

Abstract
Potassium chloride (KCl)-depolarization has been used to study the properties of L-type Ca2+ channel-mediated signal transduction in hippocampal neurons. Calcium influx through L-type Ca2+ channels stimulates a second messenger pathway that transactivates genes under the regulatory control of the Ca2+- and cyclic AMP-responsive element (CRE). Here, we show that in striatal neurons, but not in hippocampal neurons, CRE binding protein (CREB) phosphorylation and CRE-mediated gene expression after KCl-depolarization depends on functional NMDA receptors. This difference in NMDA receptor dependence is not due to different properties of L-type Ca2+ channels in either neuronal type, but rather to different neuron-intrinsic properties. Despite this variation, the second messenger pathway activated by KCl requires Ca2+ /calmodulin (CaM) kinase for CREB phosphorylation in both neuronal types. We conclude that depolarization by KCl works differently in striatal and hippocampal neurons.
Keywords: CREB, Striatum, L-type Ca2+ channel, Potassium chloride depolarization, NMDA receptor, c-fos
1. Introduction
Calcium is an important second messenger in signal In neurons, extracellular Ca2+ transduction pathways. enters via two main routes, L-type Ca2+ channels and N-methyl-d-aspartate (NMDA) receptors, to stimulate various kinases and phosphatases which regulate protein phosphorylation and gene expression. The cyclic AMP-and Ca2+-responsive element binding protein, CREB, is a transcription factor that is phosphorylated in response to Ca2+ influx [4,16,43,45]. CREB is constitutively bound at the cyclic AMP- and Ca2+-responsive element (CRE), and initiates transcription when phosphorylated on the Ser133 residue [15,16]. Among the neuronal genes activated by Ser133 phospho-CREB are neuropeptides [7,10,16,21,22] and the immediate early gene c-fos [39,40,44].
Studies on transcriptional activation of c-fos in hippocampal cultures have shown that Ca2+-entry stimulated by glutamate, and Ca2+-entry stimulated by potassium chloride (KCl) leads to the activation of two distinct signaling pathways which target two different enhancer elements [1]. Glutamate, through its interaction with NMDA receptors, leads to c-fos induction via the serum response element (SRE), while KCl, through its effect on L-type Ca 2+ channels, induces c-fos expression via the Moreover, activation of Ca2+ /calmodulin (CaM) CRE [1]. kinase is important for KCl-mediated, but not NMDA receptor-mediated induction of the c-fos gene [1].
In the striatum, signal transduction and gene expression are important mechanisms of neuronal adaptation to disorders of the brain, therapeutic drug treatments and drug addiction [5,11,18,24,33,34]. Similarly to Bading et al. [1], we used KCl-depolarization in primary striatal cultures to activate L-type Ca2+ channels in an effort to differentiate this pathway from glutamate/NMDA receptor signal transduction pathways. Surprisingly, we found that in primary striatal culture CREB phosphorylation and gene expression following KCl-depolarization relies on the function of NMDA receptors as much as of L-type Ca2+ channels. Moreover, CaM kinase is important for Ca2+-mediated CREB phosphorylation in striatal neurons independent of the route of Ca2+ entry. This is in contrast to hippocampal cultures, where Ca2+ signals evoked by activation of NMDA receptors and by KCl are propagated to the nucleus by two pharmacologically distinguishable pathways [1]. Our data suggest that KCl-depolarization utilizes different signal transduction pathways in striatal and hippocampal neurons.
2. Materials and methods
2.1. Primary striatal and hippocampal cultures
The establishment and maintenance of primary striatal cultures is described in detail elsewhere [23,37]. Neurons were obtained from striatum and nucleus accumbens at E18. After dissection, cells were maintained in defined medium (50% F12/DMEM, 50% DMEM, 1×B27, 10 units/ml penicillin, 10 μg/ml streptomycin, 25 mM HEPES (pH 7.2); Gibco BRL, Grand Island, NY) for 6 days before experiments were performed. As determined by HPLC analysis, glutamate levels in the medium on the day of the experiments ranged from 1 to 5 μM, glycine levels were unchanged at 325 μM. All experiments were performed in duplicate and repeated at least once in an independent dissection. Primary hippocampal cultures were derived from E18 rat embryos and maintained in culture using the same defined medium as for primary striatal culture. Hippocampal cultures were dissected in 25 μM kynurenic acid to protect against excitotoxic cell death. Kynurenic acid was removed before plating of the neurons. All experiments in hippocampal culture were performed in the presence of 1 μM TTX (RBI). All experiments in striatal culture were at least once performed in the presence of TTX, with no difference in the results to experiments without TTX.
2.2. Drugs
FPL 64176, nifedipine, MK 801, GYKI 52466, DNQX, TTX, Ro 05-3663, and Phaclofen were purchased from RBI (Natick, MA). KCl, bicuculline and picrotoxin were purchased from Sigma (St. Louis, MO). Recombinant human brain derived neurotrophic factor (BDNF) was purchased from UBI (Lake Placid, NY). Cultures were preincubated with antagonists for 20 min.
2.3. Immunoblots
Primary rat striatal cultures were harvested in boiling sample buffer (62.5 mM Tris–HCl (pH 6.8), 20% glycerol, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 0.025% bromophenol blue), sonicated and centrifuged for 10 min. Equal volumes of the lysates were separated on a 12% polyacrylamide gel and transferred to nylon mem branes. The protocol for the immunoblots has been de scribed in detail [37,38]. The Ser133-CREB antiserum and the CREB antiserum were purchased from UBI (Lake Placid, NY). Phospho-CREB bands ran close to the 43 kDa standard. To ensure that equal concentrations of protein were loaded, immunoblots were either stripped and probed with a CREB antiserum or samples were re-run and immunoblots developed with the CREB antiserum.
2.4. Calcium phosphate transfections
Transfection of primary striatal cells was performed as previously described [37,50]. E18 striatal neurons were transfected on 4 DIV. Transfection was carried out in DMEM medium for 80 min, followed by a 2% DMSO shock. For all transfections, 6 μg of total DNA were used per 35-mm well. Forty-eight hours after transfection, cells were treated with the respective drugs for 6 h. The 3×CRE-luciferase construct has a DNA sequence containing three Ca2+- and cyclic AMP-responsive element binding protein (CREB)-binding sites (CRE; sequence: TGACGTCA), fused to a minimal Rous sarcoma virus promoter (enhancer-less) in the pA3Pluc vector [27] which contains a luciferase reporter gene. The 3×CRE-luciferase construct was provided by Susan E. Lewis. The serum response element (SRE)-luciferase construct contains two SRE sites and has been described previously [20].
2.5. Luciferase assay
Luciferase assay was performed using the Promega Luciferase Assay kit (Promega, Madison, WI) [37]. Luciferase activity was measured using a luminometer (EG&G Berthold, Oak Ridge, TN) and light intensity expressed as relative light units (RLU).
2.6. RNA blots
The protocol used for RNA blots was previously described [23]. Cultures were treated for 40 min. A c-fos riboprobe was made with a riboprobe kit (Promega). All blots were stripped and re-probed with an internal loading control, cyclophilin [8].
2.7. Statistical analyses
Autoradiographic films were scanned with the Hewlett Packard Scan Jet. Due to the narrow range of film (approximately one order of magnitude), the data obtained are semi-quantitative. Data are presented as fold induction over untreated control±S.E.M. Control was arbitrarily set to 1. Northern blots were developed with a phosphor- imager (Molecular Dynamics, Sunnyvale, CA). Data were analyzed with one-way analyses of variance. The Tukey– Kramer HSD (honestly significant difference) was used to analyze differences between the groups. The JMP com puter program (SAS Institute, Cary, NC) was used for data analysis.
3. Results
3.1. KCl-mediated CREB phosphorylation and gene expression are blocked by glutamate antagonists in the striatum
Phosphorylation of CREB on Ser133 was determined in immunoblots with an antibody raised against Ser133 phospho-CREB [14]. The specific band was detected as a doublet with varying degrees of resolution at around 43 kDa. Since it is well known that CREB has various splice variants, in particular CREBα and CREBΔ [2,17,41,43], and since both bands of the doublet were regulated similarly in our experiments, we were not further concerned about their resolution.
Depolarization with KCl (20 mM) and activation of L-type Ca 2+ channels with FPL 64176 (20 μM) induced Ser133 CREB phosphorylation in primary striatal cultures. On average, cells treated with FPL 64176 showed a fourfold induction, while cells treated with KCl showed a threefold induction over untreated controls (Table 1), a difference which did not reach statistical significance. Surprisingly, KCl-mediated CREB phosphorylation was blocked by the NMDA antagonist, MK 801 (1 μM) (Table 1, Fig. 1A), while FPL 64176-mediated CREB phosphoryl ation remained unaffected (Table 1, Fig. 1A). This dif ferential involvement of NMDA receptors was independent of KCl or FPL 64176 concentrations, since higher con centrations of KCl (20 and 90 mM; Fig. 1C) or lower concentrations of FPL 64176 (0.5, 5 and 20 μM; Fig. 1D) did not change the effect of MK 801. Pretreatment with the L-type Ca2+ channel blockers, nifedipine (20 μM, Table 1, Fig. 1B) and verapamil (20 μM, data not shown), caused a decrease of both KCl- and FPL 64176-mediated CREB phosphorylation to basal levels. Since KCl could cause neurotransmitter release by initiating Na+-dependent depo larization, we performed all experiments at least once in the presence of TTX (1 and 2 μM). Unlike in hippocampal cultures, in striatal cultures TTX did not affect the results (not shown).
Table 1.
The effect of an L-type Ca2+ channel blocker, ionotropic glutamate receptor antagonists, and GABA receptor antagonists on KCl-depolarization- and L-type Ca2+ channel-mediated Ser133 CREB phosphorylation in striatal neuronsa
| Antagonist | Treatment |
|||||
|---|---|---|---|---|---|---|
| Antagonist | KCl | Antagonist & KCl | FPL 64176 | Antagonist & FPL 64176 | n = | |
| nifedipine | 1.2±0.23 | 3.2±0.27 | 1.4±0.09* | 3.8±0.56 | 1.2±0.21* | 8 |
| MK 801 | 0.5±0.11 | 3.6±0.66 | 1.4±0.23* | 4.1±0.9 | 3.5±0.57 | 8 |
| GYKI 52466 | 0.8±0.28 | 3.2±0.33 | 2.0±0.16* | 3.9±0.39 | 3.7±0.24 | 2 |
| DNQX/MK 801 | 0.4±0.11 | 2.4±0.18 | 0.6±0.06* | 3.4±0.36 | 3.3±0.33 | 4 |
| Ro 05-3663 | 1.0±0.10 | 3.0±0.19 | 2.1±0.22* | 4.0±0.33 | 3.6±0.33 | 6 |
| Ro 05-3663/MK 801 | 0.4±0.04 | 3.0±0.19 | 1.0±0.14* | 4.0±0.33 | 3.5±0.25 | 6 |
| bicucullin | 1.4±0.06 | 4.5±0.39 | 3.1±0.25* | 4.5±0.32 | 4.0±0.39 | 2 |
| bicucullin/MK 801 | 0.8±0.24 | 4.5±0.39 | 0.8±0.11* | 4.5±0.32 | 3.7±0.29 | 2 |
| picrotoxin | 0.9±0.05 | 4.5±0.39 | 2.7±0.16* | 4.5±0.32 | 3.8±0.26 | 2 |
| picrotoxin/MK 801 | 0.4±0.14 | 4.5±0.39 | 1.1±0.31* | 4.5±0.32 | 3.5±0.44 | 2 |
| phaclofen | 0.6±0.07 | 3.0±0.19 | 1.6±0.19* | 4.0±0.33 | 3.1±0.25 | 6 |
| phaclofen/MK 801 | 0.3±0.05 | 3.0±0.19 | 0.7±0.09* | 4.0±0.33 | 2.3±0.29* | 6 |
Data are fold induction over untreated control±SEM of imunoblots that were developed with Ser133-phospho-CREB antibody. Control was arbitrarily set to 1. KCL (20 mM)-mediated CREB phosphorylation is blocked by nifedipine (20 μM) and MK 801 (1 μM), and partly blocked by GYKI 52466 (50 μM). FPL 64176 (20 μM)-mediated CREB phsophorylation is not blocked by MK 801. GABA antagonists (Ro 05-3663, 100 μM; bicucullin, 100 μM; picrotoxin, 100 μM; phaclofen 100 μM) partly block CREB phosphorylation after treatment with KCl (see also Figs. 1 and 3).
Asterisks indicate statistical significant differences to treatment with stimulants (Tukey–Kramer HSD). In each experiment, all conditions (e.g. control, antagonist, agonist, antagonist & agonist) were run within one gel and related to each other. Fold-induction is pooled from a number of experiments (see ‘n = ’ at right).
Fig. 1.

In primary striatal culture, Ser133 CREB phosphorylation mediated by KCl-depolarization and by an L-type Ca2+ channel agonist is differently affected by the NMDA antagonist MK 801. (A) Pretreatment of primary striatal cultures with the NMDA antagonist, MK 801 (1 μM) does block CREB phosphorylation induced by KCl-depolarization (20 mM), but does not block CREB phosphorylation induced by treatment with FPL 64176 (20 μM). Levels of CREB protein were unchanged (lower panel). (B) Pretreatment of primary striatal cultures with nifedipine (20 μM) blocks depolarization-mediated CREB phosphorylation after treatment with KCl (20 mM) and L-type Ca2+ channel-mediated CREB phosphorylation after treatment with FPL 64176 (20 μM). Levels of CREB protein were unchanged (lower panel). All data are shown in duplicates. (C,D) Statistical analysis of the effect of MK 801 on Ser133 CREB phosphorylation by KCl or FPL 64176 in primary striatal neurons demonstrates that MK 801 blocks KCl-mediated (20 mM or 90 mM) Ser133 CREB phosphorylation (C), but not FPL 64176-mediated (0.5 μM, 5 μM or 20 μM) Ser133 CREB phosphorylation (D). n=6 samples for each treatment, level of induction normalized to control levels. Average fold induction±S.E.M. Asterisks mark significant inhibitions of the induction of CREB phosphorylation.
In hippocampal cultures raised identically to striatal cultures, neither KCl- nor FPL 64176-mediated CREB phosphorylation was attenuated by pretreatment with MK 801 (Fig. 2A–D), while nifedipine blocked CREB phos phorylation by both compounds (Fig. 2B, FPL 64176 not shown). This finding is in agreement with previous reports [1], and together with our striatal data suggests a different signal transduction mechanism by which KCl mediates CREB phosphorylation in these two neuronal populations. All experiments in hippocampal cultures were performed in the presence of TTX (1 μM).
Fig. 2.

In hippocampal culture, Ser133 CREB phosphorylation after KCl-depolarization is not blocked by the NMDA antagonist MK 801. (A) Pretreatment of primary hippocampal cultures with the NMDA antagonist, MK 801 (1 μM) did not block CREB phosphorylation induced by KCl-depolarization (20 mM or 90 mM). Levels of CREB protein were unchanged. (B) Pretreatment of primary hippocampal cultures with nifedipine (20 μM) blocked KCl-depolarization-mediated CREB phosphorylation (20 mM). Levels of CREB protein were unchanged. All data are shown in duplicate. (C,D) Statistical analysis of the effect of MK 801 on Ser133 CREB phosphorylation by KCl (C) or FPL 64176 (D) shows that MK 801 blocks neither KCl-, nor FPL 64176-mediated Ser133 CREB phosphorylation in primary hippocampal neurons. n=6 samples for each treatment, level of induction normalized to an internal control. Average fold induction±S.E.M.
In the striatum, AMPA/kainate receptors also play a role in KCl-mediated CREB phosphorylation but not in FPL 64176-mediated CREB phosphorylation (Table 1). Treat ment of primary striatal cultures transfected with a CRE- luciferase construct gave similar results (Fig. 4). The dependency on AMPA/kainate receptors was determined with the selective AMPA/kainate antagonist GYKI 52466 (50 μM), since the more common quinoxalinediones such as 6,7-dinitroquinoxaline-2,3-dione (DNQX) interact also with the glycine site of the NMDA receptor [36]. A combination of DNQX (50 μM) and MK 801 (1 μM), which inhibits all ionotropic glutamate receptors, did not affect FPL 64176-mediated CREB phosphorylation but blocked KCl-mediated CREB phosphorylation (Table 1), further supporting a dichotomy between L-type Ca2+ channel-mediated (by FPL 64176) and KCl-mediated Ser133 CREB phosphorylation in striatal neurons.
Fig. 4.

Striatal neurons regulate a 33CRE-luciferase construct in a pattern similar to CREB phosphorylation. Neurons were transfected with the 33CRE-luciferase construct and treated with KCl (20 mM, left) or FPL 64176 (20 μM, right). KCl-mediated 3×CRE-luciferase induction was blocked by nifedipine (20 μM), MK 801 (1 μM) and GYKI 52466 (50 μM), while FPL 64176-mediated 3×CRE-luciferase induction was blocked by nifedipine only. Average fold induction±S.E.M., n=9 for all conditions. Asterisks mark statistically significant inhibitions of the induction of the 3×CRE-luciferase construct.
The endogenously expressed c-fos gene, which is reg ulated by CREB, was activated by KCl and blocked by MK 801 in striatal neurons (Fig. 3). In addition, the induction by KCl of a transfected luciferase construct under the control of three CREs was blocked by MK 801, GYKI 52466 and nifedipine, while FPL 64176 mediated induction was not blocked by MK 801 or GYKI 52466 (Fig. 4).
Fig. 3.
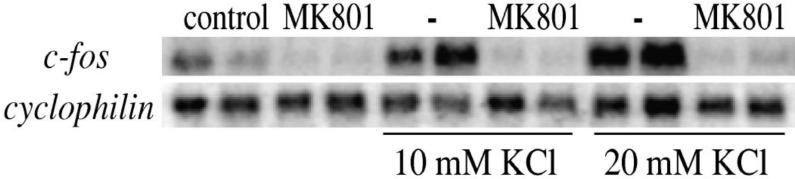
Potassium chloride depolarization-induced c-fos gene expression in striatal neurons is dependent on functional NMDA receptors. (Upper) Treatment with MK 801 (1 μM) blocks c-fos induction mediated by 10 mM or 20 mM KCl. (Lower) Cyclophilin was used as internal loading control. Data are shown in duplicate and were repeated twice.
In Ca 2+-free medium, KCl- and FPL 64176-mediated CREB phosphorylation was significantly attenuated (Fig. 5), confirming the important role of Ca2+ for the signal transduction of both compounds.
Fig. 5.

Calcium ions are important for KCl and FPL 64176-mediated CREB phosphorylation. (A) In Ca2+-free medium, neither KCl nor FPL 64176 induce Ser133 CREB phosphorylation (upper panel). CREB protein levels were unchanged (lower panel). (B) Statistical analysis confirms the block of CREB phosphorylation in Ca2+-free medium (n=4). Average fold induction±S.E.M. is shown.
3.2. GABA receptors are not inhibitory in primary striatal culture
Since KCl can induce a nuclear signal transduction pathway independent of NMDA receptor function in primary hippocampal cultures [1], we examined whether the dependence on functional NMDA receptors observed in striatal neurons is due to the presence of inhibitory GABA receptors. To examine the inhibitory potential of GABA receptors on KCl-depolarization, we added either the and GABAA receptor antagonists Ro 05-3663, bicuculline picrotoxin, or the GABAB receptor antagonist phaclofen to our experiments (Fig. 6 and Table 1). Neither antagonist facilitated KCl or FPL 64176-mediated CREB phosphoryl ation; in fact, GABA antagonists inhibited KCl-mediated CREB phosphorylation (Fig. 6 and Table 1). Thus GABA receptor antagonist did not override the ability of MK 801 to block KCl-mediated CREB phosphorylation.
Fig. 6.

Antagonists of the GABA receptor do not eliminate the dependency of KCl-mediated Ser133 CREB phosphorylation on NMDA receptors in striatal neurons. Primary striatal cultures were treated with Ro 05-3663 (100 μM) or phaclofen (100 μM) before treatment with KCl (20 mM; top) or FPL 64176 (20 μM; bottom). Neither GABA antagonist, alone or in combination with other drugs, increased CREB phosphorylation. On the contrary, a modest reduction in CREB phosphorylation was observed. P-CREB, immunoblots developed with the Ser133 antiserum; CREB, immunoblots developed with the CREB antiserum. Some control treatments shown in single lanes, FPL 64176- and KCl treatments in duplicates. See Table 1 for statistical analysis.
3.3. CaM kinase and the CRE, but not the SRE, play an important role in KCl and FPL 64176-mediated CREB phosphorylation and gene expression in striatal neurons
The c-fos SRE has been shown to mediate c-fos transcription in response to activation of L-type voltage-sensitive Ca2+ channels [30,31]. We therefore tested the regulation of a transfected SRE-luciferase construct by KCl and FPL 64176 in primary striatal culture (Fig. 7).
Fig. 7.

The SRE enhancer is not involved in KCl or FPL 64176-mediated gene expression in primary striatal culture. Primary striatal cultures were transfected with an SRE-luciferase construct and treated with KCl (20 mM), FPL 64176 (20 μM) or BDNF (50 ng/ml). Neither KCl nor FPL 64176 induced the SRE-luciferase construct, while BDNF led to a 15-fold induction. Asterisk marks significant induction. Average fold induction±S.E.M. is shown. n=6 for KCl and FPL 64176, n=3 for BDNF.
The SRE-construct was not induced in striatal neurons by either KCl or FPL 64176. The functional integrity of the SRE construct was confirmed by the 15-fold induction by BDNF (Fig. 7).
The CaM kinase pathway and CaM kinase IV have been shown to transactivate genes through the CRE enhancer [1,4,9,13,45]. In primary striatal cultures, KN62, an inhib itor of CaM kinase, blocked KCl and FPL 64176-mediated CREB phosphorylation, thus demonstrating a role for CaM kinase (Fig. 8A,B). KN62 partially blocked glutamate- mediated CREB phosphorylation (Fig. 8C), and fully blocked NMDA-mediated CREB phosphorylation (not shown).
Fig. 8.

The CaM kinase inhibitor KN62 (30 μM) blocks KCl-, FPL 64176-, and glutamate-mediated Ser133 CREB phosphorylation in striatal culture. (A) Ser133 CREB phosphorylation (P-CREB) and CREB levels (CREB) in KCl (20 mM)-treated primary striatal cultures. (B) Ser133 CREB phosphorylation (P-CREB) and CREB levels (CREB) in FPL 64176 (20 μM)-treated primary striatal cultures. (C) Ser133 CREB phosphorylation in glutamate (50 μM)-treated primary striatal cultures. All treatments are shown in duplicate. All experiments were repeated at least once.
4. Discussion
4.1 KCl induces Ser133 CREB phosphorylation and CRE-mediated gene expression through the combined activation of L-type Ca2+ channels and NMDA receptors
In hippocampal cultures, KCl treatment induces c-fos transcription primarily by stimulating Ca2+ influx through L-type Ca2+ channels [1]. In striatal cultures, KCl induces CREB phosphorylation and CRE-mediated gene expres sion by stimulating Ca2+ influx through NMDA receptor L-type Ca2+ channels. If NMDA receptors or channels and L-type Ca2+ channels are blocked, KCl cannot induce CREB phosphorylation or CRE-mediated gene expression in striatal neurons. The differential effect of KCl observed in striatal and hippocampal neurons does not appear to be due to the particular properties of their L-type Ca2+ channels, since an agonist of L-type Ca2+ channels exerts similar effects on both neuronal populations. Nor is it due to the particular concentration of KCl used, since increas ing concentrations of KCl also fail to induce CREB phosphorylation independently of NMDA receptors. Since Ca2+ channel agonist FPL 64176 induces the L-type CREB phosphorylation in striatal neurons independent of NMDA receptors, we conclude that KCl does not have a strong effect on L-type Ca2+ channels and needs to recruit additional routes of Ca2+ influx or depolarization.
Similarities of KCl-mediated CREB phosphorylation with NMDA receptor-mediated CREB phosphorylation are demonstrated by the block of CREB phosphorylation in the presence of NMDA antagonists, but also by a similar dependency on AMPA/kainate antagonists. As has been shown for NMDA-mediated CREB phosphorylation in striatal neurons (compare data presented here with [37], KCl-mediated CREB phosphorylation and 3×CRE-lucifer-ase expression depend partly on AMPA/kainate receptor activity. This dependency may be due to the interaction of AMPA/kainate receptors and NMDA receptors. At resting potential, NMDA receptor channels are blocked by Mg2+ ions, that are dislodged upon membrane depolarization [28]. The initial depolarization necessary for NMDA receptor activation may be facilitated by AMPA/kainate receptors. By comparison, activation of L-type Ca2+ channels with FPL 64176 confers CREB phosphorylation and CRE-mediated luciferase transcription independent of AMPA/kainate receptor activity. Even a complete inhibi tion of all ionotropic glutamate receptors has no effect on FPL 64176-mediated CREB phosphorylation.
The pharmacological profiles of CREB phosphorylation after KCl-depolarization and after NMDA receptor [37] stimulation are identical, but differ from the profile of L-type Ca2+ channel-activation. Since L-type Ca2+ chan nels are activated by strong depolarization [49], we hypothesize that in primary striatal cultures the increase in membrane potential after exposure to KCl is not strong enough to activate L-type Ca2+ channels, but is sufficient to open other ion channels thus facilitating the opening of NMDA receptors. Depolarization provided by KCl, to gether with ambient levels of glutamate and glycine in the medium allow ion flux through the NMDA receptor channel (Fig. 9). The resultant depolarization opens L-type Ca2+ channels, a process that is very effective in stimulating the signal transduction pathway that causes phos phorylation of CREB [29]. This dependency on ionotropic glutamate receptors renders KCl unsuited for the study of L-type Ca 2+- channel-mediated signal transduction in pri mary striatal culture.
Fig. 9.

The proposed signal transduction pathway induced by L-type Ca2+ channels and by KCl-depolarization in primary striatal culture. L-Type Ca2+ channels, stimulated by FPL 64176, activate an independent signal transduction pathway that translocates to the nucleus to stimulate Ser133 CREB phosphorylation (left diagram). While FPL 64176 may aid NMDA receptor function, this is not needed for signal transduction. In contrast, KCl depends on the combined activation of the NMDA receptor channel and of the L-type Ca2+ channel to successfully transduce a Ca2+ signal to the nucleus (right diagram). We suggest a sequential activation, i.e. that KCl depolarizes the neuron which (1a) leads to the removal of Mg2+ ions from the NMDA receptor channel (2), and the activation of NMDA receptors with the aid of glutamate and glycine in the medium. AMPA/kainate receptors may help to facilitate the removal of the Mg2+ block (1b). Ion influx through the NMDA receptor channel amplifies the depolarization and causes the opening of L-type Ca2+ channels along the dendrites (3a–c). Ca2+ entering through L-type Ca2+ channels at the cell body stimulates second messenger pathways to activate CREB phosphorylation and gene expression in the nucleus (4). While this hypothesis is in agreement with the interaction of NMDA receptors and L-type Ca2+ channels in primary striatal culture [37], the evidence does not exclude the need for a simultaneous activation of NMDA receptors and L-type Ca2+ channels to allow for enough Ca2+-entry to stimulate second messenger pathways. However, orthodromic inputs that engage the activation of synaptic receptors have been shown to be strong stimulators of L-type Ca2+ channels, which in turn are effective activators of CREB phosphorylation [29].
4.2. c-fos expression by KCl-depolarization depends on functional NMDA receptors
The c-fos gene has two distinct regulatory elements that Ca 2+-activated transcription, the CRE and the mediate SRE enhancer [1,12,39]. In hippocampal cultures, gluta- mate mediates c-fos transcription via the NMDA receptor and the SRE, while KCl mediates c-fos transcription via L-type Ca2+ channels and the CRE [1]. Our data suggest that in primary striatal culture, KCl- and glutamate-me diated second messenger pathways converge upstream of phosphorylation of the transcription factor CREB. Since a dichotomy between CREB phosphorylation and the induc tion of c-fos expression is imaginable, we examined whether KCl is also capable to induce c-fos expression in the presence of MK 801, when CREB phosphorylation is blocked. However, we found that NMDA antagonists blocked c-fos expression after KCl-depolarization, analo gous to CREB phosphorylation and CRE-mediated lucifer- ase expression.
4.3. GABA receptors are excitatory in primary striatal culture and cannot account for the dependency of KCl-mediated CREB phosphorylation on NMDA receptors
Unlike hippocampal neurons, most striatal neurons are GABAergic [46]. Since neurons in culture synapse onto each other [25], we investigated whether the inhibitory neurotransmitter GABA, which may be released by KCl [32], increases the threshold for depolarization and thus necessitates the combined activation of NMDA receptors and L-type Ca2+ channels in striatal neurons. However, antagonist for the ligand-gated GABAA receptor channel or the G protein-coupled GABAB receptor could not overcome the requirement for functional NMDA receptors in KCl-mediated gene expression. In fact, GABA antagonists partially blocked KCl-mediated CREB phosphorylation, suggesting that in embryonic striatal cultures GABA may not be an inhibitory neurotransmitter. Indeed, it has been demonstrated previously that in early neuronal development both GABA receptor subtypes raise cytosolic Ca2+ levels and induce neural excitability [3,19,35,42,48]. We therefore conclude that GABA neurotransmission in striat al cultures does not increase the depolarization-threshold, nor does it account for the difference with hippocampal cultures in KCl-depolarization-induced signal transduction. Rather, there are intrinsic differences between striatal and hippocampal neurons that are responsible for the differen tial effect of KCl. For example, the density of L-type Ca2+ channels is different in either neuronal type [6,26].
4.4. The CaM kinase pathway is critical for signal transduction mediated by KCl, FPL 64176 or glutamate in striatal neurons
In primary striatal cultures, inhibition of the CaM kinase pathway blocked CREB phosphorylation mediated by KCl, FPL 64176 and by glutamate. The significance of the CaM kinase pathway for Ca2+-mediated CREB phosphorylation in neurons has been demonstrated in other paradigms such as hippocampal cultures [4,9,45,47]. Therefore, the CaM kinase pathway cannot be used to differentiate between CREB phosphorylation mediated by L-type Ca2+ channels, and CREB phosphorylation mediated by glutamate or KCl.
In conclusion, we show that in primary striatal cultures the signal transduction pathway activated by KCl-depolar ization has the same pharmacological profile as the signal transduction pathway activated by NMDA receptors [37], and is partly divergent from the signal transduction pathway activated by an L-type Ca2+ channel agonist. signal transduction pathways converge prior to Ser133 Both CREB phosphorylation and stimulate CaM kinase and CRE-mediated gene expression (Fig. 9). Due to its depen dence on NMDA receptors, KCl-depolarization is not suited to study the role of L-type Ca2+ channels in the striatum.
Acknowledgements
This work was supported by the National Alliance for Research on Schizophrenia and Depression (C.K.), and a National Institute of Drug Abuse grant DA07134 (C.K.). The authors would like to thank David Ginty for providing the SRE construct.
Abbreviations
- CRE
Ca2+- and cyclic AMP-responsive element
- CREB
Ca 2+- and cyclic AMP-responsive element binding protein
- NMDA
N-methyl-d-aspartate
- SRE
serum response element
References
- 1.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 2.Berkowitz LA, Gilman MZ. Two distinct forms of active tran scription factor CREB (cAMP response element binding protein) Proc. Natl. Acad. Sci. USA. 1990;87:5258–5262. doi: 10.1073/pnas.87.14.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berninger B, Marty S, Zafra F, da Penha Berzaghi M, Thoenen H, Lindholm D. GABAergic stimulation switches from enhancing to repressing BDNF expression in rat hippocampal neurons during maturation in vitro. Development. 1995;121:2327–2335. doi: 10.1242/dev.121.8.2327. [DOI] [PubMed] [Google Scholar]
- 4.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: A Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 5.Bravi D, Mouradian MM, Roberts JW, Davis TL, Sohn YH, Chase TN. Wearing-off fluctuations in Parkinson's disease: contri bution of postsynaptic mechanisms. Ann. Neurol. 1994;36:27–31. doi: 10.1002/ana.410360108. [DOI] [PubMed] [Google Scholar]
- 6.Chin H, Smith MA, Kim HL, Kim H. Expression of dihydro-pyridine-sensitive brain calcium channels in the rat central nervous system. FEBS Lett. 1992;299:69–74. doi: 10.1016/0014-5793(92)80103-n. [DOI] [PubMed] [Google Scholar]
- 7.Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danielson PE, Forss-Petter S, Brow MA, Calavetta L, Douglass J, Milner RJ, Sutcliffe JG. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- 9.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 10.Douglass J, McKinzie AA, Pollock KM. Identification of multiple DNA elements regulating basal and protein kinase A-induced transcriptional expression of the rat prodynorphin gene. Mol. Endocrinol. 1994;8:333–344. doi: 10.1210/mend.8.3.8015551. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald LW, Deutch AY, Gasic G, Heinemann SF, Nestler EJ. Regulation of cortical and subcortical glutamate receptor subunit expression by antipsychotic drugs. J. Neurosci. 1995;15:2453–2461. doi: 10.1523/JNEUROSCI.15-03-02453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J. Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- 13.Ginty DD, Glowacka D, Bader DS, Hidaka H, Wagner JA. Introduction of immediate early genes by Ca2+ influx requires cAMP-dependent protein kinase in PC12 cells. J. Biol. Chem. 1991;266:17454–17458. [PubMed] [Google Scholar]
- 14.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez GA, Menzel P, Leonard J, Fischer WH, Mon tminy MR. Characterization of motifs which are critical for activity of the cyclic AMP-responsive transcription factor CREB. Mol. Cell. Biol. 1991;11:1306–1312. doi: 10.1128/mcb.11.3.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 17.Hoeffler JP, Meyer TE, Waeber G, Habener JF. Multiple adenosine 3′,5′-monophosphate response element DNA-binding proteins generated by gene diversification and alternative exon splicing. Mol. Endocrinol. 1990;4:920–930. doi: 10.1210/mend-4-6-920. [DOI] [PubMed] [Google Scholar]
- 18.Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am. J. Psychiatry. 1996;153:151–162. doi: 10.1176/ajp.153.2.151. [DOI] [PubMed] [Google Scholar]
- 19.Ito Y, Ishige K, Zaitsu E, Anzai K, Fukuda H. gamma-Hydroxybutyric acid increases intracellular Ca2+ concentration and nuclear cyclic AMP-responsive element- and activator protein 1 DNA-binding activities through GABAB receptor in cultured cerebellar granule cells. J. Neurochem. 1995;65:75–83. doi: 10.1046/j.1471-4159.1995.65010075.x. [DOI] [PubMed] [Google Scholar]
- 20.Janknecht R, Ernst WH, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993;12:5097–5104. doi: 10.1002/j.1460-2075.1993.tb06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konradi C, Cole RL, Green D, Senatus P, Leveque JC, Pollack A, Grossbard SJ, Hyman SE. Analysis of the proenkephalin second messenger-inducible enhancer in rat striatal cultures. J. Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 22.Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc. Natl. Acad. Sci. USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends upon postsynaptic NMDA receptors and calcium. J. Neuro sci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kornhuber J, Riederer P, Reynolds GP, Beckmann H, Jellinger K, Gabriel E. 3H-spiperone binding sites in post-mortem brains from schizophrenic patients: relationship to neuroleptic drug treatment, abnormal movements, and positive symptoms. J. Neural Transm. 1989;75:1–10. doi: 10.1007/BF01250639. [DOI] [PubMed] [Google Scholar]
- 25.Kowalski C, Crest M, Vuillet J, Pin T, Gola M, Nieoullon A. Emergence of a synaptic neuronal network within primary striatal cultures seeded in serum-free medium. Neuroscience. 1995;64:979–993. doi: 10.1016/0306-4522(94)00453-c. [DOI] [PubMed] [Google Scholar]
- 26.Ludwig A, Flockerzi V, Hofmann F. Regional expression and cellular localization of the alpha1 and beta subunit of high voltage-activated calcium channels in rat brain. J. Neurosci. 1997;17:1339–1349. doi: 10.1523/JNEUROSCI.17-04-01339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxwell IH, Harrison GS, Wood WM, Maxwell F. A DNA cassette containing a trimerized SV40 polyadenylation signal which efficiently blocks spurious plasmid-initiated transcription. Biotechniques. 1989;7:276–280. [PubMed] [Google Scholar]
- 28.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg 21 of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 29.Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J. Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca21 / calmodulin-dependent kinase. Mol. Cell. Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misra RP, Bonni A, Miranti CK, Rivera VM, Sheng M, Greenberg ME. L-type voltage-sensitive calcium channel activation stimulates gene expression by a serum response factor-dependent pathway. J. Biol. Chem. 1994;269:25483–25493. [PubMed] [Google Scholar]
- 32.Moroni F, Bianchi C, Tanganelli S, Moneti G, Beani L. The release of gamma-aminobutyric acid, glutamate, and acetylcholine from striatal slices: a mass fragmentographic study. J. Neurochem. 1981;36:1691–1699. doi: 10.1111/j.1471-4159.1981.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 33.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 34.Nisbet AP, Foster OJ, Kingsbury A, Eve DJ, Daniel SE, Marsden CD, Lees AJ. Preproenkephalin and preprotachykinin mes senger RNA expression in normal human basal ganglia and in Parkinson’s disease. Neuroscience. 1995;66:361–376. doi: 10.1016/0306-4522(94)00606-6. [DOI] [PubMed] [Google Scholar]
- 35.Parramon M, Gonzalez MP, Herrero MT, Oset-Gasque MJ. GABAB receptors increase intracellular calcium concentrations in chromaffin cells through two different pathways: their role in catecholamine secretion. J. Neurosci. Res. 1995;41:65–72. doi: 10.1002/jnr.490410108. [DOI] [PubMed] [Google Scholar]
- 36.Patel J, Zinkand WC, Klika AB, Mangano TJ, Keith RA, Salama AI. 6,7-Dinitroquinoxaline-2,3-dione blocks the cytotoxicity of N-methyl-D-aspartate and kainate, but not quisqualate, in cortical cultures. J. Neurochem. 1990;55:114–121. doi: 10.1111/j.1471-4159.1990.tb08828.x. [DOI] [PubMed] [Google Scholar]
- 37.Rajadhyaksha A, Barczak A, Macias W, Leveque J-C, Lewis S, Konradi C. L-type Ca2+ channels are essential fir glutamatemediated CREB phosphorylation and c-fos gene expression in striatal neurons. J. Neurosci. 1999;19:6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajadhyaksha A, Leveque JC, Macias W, Barczak A. Konradi, Molecular components of striatal plasticity: The various routes of cyclic AMP pathways. Dev . Neurosci. 1998;20:204–215. doi: 10.1159/000017314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran T. Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 40.Runkel L, Shaw PE, Herrera RE, Hipskind RA, Nordheim A. Multiple basal promotor elements determine the level of human c-fos transcription. Mol. Cell. Biol. 1991;11:1270–1280. doi: 10.1128/mcb.11.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruppert S, Cole TJ, Boshart M, Schmid E, Schutz G. Multiple mRNA isoforms of the transcription activator protein CREB: generation by alternative splicing and specific expression in primary spermatocytes. EMBO J. 1992;11:1503–1512. doi: 10.1002/j.1460-2075.1992.tb05195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segal M. GABA induces a unique rise of [Ca]i in cultured rat hippocampal neurons. Hippocampus. 1993;3:229–238. doi: 10.1002/hipo.450030214. [DOI] [PubMed] [Google Scholar]
- 43.Shaywitz AJ, Greenberg ME. CREB: A stimulus-induced tran scription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 44.Sheng M, Dougan ST, McFadden G, Greenberg ME. Calcium and growth factor pathways of c-fos transcriptional activation require distinct upstream regulatory sequences. Mol. Cell. Biol. 1988;8:2787–2796. doi: 10.1128/mcb.8.7.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(21)- regulated transcription factor phosphorylated by calmodulin-depen dent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 46.Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 1990;13:259–265. doi: 10.1016/0166-2236(90)90106-k. [DOI] [PubMed] [Google Scholar]
- 47.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+ /calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 48.Takebayashi M, Kagaya A, Hayashi T, Motohashi N, Yamawaki S. gamma-aminobutyric acid increases intracellular Ca2+ concentration in cultured cortical neurons: role of Cl— transport. Eur. J. Pharmacol. 1996;297:137–143. doi: 10.1016/0014-2999(95)00734-2. [DOI] [PubMed] [Google Scholar]
- 49.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 50.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]