thamilselvan V, Basson MD. Pressure activates colon cancer cell adhesion by inside-out focal adhesion complex and actin cytoskeletal signaling. Gastroenterology. 2004;126:8–18. doi: 10.1053/j.gastro.2003.10.078. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Z, Liu Y, Li K, Liu J, Wang H, Sun B, Xiong Z, Jiang H, Zheng J, Hu Z. Protein tyrosine phosphatase receptor U (PTPRU) is required for glioma growth and motility. Carcinogenesis. 2014;35:1901–10. doi: 10.1093/carcin/bgu123. [DOI] [PubMed] [Google Scholar]
- 30.Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 31.Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, Mikolka P, Pospisilova T, Spoustova T, Weis M, Paznekas WA, Wolf JH, Gutkind JS, Wilcox WR, Kozubik A, Jabs EW, Bryja V, Salazar L, Vesela I, Balek L. Receptor tyrosine kinases activate canonical WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct β-catenin phosphorylation. PLoS One. 2012;7:e35826. doi: 10.1371/journal.pone.0035826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol. 2001;21:5857–5868. doi: 10.1128/MCB.21.17.5857-5868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nabais S, Machado JC, Lopes C, Seruca R, Carneiro F, Sobrinho-Simões M. Patterns of beta-catenin expression in gastric carcinoma: clinicopathological relevance and mutation analysis. Int J Surg Pathol. 2003;11:1–9. doi: 10.1177/106689690301100102. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, Wang ZJ. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 2007;104:4060–4064. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–20443. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 37.van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL. Proline-rich tyrosine kinase 2 (Pyk2) mediates vascular endothelial-cadherin-based cell-cell adhesion by regulating beta-catenin tyrosine phosphorylation. J Biol Chem. 2005;280:21129–21136. doi: 10.1074/jbc.M500898200. [DOI] [PubMed] [Google Scholar]
- 38.Ciccarelli C, Marampon F, Scoglio A, Mauro A, Giacinti C, De Cesaris P, Zani BM. p21WAF1 expression induced by MEK/ERK pathway activation or inhibition correlates with growth arrest, myogenic differentiation and onco-phenotype reversal in rhabdomyosarcoma cells. Mol Cancer. 2005;4:41. doi: 10.1186/1476-4598-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tombes RM, Auer KL, Mikkelsen R, Valerie K, Wymann MP, Marshall CJ, McMahon M, Dent P. The mitogen-activated protein (MAP) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem J. 1998;330:1451–1460. doi: 10.1042/bj3301451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics. 2009;10:143. doi: 10.1186/1471-2164-10-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeong HM, Kwon MJ, Shin YK. Overexpression of Cancer-Associated Genes via Epigenetic Derepression Mechanisms in Gynecologic Cancer. Front Oncol. 2014;4:12. doi: 10.3389/fonc.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagano M, Hoshino D, Koshikawa N, Akizawa T, Seiki M. Turnover of focal adhesions and cancer cell migration. Int J Cell Biol. 2012;2012:310616. doi: 10.1155/2012/310616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li D, Ding J, Wang X, Wang C, Wu T. Fibronectin promotes tyrosine phosphorylation of paxillin and cell invasiveness in the gastric cancer cell line AGS. Tumori. 2009;95:769–779. doi: 10.1177/030089160909500621. [DOI] [PubMed] [Google Scholar]
- 46.Lipinski CA, Tran NL, Menashi E, Rohl C, Kloss J, Bay RC, Berens ME, Loftus JC. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia. 2005;7:435–445. doi: 10.1593/neo.04712. [DOI] [PMC free article] [PubMed] [Google Scholar]