

Abstract
The present study deals with the functional interaction of antipsychotic drugs and NMDA receptors. We show that both the conventional antipsychotic drug haloperidol and the atypical antipsychotic drug clozapine mediate gene expression via intracellular regulation of NMDA receptors, albeit to different extents. Data obtained in primary striatal culture demonstrate that the intraneuronal signal transduction pathway activated by haloperidol, the cAMP pathway, leads to phosphorylation of the NR1 subtype of the NMDA receptor at 897Ser. Haloperidol treatment is likewise shown to increase 897Ser–NR1 phosphorylation in rats in vivo. Mutation of 896Ser and 897Ser to alanine, which prevents phosphorylation at both sites, inhibits cAMP-mediated gene expression. We conclude that antipsychotic drugs have the ability to modulate NMDA receptor function by an intraneuronal signal transduction mechanism. This facilitation of NMDA activity is necessary for antipsychotic drug-mediated gene expression and may contribute to the therapeutic benefits as well as side effects of antipsychotic drug treatment.
Keywords: haloperidol, clozapine, D2 receptors, NMDA, c-fos, proenkephalin, striatum, CREB, tardive dyskinesia
Although most antipsychotic drugs interact directly with specific receptors such as dopamine receptors in the brain, the mechanism that conveys their therapeutic effect has remained elusive. Over the last few years the interaction of many antipsychotic drugs with NMDA receptor function has been demonstrated in various experimental paradigms. This interaction could be particularly relevant for our understanding of the mechanism of action of antipsychotic drugs. The conventional antipsychotic drug haloperidol and the atypical antipsychotic drug clozapine facilitate NMDA receptor function at clinically relevant concentrations (Banerjee et al., 1995), whereas NMDA antagonists have been shown to prevent haloperidol-induced catalepsy in rats (Yoshida et al., 1991; Moore et al., 1993; Kaur et al., 1997). Gene expression mediated by haloperidol is blocked by the NMDA antagonist MK 801, as well as by D2 agonists (Dragunow et al., 1990; Robertson and Fibiger, 1992; Ziolkowska and Hollt, 1993; Robertson et al., 1994;Boegman and Vincent, 1996).
The manner in which antipsychotic drugs interact with NMDA receptors is not entirely understood. Here we investigate this interaction by examining how haloperidol and clozapine interact functionally with NMDA receptors to mediate chronic and acute gene expressionin vivo in the rat striatum and ex vivo in primary striatal culture. Gene expression may be important for some of the therapeutic actions of antipsychotic drugs (Hyman, 1993).
We chose to analyze the regulation of two genes in the striatum: the immediate early gene c-fos, which is rapidly induced in neurons stimulated with antipsychotic drugs (Dragunow et al., 1990;Nguyen et al., 1992; Konradi and Heckers, 1995), and theproenkephalin gene, which is selectively induced in D2 receptor-expressing neurons (Le Moine et al., 1990) in response to haloperidol treatment (Hong et al., 1985; Auchus and Pickel, 1992; Konradi et al., 1993).
The protein kinase A (PKA)–cAMP signal transduction pathway is critical to haloperidol's activation of gene expression; for example, haloperidol is unable to stimulate gene expression in PKA-deficient mice (Adams et al., 1997). The signal transduction pathway activated by the atypical antipsychotic drug clozapine is less well understood, however. Clozapine is known to bind to dopamine D4 receptors, serotonin receptors, and D1 receptors (Van Tol et al., 1991; Farde et al., 1992; Meltzer, 1994), and like haloperidol it induces c-fosgene expression in the striatum (Merchant et al., 1994), albeit to a lesser extent than haloperidol.
The NMDA receptor is a heteromultimer of subunits from the NR1 and NR2 families (Hollmann and Heinemann, 1994). The activity of this receptor can be modulated by kinases and phosphatases (Chen and Huang, 1992;Wang and Salter, 1994; Leonard and Hell, 1997; Westphal et al., 1999). PKA specifically phosphorylates 897Ser of the NR1 subunit (Tingley et al., 1997). Although it has been demonstrated that PKA can regulate NMDA receptor function (Harada et al., 1991; Roche et al., 1994; Raman et al., 1996), there is no evidence as of yet that phosphorylation of897Ser-NR1 by PKA is biologically relevant.
We report here that NMDA receptor activity is essential for the regulation of gene expression by clozapine and haloperidol in the striatum. Moreover, we provide an intracellular mechanism by which antipsychotic drugs recruit NMDA receptors to the signal transduction pathway, which involves phosphorylation of the NR1 subtype of the NMDA receptor at 897Ser.
MATERIALS AND METHODS
Animals. Male Sprague Dawley rats (200–250 gm) were used for all experiments. They were housed four to a cage on a 12 hr light/dark cycle. Animals were killed by rapid decapitation for the preparation of RNA, or they were perfused under deep anesthesia with pentobarbital (70 mg/kg) for immunohistochemistry. All experiments were repeated at least once.
Drugs and drug paradigms. Haloperidol, clozapine, dizocilpine maleate [(+) MK 801], and d-cycloserine (DCS) were obtained from Research Biochemicals (Natick, MA); forskolin was obtained from Sigma (St. Louis, MO). All rats received the same number of injections, administered intraperitoneally, with either drug or 0.9% saline in chronic and acute paradigms. Pretreatment (MK 801 or DCS) was for 20 min, after which the rats were treated with antipsychotic drugs. For chronic treatment paradigms of rats, see Table1. In the acute treatment paradigm, rats were killed 40 min after antipsychotic drug treatment for RNA analysis, and 2 hr after antipsychotic drug treatment for protein analysis (immunocytochemistry and immunoblots). For the concurrent analysis of RNA and protein, rats were killed 1 hr after antipsychotic drug treatment (see Fig. 2).
Table 1.
Treatment schedule for chronically treated rats
| Group | Day 1–day 11 | Day 12 | ||
|---|---|---|---|---|
| 0 min | 20 min | 0 min | 20 min | |
| Control | Saline | Saline | Saline | Saline |
| Acute haloperidol | Saline | Saline | Saline | Haloperidol |
| Chronic haloperidol | Saline | Haloperidol | Saline | Haloperidol |
| Chronic MK 801 and chronic haloperidol (I) | MK 801 | Haloperidol | MK 801 | Haloperidol |
| Chronic MK 801 and chronic haloperidol (II) | MK 801 | Haloperidol | Saline | Haloperidol |
All rats received two injections per day, 20 min apart. Rats were killed on day 12, 40 min after the last injection.
Fig. 2.

MK 801 attenuates Fos protein induction after haloperidol administration. A, An immunoblot of rat striata with the M-peptide antiserum shows an attenuation of haloperidol (1 mg/kg)-induced Fos expression by MK 801 (1 mg/kg). At least two regulated bands are observed between the 53 and 78 kDa size markers. The uppermost band (arrow onleft) is the same size as a band observed with a Fos antiserum. B, mRNA induction in the contralateral striata of the animals shown in A. All treatments are shown in triplicate.
Immunohistochemistry. Two hours after the final intraperitoneal injection, animals were perfused under deep pentobarbital anesthesia with 20 ml of 0.9% saline, followed by 300 ml of 4% paraformaldehyde in PBS. Brains were cryoprotected in 30% sucrose for 24 hr, sliced into 45 μm sections on a freezing microtome, and preincubated free floating for 0.5 hr in PBS containing 1% hydrogen peroxide and 0.3% Triton X-100. Normal goat serum (3%) with 0.3% Triton X-100 in PBS served as buffer in all subsequent steps. Primary Fos antiserum (Ab-2 and Ab-5; Oncogene Research Products, Cambridge, MA) was diluted 1:5000, with 24 hr incubation time at 4°C, followed by an overnight incubation with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), diluted 1:500. An incubation for 2 hr with Avidin DH and biotinylated peroxidase, diluted 1:100, was followed by a change of buffer to Tris–HCl, pH 7.6, and development of the antibody complex with 3,3′diaminobenzidine (DAB; 50 mg/100 ml Tris, pH 7.6) and 0.03% hydrogen peroxide.
Morphometry. Fos-positive nuclei of sections stained with Fos (Ab-2) were counted. Fos (Ab-5) had a higher sensitivity and reacted with more nuclei than Fos (Ab-2) in all conditions but had the same relative staining pattern as Fos (Ab-2). A comparative count of saline- and haloperidol-treated rats with both antibodies revealed a similar relative induction of Fos-positive nuclei by haloperidol. A cell area of 500 × 500 μm was counted in six sections, spanning evenly from +2.0 to −1.0 bregma for the striatal counts, and from +2.7 to +0.5 bregma for the accumbal counts (Paxinos and Watson, 1986). Lateral striatum counts were performed between 3 and 5 dorsoventral (DV) and 3 and 5 mediolateral (ML), and medial striatum counts were performed between 5.5 and 7 DV and 1.5 and 3 ML (Paxinos and Watson, 1986).
Primary striatal cultures. Striata were dissected under a stereomicroscope from 18-d-old Sprague Dawley rat fetuses. Tissue was resuspended in 2 ml of defined medium [50% F12/DMEM and 50% DMEM (Cellgro, Herndon, VA) with the following supplements per liter of medium: 4 gm dextrose, 1× B27 (Life Technologies, Gaithersburg, MD), 10 ml of penicillin–streptomycin liquid (Sigma, St. Louis, MO), and 25 mm HEPES]. The tissue was mechanically dissociated with a fire-narrowed Pasteur pipette; the cells were then resuspended in defined medium to 106 cells/ml and plated in six-well plates (Becton Dickinson, Franklin Lakes, NJ) at 2.4 × 106 cells per well. Plates were pretreated with 2 ml of 1:500 diluted sterile solution of polyethylenimine (Sigma) in water for 24 hr, washed twice with sterile water, coated with 2.5% serum-containing PBS solution for at least 4 hr, and aspirated just before plating. All experiments were performed with cells 6–8 d in culture and repeated at least once in an independent dissection.
Transfection. Transfection of primary striatal neurons was performed on 4 d in vitro following the protocol of Xia et al. (1996) with modifications described byRajadhyaksha et al. (1999). The proenkephalinconstruct used, pENKAT12, contained the human proenkephalinminimal enhancer (ENKCRE-1 and ENKCRE-2) fused to chloramphenicol acetyltransferase (CAT), and included 1.2 kb of the 3′ flanking sequence (Comb et al., 1986, 1988). Steven E. Hyman (National Institute of Mental Health) provided the pENKAT12 construct. The3xCRE-luciferase construct had a DNA sequence containing three Ca2+- and cAMP-responsive element (CRE) binding protein (CREB)-binding sites (sequence: TGACGTCA), fused to a minimal Rous sarcoma virus promoter (enhancer-less) in the pA3Pluc vector (Maxwell et al., 1989) that contains a luciferase reporter gene. The 3xCRE–luciferase construct was provided by Susan E. Lewis (Massachusetts General Hospital). The NR1 wild-type and896/897serine to alanine NR1 constructs were cloned into the pRK5 mammalian expression vector (Tingley et al., 1993) and were provided by Richard Huganir (Johns Hopkins University School of Medicine) (Ehlers et al., 1995). In the cotransfection experiments, a fourfold molar excess of the NR1 constructs or the control vector over the 3xCRE-luciferase construct was used. In all experiments, the total amount of DNA added was 6 μg per well of six-well plates.
Forty-eight hours after transfection, cells were treated with the respective drugs for 6 hr. CAT assays were performed according to the Promega Protocols and Applications Guide. The luciferase assay was performed using the Promega luciferase assay kit (Promega, Madison, WI) and has been described by Rajadhyaksha et al. (1999).
Northern blot analysis of brain samples. Total striatal RNA (7–10 μg), prepared as described (Berger and Chirgwin, 1989), was size-separated on a 1.2% denaturing agarose gel (1 mparaformaldehyde) in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer (20 mm MOPS, pH 7.0, 5 mm sodium acetate, and 1 mmEDTA), electroblotted onto a nylon membrane (GeneScreen; DuPont, Billerica, MA), and hybridized with a32P-labeled RNA probe (Gemini system, Promega). Cyclophilin mRNA was used as an unregulated internal reference probe to control for loading differences (Danielson et al., 1988). Membranes were exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA) and analyzed with the IP lab imaging software. The c-fos plasmid (rat) was a gift from T. Curran (St. Jude Children's Research Hospital), and theproenkephalin plasmid (rat) was a gift from D. Borsook (Massachusetts General Hospital).
Northern blot analysis of primary striatal neurons. Cells were treated for 6 hr to study proenkephalin gene regulation and for 40 min to study c-fos gene regulation. Medium was aspirated and striatal neurons were lysed in 500 μl of lysis buffer (50 mm Tris, pH 8.0, 100 mmNaCl, 5 mm MgCl2, and 0.5% NP40). After a 5 min incubation on ice, lysates were transferred into microcentrifuge tubes and centrifuged for 2 min at 14,000 rpm at 4°C; the supernatant was transferred and SDS was added to a final concentration of 0.2%. Cells were extracted with phenol, followed by a chloroform extraction and ethanol precipitation. RNA was size-separated and hybridized exactly like RNA from brain samples (see above).
Immunoblots. The M-peptide antiserum was used for Fos immunoblots and was provided by Michael J. Iadarola (National Institutes of Health); the antiserum against phosphorylated897Ser of the NR1 subtype of the NMDA receptor (897Ser–NR1) was provided by Richard L. Huganir (Johns Hopkins University School of Medicine) and is commercially available from UBI (Lake Placid, NY); the NR1 antiserum,133Ser-CREB antiserum, and CREB antiserum were purchased from UBI. The M-peptide antiserum was diluted 1:5000, and the secondary antibody was diluted 1:20,000. Blots were also developed with a Fos antiserum from UBI (diluted 1:500; secondary antibody diluted 1:10,000; data not shown). For both antisera, a band of identical size was observed between the 53 and 78 kDa size markers. Dilutions for anti-897Ser–NR1, anti-NR1, anti-133Ser–CREB, and anti-CREB were 1:1000. The secondary antibody for these antisera was diluted 1:10,000. The method used for immunoblots has been described previously (Rajadhyaksha et al., 1998).
Data analysis. Northern blots were analyzed with a PhosphorImager (Molecular Dynamics) that has a range of five orders of magnitude. Data were normalized to the saline-treated controls of each experiment, and because they are based on relative numbers, they are semi-quantitative. Data were analyzed with one-way ANOVAs. The Tukey–Kramer honestly significant difference (HSD) was used to analyze differences between the groups, whereas the Dunnett's test was used for comparisons of treatment groups with controls. The JMP computer program (SAS Institute, Cary, NC) was used for data analysis.
RESULTS
Haloperidol-mediated c-fos gene and Fos protein induction is blocked by the NMDA antagonist MK 801 and enhanced byd-cycloserine, a partial agonist at the glycine site of the NMDA receptor
Haloperidol treatment led to c-fos mRNA induction in the rat striatum 30 min after injection in a dose-dependent manner (haloperidol 0.3 mg/kg: average fold induction ± SEM 3.3 ± 0.34, n = 22, analyzed in six separate blots; haloperidol 1 mg/kg: average fold induction ± SEM 6.5 ± 1.3, n = 9, analyzed in three separate blots) (Fig.1A). A higher concentration of haloperidol (2 mg/kg) yielded c-fos levels comparable to 1 mg/kg (data not shown). The c-fos induction was significantly attenuated by pretreatment for 20 min with the NMDA antagonist MK 801 (Fig. 1B,C; see Fig. 4A,B). All changes in mRNA levels were reflected subsequently in Fos protein expression. Fos protein expression was examined with the Fos/Fra specific M-peptide antiserum in an immunoblot (Fig. 2). Samples for the immunoblot were collected 1 hr after injection of haloperidol (Fig. 2A), with the contralateral striatum used for c-fos mRNA analysis (Fig.2B) (note that for this comparison c-fosmRNA was analyzed 1 hr after treatment with haloperidol).d-cycloserine, a partial agonist at the glycine site of the NMDA receptor (Johnson and Ascher, 1987; Henderson et al., 1990), significantly enhanced c-fos expression after treatment with 0.3 mg/kg haloperidol (Figs.3A,4C) but not after treatment with 1 mg/kg haloperidol (Figs. 3B,4D).
Fig. 1.

MK 801 inhibits haloperidol-mediatedc-fos gene expression. A, Haloperidol (0.3 and 1 mg/kg) induced c-fos gene expression in the striatum in a dose-dependent manner. B,c-fos induction after treatment with 0.3 mg/kg haloperidol (i.p.) was completely inhibited by MK 801 (1 mg/kg).C, c-fos induction after treatment with 1 mg/kg haloperidol was partially inhibited by MK 801. RNA blots with duplicate samples are shown. For a statistical analysis of multiple experiments, see Figure 4, A and B.
Fig. 4.

Statistical analysis demonstrates an involvement of NMDA receptors in haloperidol-mediated c-fos mRNA expression in the rat striatum. A, c-fosinduction by 0.3 mg/kg haloperidol (H 0.3) was completely blocked by the NMDA antagonist MK 801 (1 mg/kg). The average fold induction ± SEM of 12 experiments is shown.B, c-fos induction by 1 mg/kg haloperidol (H1) was significantly blocked by MK 801 (MK/H 1). The average fold induction ± SEM of three experiments is shown. C, d-cycloserine (DCS), a partial agonist at the glycine site of the NMDA receptor enhanced at 1 mg/kg haloperidol (0.3 mg/kg)-mediatedc-fos induction (compare H 0.3 withDCS 1 H 0.3) but had no significant effect at 5 mg/kg (compare H 0.3 with DCS 5 H 0.3).c-fos levels after DCS alone were equivalent to control levels at all concentrations used. The average fold induction ± SEM of nine (DCS 1 mg/kg) and six (DCS 5 mg/kg) striata is shown.D, The increase of haloperidol (1 mg/kg)-mediatedc-fos expression after treatment with DCS (1 mg/kg) did not reach significance. The average fold induction ± SEM of four striata is shown. All data are compared with c-foslevels in saline-treated rats, which were arbitrarily set to onefold induction. Asterisks indicate statistically significant differences with haloperidol-treated rats.
Fig. 3.

d-cycloserine promotes haloperidol-mediated c-fos expression. A, In multiple experiments, a trend toward increased c-fosexpression after cotreatment with haloperidol (0.3 mg/kg) and DCS (0.5 and 1 mg/kg) was observed. This trend was significant for 1 mg/kg DCS (see Fig. 4C). B, The trend toward increased c-fos expression after cotreatment with haloperidol (1 mg/kg) and DCS was variable and not significant in multiple experiments (see Fig. 4D).
MK 801 inhibits clozapine-mediated c-fos expression in the striatum
Clozapine (20 mg/kg) induced c-fos expression in the striatum, although to a lesser extent than that observed with haloperidol (average fold induction ± SEM 2.12 ± 0.3) (Fig.5), and at a higher dose than used in standard clinical practice. At standard prescription levels (5 mg/kg), clozapine failed to induce c-fos expression in the striatum (data not shown). The NMDA antagonist MK 801 blocked thec-fos expression induced by a high dose (20 mg/kg) of clozapine (Fig. 5A,B). DCS (0.5, 1, and 5 mg) had no significant effect on c-fos expression mediated by 20 mg/kg (Fig. 5C,D), or 5 mg/kg (data not shown) clozapine.
Fig. 5.

NMDA receptors play a role in clozapine-mediatedc-fos expression. A, c-fosexpression after treatment with the atypical antipsychotic drug clozapine (20 mg/kg) was inhibited by MK 801 (1 mg/kg).B, Average fold induction ± SEM ofc-fos after treatment with clozapine (20 mg/kg), and the inhibition of c-fos induction by MK 801, of 10 striata.C, No significant increase of clozapine-mediatedc-fos expression by DCS (1 mg/kg) was observed.D, The average fold induction ± SEM ofc-fos after treatment with clozapine (20 mg/kg) was not significantly changed by 0.5 mg/kg DCS (n = 12) or 1 mg/kg DCS (n = 8). Data in B andD are compared with c-fos levels in saline-treated rats, which were arbitrarily set to onefold induction.Asterisks indicate statistically significant differences with clozapine-treated rats.
Fos expression in the striatum after antipsychotic drug treatment is specific for drug type and drug concentration
Analysis of the anatomical pattern of Fos-positive neurons revealed that all three treatments (0.3 mg/kg haloperidol, 1 mg/kg haloperidol, and 20 mg/kg clozapine) caused different patterns of Fos staining in the striatum (Figs. 6,7A–C) (Robertson et al., 1994). Clozapine at a concentration of 20 mg/kg was particularly effective in inducing Fos protein in the medial striatum, whereas no Fos-positive nuclei were seen after treatment with 5 mg/kg clozapine (data not shown). Treatment with a high concentration of haloperidol (1 mg/kg) caused a pronounced increase in Fos-positive nuclei in the lateral striatum (Figs. 7A–C, 8).
Fig. 6.
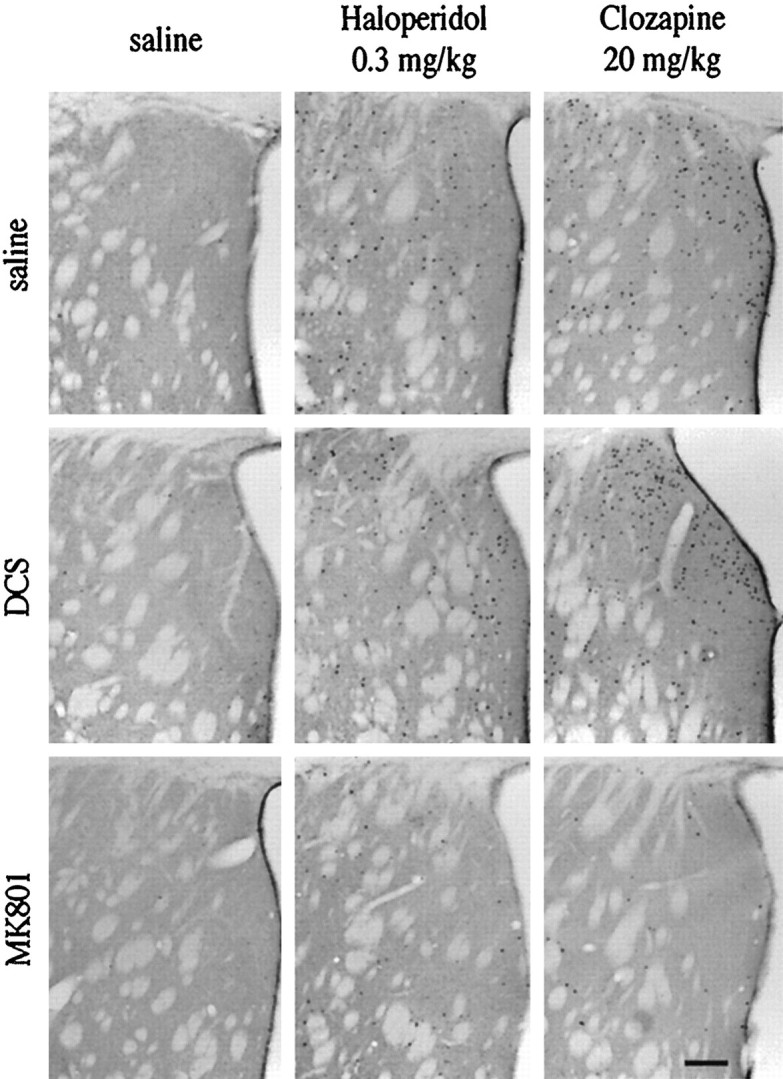
Immunocytochemical analysis of Fos-positive nuclei in the striatum after treatment with haloperidol (0.3 mg/kg) and clozapine (20 mg/kg). DCS (1 mg/kg) did not change the number of Fos-positive nuclei, whereas MK 801 caused a significant reduction (see also Fig. 8). No Fos-positive nuclei were observed after treatment with saline. Scale bar, 0.25 mm.
Fig. 7.

All treatments (haloperidol 0.3 and 1 mg/kg, clozapine 20 mg/kg) induce Fos levels in the striatum with variations in anatomical distribution. A, Fos protein staining in the lateral striatum after treatment with haloperidol (0.3 and 1 mg/kg) or with clozapine (20 mg/kg). No Fos-positive nuclei were observed after treatment with saline (data not shown). Scale bar, 0.25 mm.B, Comparison of the number of Fos-positive nuclei in the nucleus accumbens, the medial striatum, and the lateral striatum after treatment with haloperidol (0.3 and 1 mg/kg, H 0.3, H 1) and with clozapine (20 mg/kg, CLZ).n = 3 for H 1 and CLZ, n = 5 for H 0.3 (see also Fig. 8). Bars in graphs present the average fold induction ± SEM of six striatal areas of three rats treated with haloperidol (1 mg/kg) or clozapine (20 mg/kg), or five rats treated with haloperidol (0.3 mg/kg). C, The ratio of Fos-positive nuclei in the medial over the lateral striatum after treatment with haloperidol (0.3 and 1 mg/kg) and clozapine (20 mg/kg). The medial striatum was particularly sensitive to clozapine and 0.3 mg/kg haloperidol, whereas the lateral striatum responded strongly to 1 mg/kg haloperidol.
MK 801 is particularly effective in preventing antipsychotic drug-induced Fos protein expression in the medial striatum and the nucleus accumbens, whereas DCS does not alter the number of Fos-positive neurons
MK 801 attenuated haloperidol-mediated (0.3 and 1 mg/kg) Fos expression in neurons of the medial striatum and nucleus accumbens, but did so in neurons of the lateral striatum only after administration of 1 mg/kg haloperidol (Figs. 6, 8). The lack of effect of MK 801 on clozapine-mediated or 0.3 mg/kg haloperidol-mediated Fos induction in the lateral striatum suggests that a small population of neurons with a high sensitivity to antipsychotic drugs regulates Fos expression independently of NMDA receptors. Even a higher concentration of MK 801 (2 mg/kg) did not block Fos induction in these neurons (data not shown). MK 801 does not appear to interfere with Fos expression in a general manner, because MK 801 caused a pronounced increase in the number of Fos-positive nuclei in the cortex (data not shown). DCS did not affect the number of Fos-positive nuclei stimulated by clozapine (20 mg/kg) or haloperidol (0.3 mg/kg) in striatal areas (Fig. 8).
Fig. 8.

Distribution of Fos-positive nuclei in the nucleus accumbens, the medial striatum, and the lateral striatum, after treatment with haloperidol (0.3 and 1 mg/kg) and clozapine (20 mg/kg), and the involvement of NMDA receptors. MK 801 reduced the number of Fos-positive nuclei in the nucleus accumbens and the medial striatum in all treatment paradigms. In the lateral striatum, MK 801 affected only haloperidol at 1 mg/kg. DCS (1 mg/kg) did not significantly affect the number of Fos-positive nuclei in any brain area examined (DCS was not used with 1 mg/kg haloperidol treatment). Bars in graphs represent the average fold induction ± SEM of six striatal areas of each rat. Number of rats per experiment is shown in the top left corner of each individual graph.Asterisks indicate statistically significant differences between agonists (haloperidol or clozapine) and agonists pretreated with MK 801. No Fos-positive nuclei were observed after treatment with saline or DCS alone.
MK 801 affects chronic regulation of c-fos andproenkephalin by haloperidol
Repeated haloperidol injection (single daily injections for 12 d) leads to an attenuated induction of c-fos on day 12 (Konradi et al., 1993; Hiroi and Graybiel, 1996) (Fig.9C). Pretreatment with MK 801 before each of the 12 daily haloperidol injections prevented the desensitization of c-fos induction (Fig.9A,C), because a single haloperidol injection after chronic coadministration of MK 801 and haloperidol (Fig. 9A,C) yieldedc-fos levels similar to acute (one time) haloperidol treatment (Fig. 9C) (see Table1 for treatment paradigms).
Fig. 9.

The NMDA antagonist MK 801 affects the chronic regulation of the c-fos and proenkephalingenes by haloperidol. A, The attenuated induction ofc-fos after chronic treatment with haloperidol was prevented by the NMDA antagonist MK 801. Rats were treated for 11 d with MK 801, haloperidol, or combined MK 801 and haloperidol. Rats that were chronically treated with MK 801 and haloperidol responded to a singular haloperidol injection similar to acute haloperidol-treated rats (C). B, Theproenkephalin gene was upregulated after chronic treatment with haloperidol. This upregulation was prevented by pretreatment with MK 801. C, Chronic treatment with haloperidol attenuated c-fos induction after the final haloperidol injection [compare HAL (d 1–12) withHAL (d 12)]). This attenuation was prevented by pretreatment with MK 801 during the chronic haloperidol administration [compare HAL-MK (d1-d11)/HAL (d 12) with HAL (d 12)]). Average fold induction ± SEM of six experiments. For treatment paradigms see Table 1.
The opioid peptide enkephalin is upregulated by chronic administration of haloperidol (Hong et al., 1985; Auchus and Pickel, 1992; Konradi et al., 1993). We therefore examined the role of MK 801 in the chronic regulation of the proenkephalin gene by haloperidol. Pretreatment with MK 801 before each haloperidol injection prevented the chronic upregulation of the proenkephalin gene (Fig.9B).
Intracellular interaction between the cAMP pathway and the NMDA receptor pathway
Regulation of the proenkephalin and c-fosgenes was further studied in primary striatal cultures. Because primary striatal cultures are not affected by brain circuitry, they can be used to study the intrastriatal, intraneuronal effect of MK 801 on cAMP-mediated gene expression. Unfortunately, D2antagonists are not effective in striatal culture (data not shown), presumably owing to the absence of dopamine. Forskolin is an adenylate cyclase stimulating agent capable of activating the cAMP pathway and therefore can be used to mimic the intraneuronal consequences of haloperidol binding to D2 receptors. Theproenkephalin gene was chosen to assess the action of forskolin on gene regulation specifically in D2receptor-expressing neurons. c-fos expression was examined as a means to compare data obtained in tissue culture with the in vivo data. Forskolin treatment led to an increase of both theproenkephalin and the c-fos mRNAs; this induction was blocked by the NMDA antagonist MK 801 (Fig.10A). A combined treatment of glutamate and low concentrations of forskolin enhanced gene expression above either treatment alone (Fig.10B). Because c-fos mRNA has a lower baseline, a faster time course, a lower half-life, and a higher level of induction than proenkephalin mRNA, a short spike of drug exposure has a demonstrable effect on c-fos levels, but presumed changes in proenkephalin levels remain below detection limits. Thus higher concentrations of forskolin or glutamate were used in the analysis of proenkephalin gene expression than that of c-fos gene expression. Moreover, glutamate is taken up rapidly in the cultures (as measured with HPLC), and higher amounts had to be added to ensure a prolonged exposure in theproenkephalin experiments. When the effect of cotreatment of forskolin and glutamate was examined (Fig. 10B), lower forskolin concentrations were used than in the NMDA antagonist experiments (Fig. 10A) to avoid reaching the upper limit of gene expression with forskolin alone.
Fig. 10.

Upregulation by forskolin of theproenkephalin and c-fos genes is prevented by MK 801 and enhanced by glutamate in primary striatal culture. A, Primary striatal cultures were treated with the adenylate cyclase-inducing agent forskolin (10 μm forproenkephalin, 5 μm forc-fos), which increased the levels of both genes. This induction was prevented by pretreatment with the NMDA antagonist MK 801 (1 μm). B, Induction ofproenkephalin and c-fos by forskolin (5 μm for proenkephalin, 2.5 μmfor c-fos) is enhanced by simultaneous treatment with glutamate (50 μm for proenkephalin, 10 μm for c-fos). All treatments are shown in duplicate and are representative of n = 4.
Phosphorylation of the transcription factor CREB on133Ser has been shown to be important for the haloperidol-mediated induction of the proenkephalin gene (Konradi et al., 1993, 1995) and the c-fos gene (Konradi and Heckers, 1995). Phosphorylation at 133Ser was also induced by forskolin and blocked by MK 801 (data not shown; see also Rajadhyaksha et al., 1998).
Results with transfected reporter gene constructs were consistent with the results obtained from the endogenously expressed genes. The pENKAT12 construct, when transfected into primary striatal culture, gave enhanced CAT activity after forskolin treatment. This forskolin-mediated induction of CAT activity was blocked by MK 801 (Fig. 11A). An enhancer-less proenkephalin construct containing 80 bp 5′ of the transcription initiation site fused to a CAT reporter gene (Comb et al., 1988) was used as a negative control and was not regulated (data not shown). Because the proenkephalin gene is regulated by CREB after haloperidol treatment in the striatum (Konradi et al., 1993,1995), we repeated the experiment with a 3xCRE-luciferasereporter construct (Fig. 11B). Like the pENKAT12 construct, activation of the 3xCRE-luciferase construct by forskolin was blocked by MK 801 (Fig. 11B). These data suggest an important role for NMDA receptor activity in D2 receptor-mediated signal transduction.
Fig. 11.

Transfected pENKAT12 and3xCRE-luciferase constructs are induced by forskolin in primary striatal cultures. MK 801 blocks this induction.A, Transfection with pENKAT12. B, Transfection with 3xCRE-luciferase. Transfected cells were treated with MK 801 (1 μm), forskolin (10 μm; FOR), or both, and CAT activity (pENKAT12) or luciferase activity (3xCRE-luc) were measured. Bars present the average fold induction ± SEM over baseline activity.Asterisks indicate statistically significant difference with forskolin treatment. Five experiments performed in triplicate were averaged.
How does the cAMP signal transduction pathway regulate NMDA receptor function? One potential mode of action is through phosphorylation of the NMDA receptor. NR1 is phosphorylated by PKA at897Ser (Tingley et al., 1997). In primary striatal culture, forskolin caused phosphorylation of897Ser-NR1 in a PKA-dependent manner (Fig.12), whereas no change in phosphorylation of 896Ser-NR1 was observed (data not shown). The PKA inhibitor H-89 blocked forskolin-mediated897Ser-NR1 phosphorylation and133Ser-CREB phosphorylation, whereas levels of NR1 and CREB proteins were unchanged (Fig.12A). The Ca2+/calmodulin kinase antagonist KN-62 did not affect forskolin-stimulated897Ser-NR1 phosphorylation (Fig.12B). NR1 phosphorylation at897Ser may serve as an important intraneuronal link between the dopaminergic (D2) and the glutamatergic second messenger pathways. In support of this theory, in vivo injection of haloperidol led to a doubling of 897Ser–NR1 phosphorylation in the striatum (Fig. 13) (fold induction ± SEM: 2.1 ± 0.2; n = 7).
Fig. 12.

Forskolin induces phosphorylation of897Ser of the NR1 receptor in a PKA-dependent manner.A, An immunoblot with an antiserum specific for897Ser–NR1 shows that forskolin (2.5 μm)-induced phosphorylation of 897Ser–NR1 is blocked by pretreatment with the PKA antagonist H-89 (20 μm) (top panel). Levels of NR1 protein were not changed (second panel).133Ser-CREB was induced by forskolin and blocked by H-89 (third panel). CREB protein levels were unchanged (bottom panel). Blot was cut at 80 kDa, and the upper part was exposed to 897Ser–NR1 antiserum, stripped, and reprobed with NR1 antiserum, whereas the lower part was exposed to133Ser-CREB antiserum, stripped, and reprobed with CREB antiserum. B, The CaM kinase antagonist KN-62 (30 μm) does not block forskolin-induced phosphorylation of897Ser-NR1.
Fig. 13.

In vivo treatment with haloperidol causes 897Ser–NR1 phosphorylation. Rats were treated with 1 mg/kg haloperidol for 15 min and killed, and their striata were quickly frozen. The top panel is an immunoblot with the897Ser-NR1 antiserum; the bottom panel is the same immunoblot stripped and exposed to an NR1 antiserum.n = 3 in each group.
897Ser phosphorylation of the NR1 receptor is important for forskolin-mediated gene expression
In primary striatal cultures, we cotransfected NR1 with a3xCRE-luciferase construct and measured luciferase activity after stimulation with forskolin. NR1 was either in its wild-type form or mutated at 896Ser and897Ser to alanine (Ehlers et al., 1995). Forskolin alone doubled the activation of the3xCRE-luciferase construct, an effect that was enhanced by excess NR1 (Fig. 14). Mutation of NR1 at 896/897Ser abolished the effect of forskolin (Fig. 14).
Fig. 14.

Mutation of 896/897Ser–NR1 to alanine blocks forskolin-mediated gene expression. Primary striatal cultures were cotransfected with 3xCRE-luciferase and control expression vector, NR1 wild-type DNA (NR1wt), or mutated 896/897S/A DNA. Forskolin (2.5 μm) activated the 3xCRE-luciferaseconstruct. This activation was enhanced by cotransfection with wild-type NR1 and completely blocked by the mutated construct.Asterisks indicate statistically significant induction.n = 6.
DISCUSSION
Haloperidol and clozapine induce gene expression via NMDA receptors
Inhibition of NMDA receptors with MK 801 significantly reduced the number of c-fos mRNA- and Fos protein-expressing neurons that were activated by antipsychotic drug treatment. Both the conventional antipsychotic drug haloperidol (Ziolkowska and Hollt, 1993) and the atypical antipsychotic drug clozapine depended on NMDA receptors for Fos expression. Only a small number of neurons demonstrated Fos expression in the presence of antipsychotic drugs that was not blocked by MK 801 and thus independent of NMDA receptors.
MK 801 affected both acute and chronic regulation of gene expression. The haloperidol-mediated increase of proenkephalin mRNA after chronic administration was blocked in the presence of MK 801, as was the chronic downregulation of c-fos gene expression. Thus, when NMDA receptors were blocked in chronic haloperidol-treated rats, neither the attenuation of c-fos induction nor the increase in proenkephalin mRNA levels normally seen after chronic treatment was observed.
To examine whether external activation of the NMDA receptor potentiates antipsychotic drug-induced c-fos expression, we used DCS. DCS had no effect when administered alone but increased the levels ofc-fos mRNA after 0.3 mg/kg haloperidol treatment. However, DCS did not increase the number of Fos-positive neurons when given with 0.3 mg/kg haloperidol. This observation indicates that DCS, when combined with haloperidol, does not stimulate Fos expression in new neurons, but rather augments Fos expression in neurons already responding to the treatment with haloperidol. Alternatively, DCS plus haloperidol (0.3 mg/kg) may lead to an increase in c-fosmRNA expression that does not lead to an increase in Fos protein expression.
Surprisingly, only haloperidol (0.3 mg/kg)-mediated, but not clozapine-mediated, c-fos expression was augmented by DCS. These results parallel studies of the effect of DCS in schizophrenia, which show that DCS improves negative symptoms and cognitive deficits when added to conventional antipsychotic agents such as haloperidol (Goff et al., 1995b, 1999) but not when added to clozapine (Goff et al., 1996). The concentrations of DCS used in the present study are comparable to the concentrations used in clinical trials (Goff et al., 1995b), and a dose–response similar to that seen clinically was observed when DCS was added to haloperidol. In both clinical observations in humans (Goff et al., 1995b) and c-fos gene expression in rats, the higher concentration of DCS (5 mg/kg) has no effect, whereas the lower concentration of DCS (1 mg/kg) significantly enhances the response to 0.3 mg/kg haloperidol. This paradoxical dose–response is attributable to the pharmacological properties of DCS. DCS is a partial agonist at the glycine site of the NMDA receptor and, at higher concentrations, displaces the full agonist glycine (Henderson et al., 1990). Thus at low levels the partial agonist DCS can act in synchrony with the full agonist glycine to increase the observed response, whereas at higher levels DCS displaces glycine to effectively blunt the response to stimulation. The lack of significant effect of DCS on c-fos gene expression mediated by 1 mg/kg haloperidol, as well as the high variance in c-fos levels observed, may have been caused by the fact that maximal cellularc-fos levels were reached with 1 mg/kg haloperidol alone. This notion was supported by the finding that a higher concentration of haloperidol (2 mg/kg; data not shown) led to an induction ofc-fos levels similar to that observed after treatment with 1 mg/kg haloperidol.
Although clozapine-mediated c-fos expression depends on NMDA receptors, it is important to note that clozapine inducesc-fos only at concentrations that are above therapeutic levels; i.e., a coincidence between therapeutic drug levels andc-fos expression occurs only with haloperidol but not with clozapine. In addition, c-fos gene expression after clozapine treatment is smaller than after haloperidol treatment and has a different anatomical distribution. Thus, although the present experiments demonstrate similar requirements for c-fosexpression induced by haloperidol or clozapine in the striatum, it does not establish an identical mechanism of action at therapeutic concentrations. Given that both drugs interact with different receptors (see introductory remarks), a distinct mechanism of action forc-fos expression is more likely than are similar requirements.
NMDA receptors are necessary to modulateproenkephalin expression by the cAMP second messenger pathway
We used primary striatal cultures to determine whether the interaction of antipsychotic drugs and NMDA receptors depends on functional brain circuitry or whether it is caused by an intraneuronal cooperation of signal transduction pathways. In striatal cultures, which lack the in vivo circuitry, we demonstrated an intraneuronal interaction between cAMP pathways and NMDA receptor-mediated signal transduction pathways. It is important to note that we used low concentrations of forskolin to mimic an induction of cAMP levels similar to G-proteins (Rajadhyaksha et al., 1998). Unlike low concentrations of forskolin, high concentrations of forskolin can activate CREB phosphorylation independent of NMDA receptors (Rajadhyaksha et al., 1998). Moderate activation of the cAMP second messenger pathway led to proenkephalin and c-fosgene induction only when NMDA receptors were functional. Thus in the presence of MK 801, low amounts of the cAMP activator forskolin could not induce either gene. Moreover, glutamate further enhanced cAMP-mediated induction of both genes. In neurons transfected with a reporter gene under the control of the proenkephalinor the CRE promoter, MK 801 also blocked induction of the reporter gene by forskolin.
Antipsychotic drugs modulate NMDA receptor function via phosphorylation of the NR1 subtype
Our results suggest that antipsychotic drugs facilitate NMDA receptor activity by an intracellular mechanism. In striatal culture, activation of the cAMP pathway led to the phosphorylation of897Ser-NR1 in a PKA-dependent manner. In concordance with this finding, systemic administration of haloperidol caused 897Ser–NR1 phosphorylationin vivo. A mutation of896/897Ser–NR1 that prevents phosphorylation by PKA and PKC (Ehlers et al., 1995) totally abolished the induction of gene expression by forskolin. Together, these data demonstrate that D2 antagonists activate the NMDA receptor via PKA-mediated phosphorylation of897Ser-NR1 (Fig.15). A recent study suggests that PKA may even be physically associated with the NR1 receptor subtype (Westphal et al., 1999).
Fig. 15.

Hypothesized interaction of D2receptors and NMDA receptors in the striatum. Inhibition of D2 receptors or stimulation with forskolin increases cAMP levels and activates the PKA–second messenger pathway. The second messenger pathway increases NMDA receptor function, e.g., via897Ser-NR1 phosphorylation, such that the NMDA receptor activates a signal transduction pathway that translocates to the nucleus and causes phosphorylation of the transcription factor CREB. Phosphorylation of CREB leads to an increase in mRNA synthesis of thec-fos and proenkephalin genes. Although CREB may be necessary for the transactivation of c-fosand the proenkephalin gene in the striatum, additional transcription factors may be involved and activated by the same signal transduction pathway. Inset, D2 receptor activity depresses PKA activity. Inhibition of D2 receptors with drugs such as haloperidol disinhibits PKA activity and leads to897Ser–NR1 phosphorylation. Activation of PKA with low levels of forskolin has a similar effect, whereas high levels of forskolin can mediate CREB phosphorylation in addition to NMDA receptor phosphorylation. Thus, high levels of forskolin can mediate gene expression independent of NMDA receptors (Rajadhyaksha et al., 1998).
Intraneuronal interaction between D2 and NMDA receptors: unifying the glutamate and dopamine hypotheses of schizophrenia
Putative schizophrenia-like behaviors in mice with reduced levels of NMDA receptors are reversed by treatment with haloperidol and clozapine (Mohn et al., 1999). In our model (Fig. 15), the second messenger pathway activated by haloperidol phosphorylates897Ser–NR1 and increases NMDA receptor activity by enhancing the receptor's sensitivity to glutamate. This mechanism, which takes place inside the neuron, does not require increased glutamatergic neurotransmission. The therapeutic benefit of D2 antagonists may be indicative of a need to bolster the glutamatergic system. However, because clozapine inducesc-fos expression mostly above therapeutic levels, it is likely that atypical antipsychotics do not achieve their therapeutic effect by the same intraneuronal mode of action.
Is tardive dyskinesia caused by glutamate neurotoxicity in the striatum?
Our data suggest that haloperidol alters NMDA receptor activity by an intraneuronal mechanism (Fig. 15). In the presence of haloperidol, the D2 receptor-expressing neurons of the striatum become more sensitive to glutamate although glutamate levels in the synaptic cleft are not altered. Haloperidol, through its facilitation of glutamatergic function, may render neurons susceptible to glutamate toxicity. In patients with normal glutamate levels, haloperidol has the potential to cause glutamate toxicity of neurons expressing D2 receptors, whereas in patients with decreased glutamate levels, haloperidol compensates for low glutamate neurotransmission (Goff et al., 1995a). In concordance with this notion, CSF aspartate concentrations are significantly elevated in schizophrenics with tardive dyskinesia and below control levels in schizophrenics who do not have tardive dyskinesia (Goff et al., 1995a). The reduced risk of tardive dyskinesia in patients treated with clozapine may be related to our finding that clozapine, at therapeutic levels, has no prominent intracellular modulatory effect on NMDA receptors.
Footnotes
This work was supported by the National Alliance for Research on Schizophrenia and Depression (C.K., S.H.) and National Institute of Drug Abuse Grant DA07134 (C.K.). We thank Michael J. Iadarola for the M-peptide antibody, Steven E. Hyman for the pENKAT12 construct, and Susan E. Lewis for the 3xCRE-luciferase construct.
Correspondence should be addressed to Dr. Christine Konradi, Laboratory of Neuroplasticity, McLean Hospital, Mailman Research Center, 115 Mill Street, Belmont, MA 02478. E-mail:konradi@mclean.harvard.edu.
REFERENCES
- 1.Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci USA. 1997;94:12157–12161. doi: 10.1073/pnas.94.22.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auchus AP, Pickel VM. Quantitative light microscopic demonstration of increased pallidal and striatal met5-enkephalin-like immunoreactivity in rats following chronic treatment with haloperidol but not with clozapine: implications for the pathogenesis of neuroleptic-induced movement disorders. Exp Neurol. 1992;117:17–27. doi: 10.1016/0014-4886(92)90106-z. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee SP, Zuck LG, Yablonsky-Alter E, Lidsky TI. Glutamate agonist activity: implications for antipsychotic drug action and schizophrenia. NeuroReport. 1995;6:2500–2504. [PubMed] [Google Scholar]
- 4.Berger SL, Chirgwin JM. Isolation of RNA. Methods Enzymol. 1989;180:3–13. doi: 10.1016/0076-6879(89)80087-4. [DOI] [PubMed] [Google Scholar]
- 5.Boegman RJ, Vincent SR. Involvement of adenosine and glutamate receptors in the induction of c-fos in the striatum by haloperidol. Synapse. 1996;22:70–77. doi: 10.1002/(SICI)1098-2396(199601)22:1<70::AID-SYN8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 7.Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323:353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- 8.Comb M, Mermod N, Hyman SE, Pearlberg J, Ross ME, Goodman HM. Proteins bound at adjacent DNA elements act synergistically to regulate human proenkephalin cAMP inducible transcription. EMBO J. 1988;7:3793–3805. doi: 10.1002/j.1460-2075.1988.tb03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danielson PE, Forss-Petter S, Brow MA, Calavetta L, Douglass J, Milner RJ, Sutcliffe JG. p1B15: a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- 10.Dragunow M, Robertson GS, Faull RL, Robertson HA, Jansen K. D2 dopamine receptor antagonists induce fos and related proteins in rat striatal neurons. Neuroscience. 1990;37:287–294. doi: 10.1016/0306-4522(90)90399-o. [DOI] [PubMed] [Google Scholar]
- 11.Ehlers MD, Tingley WG, Huganir RL. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science. 1995;269:1734–1737. doi: 10.1126/science.7569904. [DOI] [PubMed] [Google Scholar]
- 12.Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry. 1992;49:538–544. doi: 10.1001/archpsyc.1992.01820070032005. [DOI] [PubMed] [Google Scholar]
- 13.Goff DC, Tsai G, Beal MF, Coyle JT. Tardive dyskinesia and substrates of energy metabolism in CSF. Am J Psychiatry. 1995a;152:1730–1736. doi: 10.1176/ajp.152.12.1730. [DOI] [PubMed] [Google Scholar]
- 14.Goff DC, Tsai G, Manoach DS, Coyle JT. Dose-finding trial of d-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry. 1995b;152:1213–1215. doi: 10.1176/ajp.152.8.1213. [DOI] [PubMed] [Google Scholar]
- 15.Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. d-cycloserine added to clozapine for patients with schizophrenia. Am J Psychiatry. 1996;153:1628–1630. doi: 10.1176/ajp.153.12.1628. [DOI] [PubMed] [Google Scholar]
- 16.Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. A placebo-controlled trial of d-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry. 1999;56:21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- 17.Harada K, Nagatsugu Y, Ito H, Shingai R. Intracellular cAMP regulates the response to NMDA in hippocampal neurons. NeuroReport. 1991;2:673–676. doi: 10.1097/00001756-199111000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Henderson G, Johnson JW, Ascher P. Competitive antagonists and partial agonists at the glycine modulatory site of the mouse N-methyl-d-aspartate receptor. J Physiol (Lond) 1990;430:189–212. doi: 10.1113/jphysiol.1990.sp018288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiroi N, Graybiel AM. Atypical and typical neuroleptic treatments induce distinct programs of transcription factor expression in the striatum. J Comp Neurol. 1996;374:70–83. doi: 10.1002/(SICI)1096-9861(19961007)374:1<70::AID-CNE5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 21.Hong JS, Yoshikawa K, Kanamatsu T, Sabol SL. Modulation of striatal enkephalinergic neurons by antipsychotic drugs. Fed Proc. 1985;44:2535–2539. [PubMed] [Google Scholar]
- 22.Hyman SE. New insights into how antipsychotic drugs might work. Harv Rev Psychiatry. 1993;1:68–69. doi: 10.3109/10673229309017060. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 24.Kaur S, Ozer H, Starr M. MK 801 reverses haloperidol-induced catalepsy from both striatal and extrastriatal sites in the rat brain. Eur J Pharmacol. 1997;332:153–160. doi: 10.1016/s0014-2999(97)01078-9. [DOI] [PubMed] [Google Scholar]
- 25.Konradi C, Heckers S. Haloperidol-induced Fos expression in striatum is dependent upon transcription factor cyclic AMP response element binding protein. Neuroscience. 1995;65:1051–1061. doi: 10.1016/0306-4522(94)00546-h. [DOI] [PubMed] [Google Scholar]
- 26.Konradi C, Kobierski LA, Nguyen TV, Heckers S, Hyman SE. The cAMP-response-element-binding protein interacts, but Fos protein does not interact, with the proenkephalin enhancer in rat striatum. Proc Natl Acad Sci USA. 1993;90:7005–7009. doi: 10.1073/pnas.90.15.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konradi C, Cole RL, Green D, Senatus P, Leveque JC, Pollack A, Grossbard SJ, Hyman SE. Analysis of the proenkephalin second messenger-inducible enhancer in rat striatal cultures. J Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 28.Le Moine C, Normand E, Guitteny AF, Fouque B, Teoule R, Bloch B. Dopamine receptor gene expression by enkephalin neurons in rat forebrain. Proc Natl Acad Sci USA. 1990;87:230–234. doi: 10.1073/pnas.87.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell IH, Harrison GS, Wood WM, Maxwell F. A DNA cassette containing a trimerized SV40 polyadenylation signal which efficiently blocks spurious plasmid-initiated transcription. Biotechniques. 1989;7:276–280. [PubMed] [Google Scholar]
- 31.Meltzer HY. An overview of the mechanism of action of clozapine. J Clin Psychiatry. 1994;55[Suppl B]:47–52. [PubMed] [Google Scholar]
- 32.Merchant KM, Dobie DJ, Filloux FM, Totzke M, Aravagiri M, Dorsa DM. Effects of chronic haloperidol and clozapine treatment on neurotensin and c-fos mRNA in rat neostriatal subregions. J Pharmacol Exp Ther. 1994;271:460–471. [PubMed] [Google Scholar]
- 33.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 34.Moore NA, Blackman A, Awere S, Leander JD. NMDA receptor antagonists inhibit catalepsy induced by either dopamine D1 or D2 receptor antagonists. Eur J Pharmacol. 1993;237:1–7. doi: 10.1016/0014-2999(93)90085-v. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen TV, Kosofsky BE, Birnbaum R, Cohen BM, Hyman SE. Differential expression of c-fos and zif268 in rat striatum after haloperidol, clozapine, and amphetamine. Proc Natl Acad Sci USA. 1992;89:4270–4274. doi: 10.1073/pnas.89.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; San Diego: 1986. [DOI] [PubMed] [Google Scholar]
- 37.Rajadhyaksha A, Leveque JC, Macias W, Barczak A, Konradi C. Molecular components of striatal plasticity: The various routes of cyclic AMP pathways. Dev Neurosci. 1998;20:204–215. doi: 10.1159/000017314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajadhyaksha A, Barczak A, Macías W, Leveque J-C, Lewis S, Konradi C. L-type Ca2+ channels are essential for glutamate-mediated CREB phosphorylation and c-fos gene expression in striatal neurons. J Neurosci. 1999;19:6348–6359. doi: 10.1523/JNEUROSCI.19-15-06348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 40.Robertson GS, Fibiger HC. Neuroleptics increase c-fos expression in the forebrain: contrasting effects of haloperidol and clozapine. Neuroscience. 1992;46:315–328. doi: 10.1016/0306-4522(92)90054-6. [DOI] [PubMed] [Google Scholar]
- 41.Robertson GS, Matsumura H, Fibiger HC. Induction patterns of Fos-like immunoreactivity in the forebrain as predictors of atypical antipsychotic activity. J Pharmacol Exp Ther. 1994;271:1058–1066. [PubMed] [Google Scholar]
- 42.Roche KW, Tingley WG, Huganir RL. Glutamate receptor phosphorylation and synaptic plasticity. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 43.Tingley WG, Roche KW, Thompson AK, Huganir RL. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- 44.Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- 45.Van Tol HH, Bunzow JR, Guan HC, Sunahara RK, Seeman P, Niznik HB, Civelli O. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature. 1991;350:610–614. doi: 10.1038/350610a0. [DOI] [PubMed] [Google Scholar]
- 46.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 47.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser IDC, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida Y, Ono T, Kizu A, Fukushima R, Miyagishi T. Striatal N-methyl-d-aspartate receptors in haloperidol-induced catalepsy. Eur J Pharmacol. 1991;203:173–180. doi: 10.1016/0014-2999(91)90712-y. [DOI] [PubMed] [Google Scholar]
- 50.Ziolkowska B, Hollt V. The NMDA receptor antagonist MK-801 markedly reduces the induction of c-fos gene by haloperidol in the mouse striatum. Neurosci Lett. 1993;156:39–42. doi: 10.1016/0304-3940(93)90434-m. [DOI] [PubMed] [Google Scholar]