

Abstract
Due to the upcoming data deluge of genome data, the need for storing and processing large-scale genome data, easy access to biomedical analyses tools, efficient data sharing and retrieval has presented significant challenges. The variability in data volume results in variable computing and storage requirements, therefore biomedical researchers are pursuing more reliable, dynamic and convenient methods for conducting sequencing analyses. This paper proposes a Cloud-based bioinformatics workflow platform for large-scale next-generation sequencing analyses, which enables reliable and highly scalable execution of sequencing analyses workflows in a fully automated manner. Our platform extends the existing Galaxy workflow system by adding data management capabilities for transferring large quantities of data efficiently and reliably (via Globus Transfer), domain-specific analyses tools preconfigured for immediate use by researchers (via user-specific tools integration), automatic deployment on Cloud for on-demand resource allocation and pay-as-you-go pricing (via Globus Provision), a Cloud provisioning tool for auto-scaling (via HTCondor scheduler), and the support for validating the correctness of workflows (via semantic verification tools). Two bioinformatics workflow use cases as well as performance evaluation are presented to validate the feasibility of the proposed approach.
Keywords: Bioinformatics, Scientific workflow, Sequencing analyses, Cloud computing, Galaxy
1. Introduction
With the emergence of NGS (next-generation sequencing), various genome informatics ecosystems are now facing a potential tsunami of genome data that will swamp their storage systems and crush their computing clusters. Human DNA is comprised of approximately 3 billion base pairs with a personal genome representing approximately 100 gigabytes (GB) of data. By the end of 2011, the global annual sequencing capacity was estimated to be 13 quadrillion bases and counting [1]. The upcoming data deluge forces researchers to find reliable and convenient methods for storage and computing.
In the bioinformatics community, acquiring sequence data is always followed by large-scale computational analysis to process the data, validate experiment results and draw scientific insights. Therefore, investment in a sequencing instrument would normally be accompanied by substantial investment in computer hardware, skilled informatics support, and bioinformaticians competent in configuring and using specific software to analyze the data [2].
However, the need for storing and processing large-scale genome data, providing easy access to data analysis tools, enabling efficient data sharing and retrieval, integrating imaging, electrophysiological and clinical data, and supporting cross-institutional collaboration still has significant challenges.
Existing tools, such as Bioconductor [3], Bioperl [4], and EMBOSS [5], improve the accessibility of computation and facilitate bioinformatics research by decreasing IT efforts and automating data analyses workflows. But these approaches have difficulties when dealing with large datasets, which is generally common in NGS analyses; besides the software installation and programming efforts needed are often error-prone and time consuming for biomedical researchers. Moreover, most research institutes implement their applications on laboratory-hosted servers [6], and as data volume varies greatly, the capabilities and efficiency in storing and analyzing genome data are not enough to fulfill the dynamic requirements of different workflows.
To address these problems, the authors propose a Cloud-based bioinformatics workflow platform for large-scale NGS analyses [7]. This platform integrates Galaxy, a scientific workflow system for biomedical analyses, Globus Provision (GP), a tool for deploying distributed computing clusters on Cloud, and a set of supporting tools and modules to provide an overall solution for biomedical researchers. This combination of tools implements an easy to use, high performance and scalable workflow environment that addresses the needs of data-intensive applications through dynamic cluster configuration, automatic user-defined node provisioning, high speed data transfer, and automated deployment and configuration of domain-specific software.
More specifically, the contributions of this paper are summarized as follows.
We propose a novel approach for automatically deploying and configuring bioinformatics workflows in Cloud environments. The integration of scientific workflows and Cloud computing provides fast provisioning of computational and storage resources, elastic scaling and pay-as-you-go pricing. Our approach builds on GP, and supports automated deployment of all prerequisite tools and software packages required for Galaxy along with additional domain specific tools. The deployed workflow environment can respond to workload changes by adding or removing nodes from the cluster and changing instance types to balance cost and performance.
The variability in data volume results in variable computing and storage requirements for data processing. HTCondor [8] is a tool for High Throughput Computing (HTC) on large collections of distributive computing resources. By integrating Galaxy with the HTCondor scheduler, specified Galaxy jobs are executed in parallel using distributed computing nodes in a dynamic HTCondor pool. The proposed auto-scaling strategy significantly improves resource utilization and processing speed, especially for compute-intensive tools, like alignment and SNP calling.
When dealing with large-scale datasets that are common in NGS, Galaxy’s file upload and download capabilities via HTTP and FTP are often unreliable and inefficient. To meet the need for large-scale data transfer, we have integrated Galaxy with Globus Transfer, a service that provides high performance, secure and reliable data transfer, to enable efficient upload and download of large quantities of data in and out of Galaxy. Globus Transfer provides not only powerful Grid transfer capabilities to automate the task of moving files across administrative domains [9,10], but also superior and easy-to-use data management capabilities for transferring big datasets from geographically distributed sequencing centers into Cloud computing infrastructure.
To demonstrate the flexibility of our approach we have extended this framework to meet the requirements of a specific domain, by adding a set of domain-specific tools to the deployment. This paper introduces two different tools that we have wrapped and integrated into the Galaxy platform: CRData tools for executing R scripts, and CummeRbund [11] tool for analyzing Cufflinks RNA-Seq output. These new tools complement the functionality of Galaxy, and have been integrated into our forked Galaxy repository so it’s convenient to deploy a user-specific Galaxy with additional tools.
Galaxy’s workflow canvas provides a platform for assembling tools and building workflows; however building a workflow, especially a complex computational workflow, still requires a lot of domain-specific knowledge and understanding of Galaxy tools. This process is both error-prone and time consuming. Moreover it is often impossible to identify possible errors until the workflow is running. Consequently, we propose semantic verification approaches to facilitate the generation of workflows. By using semantic representations to describe the parameters, tools and workflows, and maintaining an ontology to identify the semantic annotations and appropriate constraints among them, the parameter consistency, functional consistency and reachability of workflows are validated.
The Cloud-based bioinformatics workflow platform integrates all the aforementioned tools, and provides an overall solution for deploying and configuring Galaxy system on Clouds, auto-scaling Cloud resources, enabling high-performance data transfer capabilities, providing customization of user-specific tools, and leveraging a semantic verification mechanism. The platform reduces the considerable usage barriers that existed previously, leverages Amazon EC2 with its pay-as-you-go billing model for resource usage, and provides a scalable and elastic execution environment for sequencing analyses. To validate the effectiveness of our proposed approaches, two bioinformatics workflow use cases as well as performance evaluation are presented, including CRData workflow and RNA-Seq analysis workflow.
The rest of the paper is organized as follows: Section 2 describes an RNA-Seq workflow as a motivating scenario. Section 3 briefly introduces Galaxy. Section 4 describes the tools we have integrated into Galaxy, including Globus Transfer, CRData, CummeRbund, and semantic verification tools. In Section 5, a Globus Provision-based method is proposed to automatically deploy Galaxy on Amazon Cloud. Then the system implementation, use cases and performance evaluation are depicted in Section 6. Section 7 reviews the related work of scientific workflow and Cloud computing. Finally the conclusions and future work are given in Section 8. This paper is an extension of our previous work that describes the methods used to deploy bioinformatics workflows on the Cloud [7].
2. Motivating scenario
In this section, we first introduce an RNA-Sequencing analysis workflow as a motivating scenario. RNA-Sequencing (RNA-Seq) [12] is a deep-sequencing technique used to explore and profile the entire transcriptome of any organism. Fig. 1 shows a sketch map of an RNA-Sequencing analysis workflow downloaded from the public Galaxy website (https://usegalaxy.org/workflow/list_published) for understanding the functional elements of the genome.
Fig. 1.
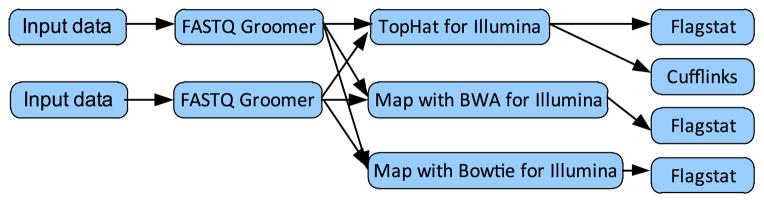
RNA-Sequencing analysis workflow.
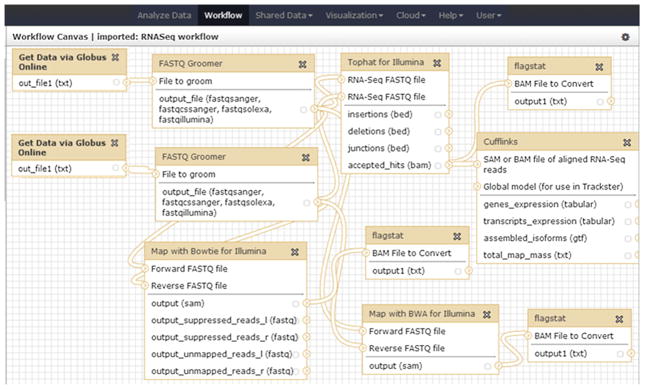
This workflow mainly contains 6 kinds of tools: FASTQ Groomer, TopHat for Illumina, Map with Bowtie for Illumina, Map with BWA for Illumina, Cufflinks and Flagstat. FASTQ Groomer offers several conversion options relating to the FASTQ format if a quality score falls outside of the target score range. TopHat for Illumina is a fast splice junction mapper for RNA-Seq reads, which aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify splice junctions between exons. Map with Bowtie for Illumina is a short read aligner designed to be ultra-fast and memory-efficient. It accepts Sanger FASTQ files and outputs SAM files. Map with BWA for Illumina is a fast light-weighted tool that aligns relatively short sequences to a large sequence database, such as the human reference genome. Cufflinks assembles transcripts, estimates their abundances, and tests for differential expression and regulation in RNA-Seq samples. Flagstat uses the SAMTools toolkit to produce simple statistics on a BAM file.
The sample input dataset includes 500,000 paired-end Illumina HiSeq reads. In traditional methods, initial sequencing data is copied on hard disks and then shipped by FedEx or other Mail services from the sequencing center to the research lab. Biomedical researchers then process these data on a single machine or local cluster in their lab, manually execute this workflow step by step, set the parameters in each script, and store the intermediate results for subsequent analysis. The resulting outputs are often shared within the lab, since the sharing of data with remote collaborators requires additional efforts to transfer data or ship hard disks.
In addition to the transport time, which can be as much as 2–3 days for shipping the disks, the whole workflow usually takes more than 50 h. The costs associated with running this workflow is primarily related to the cost of purchasing and maintaining the single machine or cluster, which is often not a trivial amount. But when data volumes are large, it is hard to execute analyses locally, both considering the computation and storage requirements.
3. Galaxy
To simplify and automate the genome analyses process, we use a scientific workflow management system called Galaxy [13] developed by the Center for Comparative Genomics and Bioinformatics (CCGB) at Pennsylvania State University, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy provides an open, Web-based platform and has been widely used by biomedical scientists for data-intensive computational analyses and data integration. With a simple Web interface, Galaxy integrates a range of NGS tools, enabling researchers to do their own custom analysis and manipulation. The main features of Galaxy comprise the following three aspects.
3.1. Web-based platform for computational analyses
Galaxy provides a simple Web interface to a set of biomedical tools, enabling researchers to do their analysis without any efforts on software installation or computer programming. Users can import datasets into their workspaces from many established data warehouses or upload their own datasets. With Galaxy’s workflow editor, various tools can be configured and composed by biomedical researchers to complete an analysis. Galaxy automatically generates history items and provenance information for each tool executed via a workflow.
3.2. User-friendly workflow publishing and data sharing
When scientific results are published, the publications should include enough information that others can repeat the experiment and get the same results. Galaxy supports reproducibility by capturing sufficient information about every step in a computational analysis, so that the analysis can be repeated in the future. This includes keeping track of all input, intermediate, and final datasets, as well as the specified parameters, and the order of each step of the analysis. Galaxy’s sharing model, public repositories, and display framework provide the biomedical researcher with means to share datasets, histories, and workflows via Web links, either publicly or privately.
3.3. Flexible biomedical tools extension
Besides the public Galaxy server, biomedical researchers can deploy their own Galaxy servers and customize them to meet particular requirements with the help of system deployers. Galaxy uses a flexible model which makes the extension and integration of tools easy. A tool can be any piece of software (written in any language) for which a command line invocation can be constructed.
To add a new tool to Galaxy, a system deployer writes a configuration file that describes how to run the tool, including detailed specification of input and output parameters. This specification allows the Galaxy framework to generate Web interfaces for tools automatically, which makes it ideal for command line averse biomedical researchers [13].
The Galaxy team maintains a public Galaxy server at the University of Pennsylvania that is used by thousands of researchers. As demand has increased, it has become harder to meet the requirements of all the researchers in terms of computer usage and data transfer. Sections 4 and 5 present our approach to deploying user specific Galaxy instances.
4. Galaxy tool integration
Although Galaxy provides a convenient platform for average researchers, challenges remain in moving large amounts of data reliably and efficiently, adding domain-specific tools for specific analyses, and providing semantic verifications for workflows and parameters. To address these challenges, we integrate the Galaxy framework with new services, including Globus Transfer for fast and secure movement of large amounts of data, CRData and CummeRbund tools for user-specific analyses, and semantic verification tools for validating the correctness of workflows.
4.1. Globus Transfer
Although Galaxy provides tools for uploading files via FTP and HTTP, these tools are often unreliable and inefficient when transferring large amounts of data, as is often the case in NGS. Files larger than 2 GB cannot be uploaded to Galaxy directly from the computers of biomedical researchers. Consequently, we have integrated Globus Transfer tools to provide a high performance, secure and reliable way of transferring large quantities of data in and out of Galaxy.
Globus Transfer is a Globus (formerly Globus Online [14]) service, which provides powerful Grid capabilities (using the GridFTP [15] protocol) to automate the tasks of moving files between sites, or “endpoints” [9,10]. By integrating Galaxy with Globus Transfer, file transfer between users and the Galaxy server is faster than was previously possible, e.g., terabytes of data can be moved in hours. Globus Transfer offers a “fire and forget” model in which biomedical researchers only need to submit their transfer request and walk away, there is no need to monitor their jobs or worry about the status of their requests. Globus Transfer handles difficult aspects of data transfer by tuning parameters to maximize bandwidth, managing security configurations, monitoring performance, retrying failures and recovering from faults automatically, and notifying users of errors and job completion.
Globus Transfer provides important data management functionality to Galaxy, addressing the challenges in moving or synchronizing large quantities of data from one place to another without installing any software. It also supports third party transfers, in which the selected endpoints are not collocated with the requesting user. This is advantageous when moving data between a sequencing center and the Galaxy server, for example.
Another benefit is that Galaxy users are able to access the large network of existing Globus endpoints as well as data sources, and transfer files between existing data sources, their own resources and the Galaxy server securely, efficiently and quickly.
As seen in Fig. 2, the Globus Transfer toolset includes three tools: (1) third party transfers between any Globus endpoints (“GO Transfer”), (2) upload to Galaxy from any Globus endpoint (“Get Data via Globus Online”) and (3) download from Galaxy to any Globus endpoint (“Send Data via Globus Online”). Each of these tools has been added as a native Galaxy tool with an associated user interface to specify properties of the transfer. The tools can be integrated in a workflow, allowing automated data download and upload as a part of a repeatedly executed workflow.
Fig. 2.

Globus Transfer tools in Galaxy.
For example, using the “GO transfer” tool (see Fig. 2), a file stored in “Source endpoint” can be transferred to “Destination endpoint”, meanwhile the file is manifested as a Galaxy dataset in the history panel available for further analysis. If the “Deadline” is specified, the job will be terminated if it is not completed within the specified time period. During execution, Galaxy invokes the Globus Transfer API (Application Program Interface) and returns the status of the job. The end-user also receives an Email notification when his job is finished.
To compare the performance of Globus Transfer with FTP and HTTP transfer in Galaxy, Table 1 shows the average transfer rate (in Mbits/s) obtained when moving data from a laptop to the Galaxy server using different methods and file sizes. The transfer rate of Globus Transfer varies with file size, from 1.8 to 37Mbits/s, while the transfer rate of FTP varies from 0.2 to 5.9 Mbits/s and HTTP is only able to achieve a transfer rate of less than 0.03 Mbits/s (up to the maximum 2 GB file size). We see that Globus Transfer outperforms FTP and HTTP significantly for all file sizes considered. Moreover, Globus Transfer offers additional benefits in terms of security and reliability, which will be further discussed in Section 5.5.
Table 1.
Comparison of average transfer rate (mbps).
| File size (bytes) | Globus Transfer | FTP | HTTP |
|---|---|---|---|
| 1.E+03 | 1.8 | 0.2 | 0.001 |
| 1.E+04 | 7 | 1 | 0.002 |
| 1.E+05 | 13 | 1.8 | 0.004 |
| 1.E+06 | 20 | 2.4 | 0.008 |
| 1.E+07 | 25 | 3 | 0.012 |
| 1.E+08 | 29 | 3.8 | 0.023 |
| 1.E+09 | 32 | 5 | 0.028 |
| 1.E+10 | 37 | 5.9 |
4.2. Domain-specific tools
To highlight the flexibility of our approach for deploying customized Galaxy environments, two different toolsets, CRData and CummeRbund, are integrated into the Galaxy framework. The CRData tools represent a custom tool deployment that provides a set of statistical tools, while the CummeRbund tool is designed to analyze Cufflinks RNA-Seq output.
4.2.1. CRData
Although Galaxy provides a range of NGS tools for biomedical scientists, it still lacks tools for executing R scripts.
R [16] is a free software environment for statistical computing and graphics. R provides a wide variety of statistical (linear and nonlinear modeling, classical statistical tests, time-series analysis, classification, clustering, etc.) and graphical (well-designed and publication-quality plots can be produced) techniques. R is highly extensible via packages. Besides the packages supplied with R distribution, many more packages are available through CRAN (http://cran.csdb.cn/web/packages/) covering a wide range of modern statistics.
CRData.org is a website that offers Web-based computational tools designed to execute BioConductor R scripts. Because the R scripts from CRData.org are commonly used in many research domains, we wrapped them into Galaxy framework as CRData toolset so that Galaxy users can directly execute these R scripts in Galaxy and use the resulting outputs for subsequent analyses.
As seen in Fig. 3, the CRData toolset consists of 35 tools with various functions. For example, the “heatmap_plot_demo.R” tool performs hierarchical clustering by genes or samples, and then plots a heatmap. The “sequenceDifferentialExperssion.R” tool (see Fig. 3) performs a two-sample test for RNA-Sequence differential expression. The “affyClassify.R” tool conducts statistical classification of affymetrix CEL files into groups. And the “sequenceCountsPerTranscript.R” tool summarizes the number of reads (presented in one or more BAM files) aligning to different genomic features retrieved from the UCSC genome browser (http://genome.ucsc.edu/).
Fig. 3.

CRData tools in Galaxy (this figure shows the interface of “sequenceDifferentialExperssion.R”).
Each CRData tool corresponds to an R script from CRData.org. The execution of a CRData tool invokes the corresponding R script, transfers input parameters and files to the script, and returns output files and figures after running R. The output is also shown in Galaxy’s history panel for subsequent analysis or download.
4.2.2. CummeRbund
CummeRbund [11] is an R package designed to aid and simplify the task of analyzing Cufflinks RNA-Seq output. The results of high-throughput RNA-Sequencing analysis are normally large datasets with high-degree relations between various data types. The aim of CummeRbund is to simplify the analysis and exploration portion of RNA-Seq data derived from the output of a differential expression analysis using Cuffdiff in order to provide fast and intuitive access to experiment results.
In order to enable the execution of CummeRbund packages in Galaxy we have wrapped CummeRbund as a Galaxy tool so that biomedical researchers can embed this tool into a workflow for RNA-Seq analyses.
As seen in Fig. 4, the input of CummeRbund tool is either a backend SQLite database file from the history, or a new SQLite database generated from Cuffdiff outputs. The SQLite database describes the relationships between genes, transcripts, transcription start sites, and CDS regions. We have implemented 8 plotting functions for commonly used visualizations, including Density, Boxplot, Scatter, Volcano, Heatmap, Cluster, Expression Plot and Expression Bar Plot. Fig. 5 is a density plot generated by CummeRbund.
Fig. 4.

CummeRbund tool in Galaxy.
Fig. 5.

Density plot generated by CummeRbund.
4.3. Semantic verification
Galaxy’s workflow canvas provides a platform for biomedical researchers to design and build workflows through which diverse Galaxy tools are configured and assembled for a wide variety of purposes. Currently, the workflow generator manually connects tools and sets the parameters of each tool. In order to create these workloads a lot of domain-specific knowledge and full understanding of Galaxy tools are required. Thus, building a workflow, especially a complex computational workflow, is a non-trivial task.
Until the workflow is running, it’s hard to identify possible errors in advance, for example, the input and output data types do not match, the value of a parameter is beyond legal range, the workflow has an isolated node/tool, etc.
To address these problems, we created semantic verification tools to facilitate the generation of workflows in Galaxy. By using semantic representations to describe the parameters, tools and workflows, useful semantic annotations are built including parameter name, data type, value range, tool name, tool function, etc. An ontology is maintained to identify the aforementioned annotations and appropriate constraints among them.
More specifically, the semantic verification tools are developed from the following three aspects.
4.3.1. Validate the parameter consistency
Parameter consistency denotes the matching of input and output parameters. From one side, the input and output data types should be equivalent; otherwise the links between them cannot be generated. From the other side, for each parameter, if there is a legal range of values, any input value beyond this range will result in a warning and helpful tips will be sent to the workflow generator. To validate the parameter consistency, we modified the tool’s configuration file, added validators to monitor the value of parameters, and check the similarity of data types of connected input and output parameters.
4.3.2. Validate the functional consistency
Functional consistency indicates the matching of connected tools with respect of functions and operations. Based on the semantic annotation of tools’ names and functions, constraints on which tools can (not) be connected are constructed by domain experts. For example, as seen in Fig. 6, CummeRbund is designed to analyze Cuffdiff’s RNA-Seq output, so a constraint “Cuffdiff links to CummeRbund” is added to our knowledge base. When a link between Cuffdiff and CummeRbund is generated in the workflow canvas, Galaxy will check the knowledge base to see whether it is allowed to build such a link. More specifically, the 11 outputs of Cuffdiff are passed to 11 inputs of CummeRbund. They can be connected only when both their data types and parameter names match, e.g., the splicing_diff parameter of Cuffdiff links to the “Splicing differential expression testing” parameter of CummeRbund. This is handled by the validator for parameter consistency.
Fig. 6.

Connection of Cuffdiff and CummeRbund.
4.3.3. Validate the reachability of workflows
The reachability of workflows refers to the ability to get from one tool to another along with the directed links between tools in this workflow graph. A tool T1 can reach tool T2 (or that T2 is reachable from T1) if there exists a sequence of adjacent links which starts with T1 and ends with T2. A workflow is reachable means that all the tools are reachable and the workflow can be completed in a limited time (not infinite time). This validator aims to prevent deadlock and isolated tools. Because some workflows are not well-designed, deadlocks may exist when tools are connected to form a cycle which cannot be completed at run time. Also, isolated tools may exist in a workflow when these tools are not connected to other tools. Although this situation is not forbidden, it is not suggested.
To avoid the above problems, we have created a validator to check the structure of workflows at build time. When deadlock or isolated tools are detected, a warning is sent to the workflow generator, who can then decide whether to modify the workflow or ignore the warning.
5. Cloud-based Galaxy platform
In Galaxy, the resources needed by scientific workflows may vary drastically during run time. It is often inefficient, both in terms of resource usage and cost, to pre-provision infrastructure for peak usage. Cloud computing provides an alternative model to elastically scale to the demands of a workflow. Accordingly, deploying Galaxy on Cloud, like Amazon EC2 (Elastic Compute Cloud) [17], provides obvious benefits such as on-demand resource configuration, usage-based pricing, better resource utilization, increased processing speed and improved user experience.
However, setting up a production instance of Galaxy is a nontrivial task that involves a number of manual installation and configuration steps for both the platform and any dependent software packages—steps that can be both error-prone and time consuming. These steps require that biomedical researchers either become IT experts or rely upon the potentially sparse IT resources provided by their institutions. Either approach tends to result in sub-optimal use of researchers’ time and expertise.
To address these problems, we have designed a Globus Provision- based method to automate the process of deploying and scaling Galaxy on Amazon EC2. GP is a tool that provides on-demand cluster reconfiguration, user-specific node provisioning, and automatic instance deployment.
This section first introduces GP, and then presents the methods for deploying Galaxy on EC2 using GP. Based on this model, the auto-scaling strategy is realized with HTCondor scheduler, and finally the system architecture and modules are illustrated in detail.
5.1. Globus Provision
Globus Provision [18] is a tool for automatically deploying a highly configurable and scalable distributed computing system that includes remote data access, job submission, and security. The system can be deployed with any subset of the tools it supports such as GridFTP [15] for high performance transfer, MyProxy [19] for user-based access management, and HTCondor [8] for job submission. As part of this configuration, GP also generates user accounts and certificates to support secure access, sets up a Network File System (NFS) and Network Information System (NIS) to provide a robust shared file system across nodes, and dynamically adds and removes software, hosts and user accounts.
GP relies on Chef [20] to configure hosts for a given topology. The topology is the specification of what will be deployed (e.g. a GridFTP server, a specific set of users, and a HTCondor cluster.). In Chef, the actions required to set up a specific piece of software are defined in a Ruby script called a recipe [20]. Similar recipes are grouped into a cookbook which includes associated configuration templates and default values. GP defines several Chef Cookbooks to handle basic host setup and configuration of each node.
Fig. 7 describes the main steps for using GP.
Fig. 7.

The main steps for using Globus Provision (the blocks with solid lines are necessary steps, while the ones with dashed lines are optional steps).
Pre-requirement: Before starting with GP, the system deployer should create an Amazon Web Services (AWS) account and install GP software.
Create/Modify recipe: The system deployer can modify existing recipes or add new recipes to install specific software and packages, run commands and conduct operations that should be performed on each host.
Define a topology: Then the system deployer should write a topology file, which is a configuration file that defines the user’s requirements of the system.
Create/Start a GP instance: Based on the topology file, GP will create and start one or more instances on Amazon EC2.
SSH to hosts if needed: When the GP instance is running, the system deployer can connect to any of its hosts defined in the topology via SSH (Secure Shell).
Stop/Terminate the GP instance: The GP instance can be stopped while not in use (to avoid paying for idle resources), and resumed at a later time. Terminated instances cannot be resumed. All the hosts are shut down and all their resources are released after termination.
Modify topology: Once an instance is running, it is possible to modify its topology, e.g., adding and removing hosts/users/domains, and adding software requirements to hosts.
Create/Update GP AMI: Although GP already provides a public Amazon Machine Image (AMI), the system deployer can also create new AMI (e.g., to use an AMI that is preloaded with required software packages such as specific bioinformatics tools) to speed up the deployment.
5.2. Deploy Galaxy on Cloud
GP provides a generic architecture for automatically configuring distributed Cloud-based infrastructure which includes many of the features required for a Galaxy deployment. For this reason we have extended GP to support configuration and deployment of a Galaxy instance with integrated Globus Transfer capabilities, user-defined domain-specific tools, and semantic verification mechanism. An extensible framework is created to support the deployment of custom Galaxy tools such as CRData tools and CummeRbund tool. The combination of default tools and user-specific tools simplifies the ability for end-users in different domains to create a domain-specific Galaxy instance suitable for supporting data-intensive applications.
To deploy a Galaxy instance on EC2 using GP we require a topology file describing the deployment. Fig. 8 gives an example topology file (galaxy.conf). The topology file defines the user’s requirements with respect to four general categories: general, domain- simple, ec2, and globusonline. In practice, the topology file is often written by system deployers, who obtain the system requirements and application scenarios by communicating with biomedical researchers.
Fig. 8.

Topology file “galaxy.conf”.
In this topology file, “users” presents the username that will be added to the list of users on the Galaxy cluster; “cluster-nodes” specifies the number of worker nodes to be deployed; “go-endpoint” defines the name of the endpoint that will be created for this cluster; “instance-type” specifies the EC2 instance type. Generally speaking, t1.micro is suitable for testing, c1.medium is good for demos, and m1.large (or larger) is used for high performance instances. The parameters “gridftp”, “condor” and “galaxy” define the required GP packages to be set up in the instance. More detailed instructions on these parameters are given in Appendix A.
Through the topology file, the requirements of biomedical researchers are well translated to GP, including which servers need to be deployed, how many cluster nodes will be created, etc.
In order to deploy a Galaxy instance with Globus Transfer tools, we have created new recipes and added them to the default cookbook of GP. One recipe (“galaxy-globus-common.rb”) is responsible for installing the common requirements for the Globus fork of Galaxy. More specifically, it creates a galaxy user, downloads Globus Transfer tools as well as Galaxy from bitbucket.org, and copies default configuration files and set-up scripts for Galaxy. The other recipe (“galaxy-globus.rb”) installs the Globus fork of Galaxy and Globus Transfer API, sets up the Galaxy database, executes set-up scripts and restarts Galaxy. After adding these two recipes, when the GP instance is started, Galaxy with Globus Transfer tools can be accessed via the URL of Galaxy host. A Globus endpoint, with the name specified in the topology file, is also created for data transfer with Globus Transfer tools.
Similarly, the other user-specific tools can be automatically deployed in Galaxy by adding new recipes. For example, to add the CRData toolset, we created “galaxy-globus-crdata.rb” recipe. It downloads and installs the necessary software and R packages on the Galaxy host. So the generated Galaxy instance includes both Globus Transfer tools and CRData tools. Moreover, the recipes created are open source so they can be reused in any combination to deploy customized Galaxy instances.
Once an instance is running, it is possible to modify its topology. The system deployer can actually edit the instance’s topology and make GP modify the running instance to match the new topology. GP will determine what changes to make, and prevent “impossible” changes. The following changes can be made by editing the topology file.
Add or remove several hosts at once. For example, instead of creating a t1.micro EC2 instance, c1.medium EC2 instances can be added.
Add, remove or modify several users at once. For example, modifying a user may include changing a user’s password or authorized SSH public key.
Add or remove entire domains.
Add software to one or several hosts.
The characteristics of GP enable complete customization of a deployment to meet real-time requirements. For example, if workflow usage is low, micro or small instances can be used, while if memory requirements of a workflow increase, the running instances can be upgraded to large or extra-large instances. In addition, when the workflow platform is not being used, it can be suspended and restarted when required, thereby reducing the overhead of running an unused or sparsely used platform.
5.3. HTCondor scheduler
Since many Galaxy tools submit CPU/memory-intensive jobs that are generally common in genome analysis, the size of the required computational capability may outpace the initially deployed Galaxy system. To improve system performance in terms of both processing speed and cost, we integrate Galaxy and HTCondor scheduler so that specified Galaxy jobs can be run through
HTCondor on remote clusters with higher performance. HTCondor is a tool for High Throughput Computing on large collections of distributive computing resources. As a specialized workload management system for compute-intensive jobs, HTCondor provides a job queuing mechanism, scheduling policy, priority scheme, resource monitoring, and resource management. Users submit their serial or parallel jobs and then HTCondor places them into a queue, chooses when and where to run the jobs based upon a policy, carefully monitors their progress, and ultimately informs the user upon completion [8].
As mentioned in Section 5.2, GP provides a mechanism to install and configure HTCondor, so we further configure Galaxy to use a HTCondor job scheduler. When submitting a Galaxy job, the executable along with command line options get passed to a HTCondor runner, then the HTCondor runner automatically assigns the job to a worker node in the HTCondor pool. So applications are run on a worker node instead of the Galaxy node, thus leveraging Cloud-based scalable computational resources for parallelizing Galaxy jobs. The number of parallel Galaxy jobs is dependent on the number of worker nodes in dynamic HTCondor pool.
Because of the pay-as-you-go pricing of Amazon EC2, we maintain a Galaxy node with medium or large instance type for general Galaxy jobs that are not compute-intensive, and create large or extra- large instances with high CPU/memory for HTCondor worker nodes when compute-intensive jobs are submitted or the job queue exceeds a predefined threshold. The HTCondor worker nodes are terminated after the execution of assigned jobs.
By configuring the tool runner of Galaxy, the system deployer can decide which tools are run through HTCondor and which tools are run locally. Moreover, they can design the strategies of autoscaling through user-specified policies. By default, we set the wait time threshold (the time a job waits in the HTCondor queue) to 5 minutes and the queue length threshold to 10 jobs. That is to say, when a HTCondor job has waited for more than 5 min, or the current queue length is bigger than 10, new HTCondor worker node will be created and assigned jobs. We also set the idle time of node as 0.5 h, i.e., when a worker node has been idle for half an hour, it will be terminated and the resource will be released. This auto-scaling strategy and the instance type of created worker nodes can be configured by the system deployer for different application scenarios to maximize the overall performance and minimize the cost.
5.4. System architecture
Fig. 9 shows the architecture of the overall Cloud-based bioinformatics workflow platform. First, the system deployer and biomedical researchers should agree upon system requirements and application scenarios. The system deployer then writes a configuration file (or topology file as seen in Fig. 8) according to the researchers’ practical requirements. The configuration file is passed to GP via its API and then processed by the other modules in GP, in which the “Configuration File Parsing Module” parses the submitted configuration file, the “Topology Management Module” maintains all the history topology files, the “Certificate Management Module” manages users’ certifications for accessing EC2 instances and using Globus Transfer tools, the “Chef Cookbook” stores a set of cookbooks and recipes for basic host setup (like creating users, installing a host certificate, etc.) and installation and configuration of Globus, Galaxy and other software. The “EC2 Deployer” interacts with the above four modules and implements the deployment of Galaxy system on EC2, including the configuration of Cloud storage and computing nodes.
Fig. 9.

Architecture of Cloud-based bioinformatics workflow platform.
The computing nodes as well as Cloud storage are then automatically configured on AWS. The core of the system is the Galaxy node that provides Galaxy applications and user interface. The “Globus Transfer Services” configures a Globus endpoint on Galaxy node and allows Globus Transfer between the Galaxy system and other Globus endpoints. When a HTCondor scheduler is configured, the Galaxy node also operates as a HTCondor head node that manages a set of HTCondor worker nodes in a dynamic HTCondor pool. The HTCondor pool grows and shrinks based on the computational characteristics of the workflows. In this model, Galaxy jobs are transparently assigned to HTCondor worker nodes for parallel execution. The “Semantic Verification Module” is responsible for the semantic verification functions described in Section 4.3. The “Security and Privacy Module” ensures the security and privacy of Galaxy usage, data transfer and sharing.
The shared file system provides a common storage accessible to Galaxy, Globus Transfer, and the Amazon EC2 nodes used for genome analysis. It can be a separate node or located in the Galaxy node. Moreover, in order to provide flexible on-demand storage, the shared file system can be extended by using Amazon Elastic Block Storage (EBS) [21] and Amazon Simple Storage Service (S3) [22]. EBS has lower latency so we use it for frequently accessed data, e.g. genome reference, NGS tools, input and output datasets. S3 has higher latency but it is independent from EC2 instances, so we use it for history data and backup of long-term data.
The Galaxy User Interface provides a Web-based UI for biomedical researchers to conduct sequencing analysis. For example, a Galaxy user can transfer datasets from a “Sequencing center” to the “Galaxy Endpoint” via Globus Transfer tools, and then take these datasets as inputs to run an RNA-Seq analysis workflow in Galaxy, and finally transfer the analysis results back to the “Sequencing center”.
5.5. Security enforcement
Security concerns are particularly important and challenging in geographically distributed systems. In order to ensure the privacy, authentication and authorization in data transfer when using Globus Transfer in Galaxy, we rely on several security mechanisms.
First the biomedical researchers need to create a Globus account (https://www.globus.org/) and register an account in Galaxy with the same username. Then the system deployer configures X.509 certificates on the Galaxy server so that the Galaxy server can submit transfer requests on behalf of individual researchers while guaranteeing security of the transfer. When submitting a transfer in Galaxy, Globus Transfer requires “activation” of selected endpoints (source endpoint and destination endpoint) using appropriate credentials. Globus Transfer manages the security credentials required to authenticate against different endpoints.
If the biomedical researchers want to use the Command-Line Interface of Globus, which provides a set of commands for directly managing Globus endpoints, the user’s SSH public key must be added to the user’s profile through the Globus website. Moreover, the biomedical researchers can access Galaxy node via SSH if their SSH public keys are deployed on the Galaxy node.
Finally the Galaxy server includes a registered Globus Endpoint, which is configured in GP for Galaxy recipes as described in the previous section. A more detailed description of the security mechanism is shown in Fig. 10.
Fig. 10.
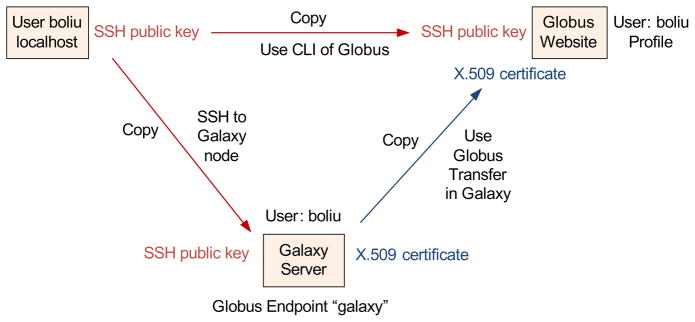
Security mechanism.
6. System implementation and evaluation
Based on the aforementioned approaches, we have implemented a production-level Galaxy instance on Amazon EC2. With GP, we used a set of recipes to launch an instance of Galaxy preloaded with Globus Transfer tools, CRData tools, CummeRbund tool along with all of the NGS tools, and integrated support for semantic verification, HTCondor scheduler and security management. This instance is provided to our end users to create and share analytical workflows using Galaxy.
After creating an AWS account and Globus account, a Galaxy instance is created on EC2 based on the properties specified in the topology file (galaxy.conf) described in Fig. 8. For more information, please refer to Appendix B.
In this section, we focus on two real-world use cases, CRData workflow and RNA-Seq analysis workflow, to examine the time and cost of execution on different Cloud deployments.
6.1. Use Case 1: CRData workflow
The first use case is a CRData workflow as shown in Fig. 11. In this case, we configure the GP instance to run a CRData tool on a small cluster and then expand the cluster dynamically to run the same workflow on a larger dataset.
Fig. 11.
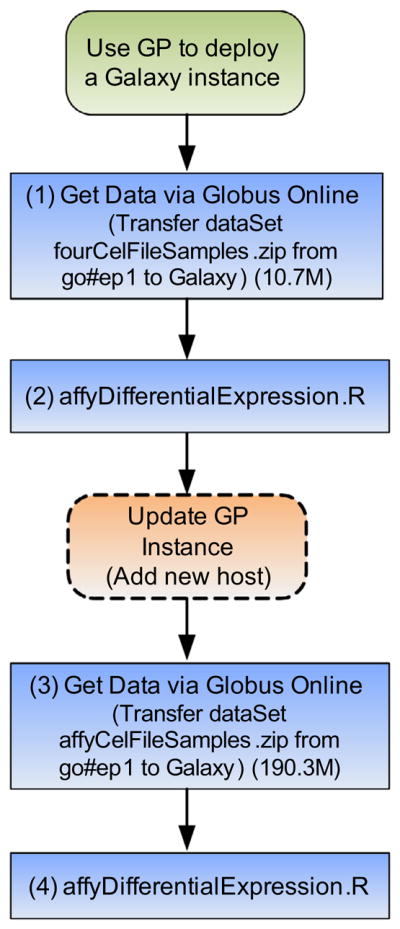
CRData workflow.
First, using the ‘Get Data via Globus Online’ tool in Galaxy, the dataset “fourCelFileSamples.zip” (10.7 MB) is transferred from a Globus endpoint to the Galaxy server. The parameters “Endpoint” and “Path” are set as follows:
Endpoint: go#ep1 (the name of the remote endpoint).
Path:/home/boliu/fourCelFileSamples.zip (the location of the file at this endpoint).
After uploading the data, the researchers can run the appropriate statistical tool by selecting the ‘CRData’ tool and ‘affyDifferentialExpression. R’, and setting the parameters as shown in Fig. 12. The tool runs the affyDifferentialExpression.R script which conducts two-group differential expression on Affymetrix CEL files. This script takes the dataset “fourCelFileSamples.zip” (uploaded in step 1) as input, and creates a “top table” of probe sets that are differentially expressed between CEL files that have been assigned to one of two groups.
Fig. 12.
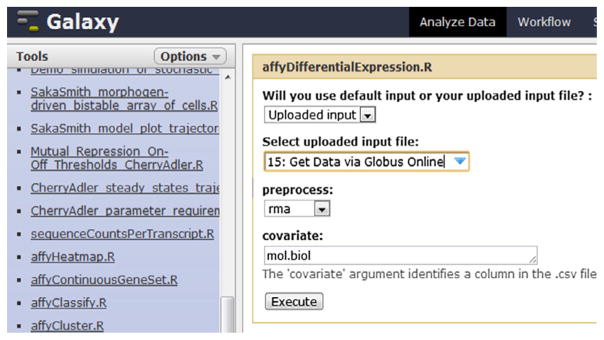
CRData tool “affyDifferentialExpression.R” (Step 3).
After execution, the output results are shown in the History panel, including both text output (Fig. 13) and figure output (Fig. 14a).
Fig. 13.

Text output of “affyDifferentialExpression.R” (Step 3).
Fig. 14.
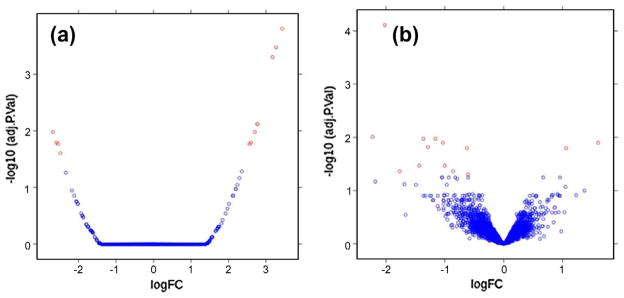
Figure output of “affyDifferentialExpression.R” in Step 3 (a) and Step 4 (b).
The input dataset used for this example is only 10.7 MB, which can be processed easily on a small EC2 instance. In the second stage, the researcher wants to process a larger dataset “affyCel-FileSamples.zip” (190.3 MB). However, this processing takes considerable time when using small EC2 instances. In order to speed up the workflow, the system deployer can update the GP instance by adding a new EC2 host. This is done by creating a new GP topology file, and requesting a new host with the instance type “c1.medium”.
The researcher can then follow the same process as outlined above by transferring the dataset “affyCelFileSamples.zip” from go#ep1 endpoint to the Galaxy server and then running the affy-DifferentialExpression.R’ tool to analyze this dataset. The output results are shown in Fig. 14b.
While the operation of updating the GP instance is optional, it does however decrease the execution time of Steps 3 and 4 from 10.7 min using a small instance to 6.9 min after adding a new medium instance. Similar improvements can be obtained using larger instances. Moreover, the same approach can be applied for concurrent execution when multiple users submit tasks for execution at the same time.
Table 2 compares the deployment time, execution time and cost of Steps 3 and 4 on different EC2 instance types. We see that significant performance improvements can be obtained when using larger instances. For example, execution time decreases to 5.4 min on a large instance and to 4.6 min on an extra-large instance. However, performance improvements are disproportionate with cost, which almost doubles for each increase in instance size. The cost for executing Steps 3 and 4 on small and extra-large instances increases from 0.007 to 0.024 dollars. This table also compares the deployment time using GP to set up a Galaxy instance with Globus Transfer tools and a set of bioinformatics tools. The deployment time is reduced from 8.8 min on a small EC2 instance to 7.2 min on a medium instance and to 4.9 min on an extra-large instance.
Table 2.
Comparison of execution time, deployment time and cost of CRData workflow.
| Instance type | Execution time (min) | Deployment time (min) | Cost (dollar) |
|---|---|---|---|
| m1.small (1 EC2 Compute Units (ECU) with 1.7 GB memory and 160 GB storage) | 10.7 | 8.8 | 0.007 |
| m1.medium (2 ECU with 3.75 GB memory and 410 GB storage) | 6.9 | 7.2 | 0.009 |
| m1.large (4 ECU with 7.5 GB memory and 2 * 420 GB storage) | 5.4 | 5.8 | 0.014 |
| m1.xlarge (8 ECU with 15 GB memory and 4 * 420 GB storage) | 4.6 | 4.9 | 0.024 |
This use case shows the ease by which the system deployer can deploy and scale their workflow environment to meet the needs of biomedical researchers for complex analyses or large-scale datasets. The GP-based approach can dynamically adjust the number of nodes and instance types at runtime, which can increase the performance of scientific workflows and potentially lower the cost of execution. However, modification of the topology still requires IT expertise and it may be difficult for many biomedical researchers to estimate the resources required by a workflow, so in practice we recommend using the auto-scaling strategy by which neither the system deployer nor biomedical researcher need to worry the execution time and cost. The next use case will demonstrate the effectiveness of our auto-scaling approach.
6.2. Use Case 2: RNA-Seq analysis workflow
Based on the RNA-Seq analysis workflow described in Fig. 1, we built a workflow in Galaxy, as seen in Fig. 15. As described above, the main steps are FASTQ Groomer, TopHat for Illumina, Map with Bowtie for Illumina, Map with BWA for Illumina, Cufflinks and Flagstat, while the first two steps have been replaced by “Get data via Globus Online” for moving data from a remote endpoint to Galaxy instance as input datasets. Similarly, to archive the output data on a storage endpoint, we can add steps “Send data via Globus Online” at the end of this workflow.
Fig. 15.
The RNA-Seq analysis workflow in Galaxy.
To evaluate the performance of the RNA-Seq workflow, we use the same input dataset that includes 500,000 paired-end Illumina HiSeq reads. Our Galaxy instance completed the whole analysis workflow in 24.8 h based on an m1.small EC2 instance. The execution time is reduced to 20.7 h on an m1.medium instance, 18.3 h on an m1.large instance, and 14.5 h on an m1.xlarge instance.
As seen in Fig. 16, when more HTCondor worker nodes are deployed, the execution time continues dropping because some steps are run in parallel. Since the workflow has at most 4 branches, the execution time is lowest when running on 4 nodes. It takes only 6.2 h when running on 4 m1.xlarge instances.
Fig. 16.

The execution time and cost of RNA-Seq workflow with different EC2 instance types.
The performance improvement also results in higher cost because larger instance is more expensive. For example, the cost varies from 0.9672 dollars (on 1 m1.small instance) to 7.688 dollars (on 4 m1.xlarge instances).
When utilizing the auto-scaling strategy mentioned in Section 5.3, that is, we use an m1.medium instance for Galaxy node and m1.large instances for HTCondor worker nodes, the execution time is 9.6 h and the cost is 4.03 dollars. HTCondor worker nodes are started only when parallel jobs are executed and the waiting time of jobs in HTCondor queue is more than 5 min. We can see that although the execution time (9.6 h) is similar to that in 3 m1.large nodes (9.5 h), the cost (4.03 dollars) is lower than 4.56 dollars. So in this case, the auto-scaling strategy is more economical and efficient. However, balancing execution time and cost must consider the practical scenario and requirements.
To further reduce the total cost, the system deployer can consider launching spot instances [23] instead of on-demand instances for HTCondor worker nodes, since AWS instance prices for spot instances are often up to 10-times cheaper than on-demand instances. However, spot instances are not stable and may be terminated by Amazon at random, therefore they are only suitable for time-flexible and interruption-tolerant tasks. We are planning to customize the HTCondor scheduler to manage spot instances as worker nodes. Besides, Amazon provides free usage tier model [24] for users to test AWS services. Notably, the free usage tier pricing model provides free 750 usage hours of an Amazon EC2 microinstance per month and 30 GB of Amazon EBS storage plus 2 million IOs and 1 GB snapshot storage. For small scale usage, users can mix free usage tier products with paid products, so that only the usage beyond the free usage tier will be charged.
7. Related work
Cloud computing offers many features that are attractive to scientific workflow developers [25–28]. Infrastructure-as-a-Server (IaaS) Cloud platforms, such as Amazon EC2, can provision the necessary resources for a workflow quickly, provide as much computation and storage capacity as needed in a pay-as-you-go manner, and elastically scale capacity as service demands change.
Cloud provisioning tools accelerate the deployment of scientific workflow systems and make it more agile. For example, Chef [20] makes it easy to deploy servers and scale applications throughout the entire infrastructure; Puppet [29] gives system managers the operational agility and insight to manage dynamic infrastructure; Eucalyptus [30] provides a software platform for the implementation of private Cloud computing, and OpenNebula [31] offers complete management of virtualized data centers to enable on-premise IaaS Clouds. However, very few researchers have used these tools to automate the deployment and scaling of scientific workflows.
Other researchers have investigated the use of scientific workflow systems in Cloud environments. Yuan et al. [32] proposed a cost-effective strategy for intermediate data storage in scientific Cloud workflow systems. Wu et al. [33] presented a market-oriented hierarchical scheduling strategy in Cloud workflow systems. Simmhan et al. [26] built the trident scientific workflow workbench for data management in the Cloud. Zhang and De Sterck [34] proposed CloudWF, a scalable and lightweight computational workflow system for Clouds. These methods focus on general scientific workflow systems, and are not suitable for large-scale biomedical analyses and data transfer.
More specifically, in bioinformatics domain, some researches have utilized Cloud computing to deliver large computational capacity and on-demand scalability. Crossbow [35] is a Cloudenabled tool that combines the aligner Bowtie and the SNP caller SOAPsnp, and uses Hadoop for parallel computing. Rainbow [36] is a Cloud-based software package that can assist in the automation of large-scale whole-genome sequencing (WGS) data analyses. It copies input datasets to Amazon S3 and utilizes Amazon’s computational capabilities to run WGS data analyses pipelines. However, the pipelines are executed by manual scripts and only have specific functions. The input data is uploaded to S3 by shipping hard drives to Amazon via FedEx and then coping the data to S3, which is time-consuming and expensive.
CloudMap [37] is a Galaxy-based pipeline that greatly simplifies the analysis of mutant genome sequences from raw FASTQ reads to mapping plots and short lists of candidate mutations. CloudBurst [38] is a parallel read-mapping algorithm for mapping next-generation sequence data to the human genome and other reference genomes for biological analyses. RSD-Cloud [39] runs a comparative genomics algorithm on Amazon EC2 for ortholog calculations across a wide selection of fully sequenced genomes. These projects focus on the solutions of specific problems by developing a tool or method, but lack a general solution for other NGS tools in analysis workflows.
The most similar work to our approach is CloudMan [40], a Cloud resource management system for individual researchers to deploy a Galaxy instance on EC2. The advantages of CloudMan lie in: (1) It has an easy to use interface, using only a web browser, to create a configured compute cluster ready to perform analysis. (2) It provides an automated method for building custom Cloud deployments. However, we use GP over CloudMan for the following reasons. (1) GP provides more flexibility in defining user-specific node configuration, and adding recipes for installing additional software (all the recipes are reusable). (2) At run-time, CloudMan nodes can only be added or removed manually, whereas GP allows modification of the whole configuration including adding and removing hosts and users, changing instance types, etc. (3) GP can integrate a HTCondor scheduler for auto-scaling and parallel computing. (4) GP makes it convenient to extend Galaxy with arbitrary tools, which in our example satisfies the requirement for high performance and reliable large-scale data transfer and support for user-specific functions in Galaxy.
8. Conclusions and future work
In this paper, an automatic and elastic method for deploying a scientific workflow system on Cloud is proposed. The integration of Galaxy workflow system and Globus Provision realizes the deployment of Galaxy on Amazon EC2 with the following features: elastic processing at runtime, pay-as-you-go style resource consumption, on-demand provisioning, user-defined recipe configuration, and automated instance deployment.
The extended Galaxy tools are integrated into our Galaxy repository so it’s convenient to deploy a user-specific Galaxy instance with additional tools. For example, the Globus Transfer tools enable transferring large-scale datasets in and out of Galaxy securely, efficiently and quickly, the CRData tools execute R scripts, the CummeRbund tool can analyze Cufflinks RNA-Seq output, and the semantic verification tools validate the parameter consistency, functional consistency, and reachability of workflows. Our Cloud based bioinformatics workflow platform integrates all the aforementioned tools and provides an overall solution for biomedical scientists to conduct large-scale NGS analyses.
To illustrate our proposed methods, two real-world bioinformatics workflows are presented. The CRData workflow demonstrates the dynamic scaling capability of our system, and the RNA-Seq workflow compare the execution time and cost on different instance types and number of worker node. These workflows show that the use of the HTCondor scheduler and auto-scaling strategy can significantly improve the performance of executing Galaxy jobs.
Our platform is based on Amazon EBS and S3 for scalable shared storage. Although Amazon guarantees the safety of users’ data, there may still be risks associated with this approach because of the lack of a suitable SLA (Service Level Agreement) and the lack of information describing the shared physical infrastructure.
Theoretically, our Galaxy platform can be deployed using alternative IaaS technologies such as OpenStack, Eucalyptus and OpenNebula. These options will be investigated in our future work. We will also continue to add features to our architecture and to integrate additional bioinformatics analyses tools into our Galaxy toolbox.
Acknowledgments
We appreciate the help of CVRG community and Globus team. We also appreciate the Galaxy Team for their support and maintenance of Galaxy. This work is supported by the NIH through the NHLBI grant, The Cardiovascular Research Grid, under contract number R24HL085343; US Department of Energy, under contract number DE-AC02-06CH11357; and US National Science Foundation, under contract OCI-534113. We are grateful to Amazon, Inc., for an award of Amazon Web Services time that facilitated early experiments. We thank Globus Genomics users for their invaluable contributions.
Appendix A. Globus Provision topology file
The main options of Globus Provision topology file (see Fig. 8) are listed as follows.
“deploy: ec2” means the system will be deployed on Amazon EC2.
“domain” specifies a single domain called simple. A topology can be divided into multiple domains, each with its own set of users, Globus services, etc.
“users” presents the username that will be added to the list of users on the Galaxy cluster.
“cluster-nodes” specifies the number of worker nodes that you wish to deploy.
“go-endpoint” defines the name of the endpoint that will be created for this cluster. The created endpoint will be shown in Globus Transfer interface for data transfer to and from the Galaxy instance.
“keypair” and “keyfile” are the user’s EC2 SSH keypair.
“ami” is the base Amazon Machine Image that GP will use to create each host in the domain. Although any recent AMI can be used, GP provides an AMI that has most of the necessary software pre-installed in it which considerably decreases the time taken to deploy an instance.
“instance-type” specifies the instance type of EC2.
“ssh-key” is the user’s key which can be used to access the Globus CLI (Command-Line Interface).
Appendix B. Globus Provision commands for managing Galaxy instance
Based on the configuration file described in Fig. 8 (galaxy.conf), a GP instance can be created with gp-instance-create command.
$ gp-instance-create -c galaxy.conf
Created new instance: gpi-02156188
This will return a GP instance ID with the form “gpi-nnnnnnnn”. Then start the instance with gp-instance-start:
$ gp-instance-start gpi-02156188
Starting instance gpi-02156188. . . done!
Started instance in 5 minutes and 43 seconds
Once the cluster has been deployed, gp-instance-describe command will return the status and hostname of each server.
When this GP instance is no longer needed, it can be stopped with gp-instance-stop or terminated with gp-instance-terminate. Stopped instances can be resumed at a later time, while terminated instances cannot be resumed again. After termination, all the hosts are shut down and all their resources are released.
$ gp-instance-terminate gpi-02156188
Terminating instance gpi-02156188. . . done!
When the GP instance is running, we can connect to any of its hosts as one of the users defined in the topology. For example, to log into Galaxy server using SSH, the following command should be run:
$ ssh user1@ec2-R-R-R-R.compute-1.amazonaws.com
Once an instance is running, it is possible to modify its topology. Changes made to the topology file will result in modifications to the running instances.
$ gp-instance-update -t newtopology.json gpi-02156188
References
- 1.Driscoll AO, Daugelaite J, Sleator RD. ‘Big Data’, Hadoop and Cloud computing in genomics. J Biomed Inform. 2013;46:774–81. doi: 10.1016/j.jbi.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Krampis K, Booth T, Chapman B, Tiwari B, Bicak M, Field D, et al. Cloud Biolinux: pre-configured and on-demand bioinformatics computing for the genomics community. BMC Bioinformatics. 2012;13:42. doi: 10.1186/1471-2105-13-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–8. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rice P, Longden I, Bleasby A. Emboss: the European molecular biology open software suite. Trends Genet. 2000;16:276–7. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 6.Rosenthal A, Mork P, Li MH, Stanford J, Koester D, Reynolds P. Cloud computing: a new business paradigm for biomedical information sharing. J Biomed Inform. 2010;43:342–53. doi: 10.1016/j.jbi.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Liu B, Sotomayor B, Madduri R, Chard K, Foster I. Deploying Bioinformatics Workflows on Clouds with Galaxy and Globus Provision. Proceedings of SC Companion: High Performance Computing, Networking Storage and Analysis. 2012:1087–95. [Google Scholar]
- 8.Thain D, Tannenbaum T, Livny M. Distributed computing in practice: the Condor experience. Concurr Comput Pract Exper. 2005;17:323–56. [Google Scholar]
- 9.Foster I. Globus online: accelerating and democratizing science through Cloud-based services. IEEE Internet Comput. 2011;15:70–3. [Google Scholar]
- 10.Allen B, Bresnahan J, Childers L, Foster I, et al. Globus online: radical simplification of data movement Via Saas, preprint CI-PP-5-0611. Computation Institute, The University of Chicago; 2011. [Google Scholar]
- 11.Cummerbund. < http://compbio.mit.edu/cummeRbund/>.
- 12.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Globus Online. < https://www.globus.org/>.
- 15.Bresnahan J, Link M, Khanna G, Imani Z, Kettimuthu R, Foster I. Globus Gridftp: what’s new in 2007. Proceedings of the first international conference on networks for grid applications; Lyon, France. pp. 1–5. [Google Scholar]
- 16.R Project. < http://www.r-project.org/>.
- 17.Amazon EC2. < http://aws.amazon.com/ec2/>.
- 18.Globus Provision. < http://globus.org/provision/>.
- 19.Basney J, Humphrey M, Welch V. The Myproxy online credential repository. Software Pract Exper. 2005;35:801–16. [Google Scholar]
- 20.Chef. < http://www.opscode.com/chef/>.
- 21.Amazon Elastic Block Store. < http://aws.amazon.com/ebs/>.
- 22.Amazon Simple Storage Service. < http://aws.amazon.com/s3/>.
- 23.Amazon EC2 Spot Instances. < http://aws.amazon.com/ec2/spot-instances/>.
- 24.Getting Started with Aws Free Usage Tier. < http://s3.amazonaws.com/awsdocs/gettingstarted/latest/awsgsg-freetier.pdf>.
- 25.Juve G, Deelman E, Vahi K, Mehta G, Berriman B, Berman BP, et al. Scientific workflow applications on Amazon Ec2. Workshop on Cloud-based services and applications in conjunction with 5th IEEE international conference on e- Science (e-Science 2009); UK: Oxford; 2009. pp. 59–66. [Google Scholar]
- 26.Simmhan Y, Barga R, Ingen CV, Lazowska E, Szalay A. Building the trident scientific workflow workbench for data management in the Cloud. Third international conference on advanced engineering computing and applications in sciences; 2009. pp. 41–50. [Google Scholar]
- 27.Hoffa C, Mehta G, Freeman T, Deelman E, Keahey K, Berriman B, et al. On the use of Cloud computing for scientific workflows. IEEE international conference on eScience; 2008. pp. 640–45. [Google Scholar]
- 28.Dornemann T, Juhnke E, Freisleben B. On-demand resource provisioning for BPEL workflows using Amazon’s Elastic Compute Cloud. IEEE/ACM international symposium on cluster computing and the grid; 2009. pp. 140–47. [Google Scholar]
- 29.Puppet. < http://puppetlabs.com/>.
- 30.Nurmi D, Wolski R, Grzegorczyk C, Obertelli G, Soman S, Youseff L, et al. The Eucalyptus open-source Cloud-computing system. 9th IEEE/ACM international symposium on cluster computing and the grid; 2009. pp. 124–31. [Google Scholar]
- 31.Mvelase P, Dlodlo N, Makitla I, Sibiya G, Adigun M. An architecture based on SOA and virtual enterprise principles: Opennebula for Cloud deployment. Proceedings of the international conference on information management & evaluation; 2012. pp. 214–22. [Google Scholar]
- 32.Yuan D, Yang Y, Liu X, Chen JJ. A cost-effective strategy for intermediate data storage in scientific Cloud workflow systems. 2010 IEEE international symposium on parallel & distributed processing (IPDPS); 2010. pp. 1–12. [Google Scholar]
- 33.Wu Z, Liu X, Ni Z, Yuan D, Yang Y. A market-oriented hierarchical scheduling strategy in Cloud workflow systems. J Supercomput. 2011;63:256–93. [Google Scholar]
- 34.Zhang C, Sterck HD. Cloud WF: a computational workflow system for Clouds based on Hadoop. Cloud Comput, Lecture Notes in Computer Science. 2009;5931:393–404. [Google Scholar]
- 35.Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with Cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao S, Prenger K, Smith L, Messina T, Fan H, Jaeger E, et al. Rainbow: a tool for large-scale whole-genome sequencing data analysis using Cloud computing. BMC Genomics. 2013;14:425. doi: 10.1186/1471-2164-14-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. Cloudmap: a Cloud-based pipeline for analysis of mutant genome sequences. Genetics. 2012;192:1249–69. doi: 10.1534/genetics.112.144204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schatz MC. Cloudburst: highly sensitive read mapping with Mapreduce. Bioinformatics. 2009;25:1363–9. doi: 10.1093/bioinformatics/btp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wall DP, Kudtarkar P, Fusaro VA, Pivovarov R, Patil P, Tonellato PJ. Cloud computing for comparative genomics. BMC Bioinformatics. 2010;11:259. doi: 10.1186/1471-2105-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afgan E, Baker D, Coraor N, Chapman B, Nekrutenko A, Taylor J. Galaxy Cloudman: delivering Cloud compute clusters. BMC Bioinformatics. 2010;11(Suppl 12):S4. doi: 10.1186/1471-2105-11-S12-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]