

Abstract
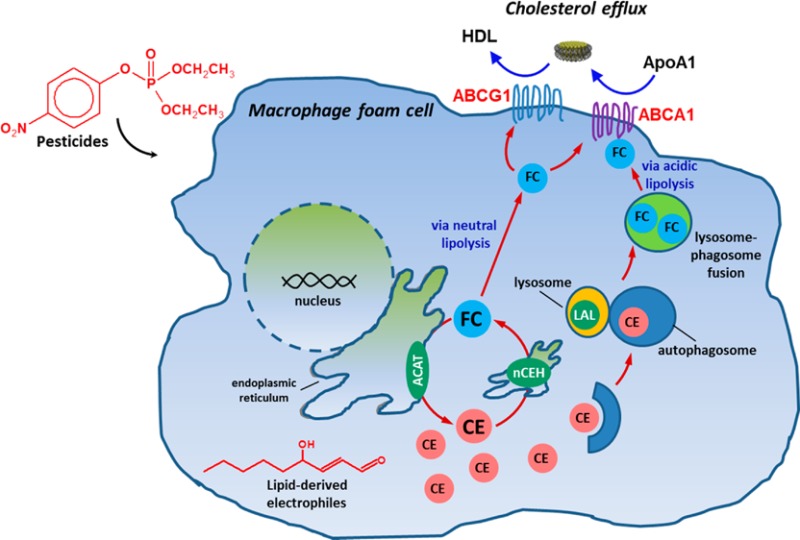
Cholesterol cycles between free cholesterol (unesterified) found predominantly in membranes and cholesteryl esters (CEs) stored in cytoplasmic lipid droplets. Only free cholesterol is effluxed from macrophages via ATP-binding cassette (ABC) transporters to extracellular acceptors. Carboxylesterase 1 (CES1), proposed to hydrolyze CEs, is inactivated by oxon metabolites of organophosphorus pesticides and by the lipid electrophile 4-hydroxynonenal (HNE). We assessed the ability of these compounds to reduce cholesterol efflux from foam cells. Human THP-1 macrophages were loaded with [3H]-cholesterol/acetylated LDL and then allowed to equilibrate to enable [3H]-cholesterol to distribute into its various cellular pools. The cholesterol-engorged cells were then treated with toxicants in the absence of cholesterol acceptors for 24 h, followed by a 24 h efflux period in the presence of toxicant. A concentration-dependent reduction in [3H]-cholesterol efflux via ABCA1 (up to 50%) was found for paraoxon (0.1–10 μM), whereas treatment with HNE had no effect. A modest reduction in [3H]-cholesterol efflux via ABCG1 (25%) was found after treatment with either paraoxon or chlorpyrifos oxon (10 μM each) but not HNE. No difference in efflux rates was found after treatments with either paraoxon or HNE when the universal cholesterol acceptor 10% (v/v) fetal bovine serum was used. When the re-esterification arm of the CE cycle was disabled in foam cells, paraoxon treatment increased CE levels, suggesting the neutral CE hydrolysis arm of the cycle had been inhibited by the toxicant. However, paraoxon also partially inhibited lysosomal acid lipase, which generates cholesterol for efflux, and reduced the expression of ABCA1 protein. Paradoxically, silencing CES1 expression in macrophages did not affect the percent of [3H]-cholesterol efflux. However, CES1 mRNA knockdown markedly reduced cholesterol uptake by macrophages, with SR-A and CD36 mRNA reduced 3- and 4-fold, respectively. Immunoblots confirmed SR-A and CD36 protein downregulation. Together, these results suggest that toxicants, e.g., oxons, may interfere with macrophage cholesterol homeostasis/metabolism.
Introduction
Organophosphorus (OP) pesticides are ubiquitous toxicants in the environment and interesting bioactive compounds to study given the ability of their metabolites to inhibit multiple serine hydrolases, including carboxylesterase 1 (CES1).1 Several commonly used OP pesticides are oxidized to electrophilic oxons, which are potent inhibitors of CES1.2 CES1 is an important xenobiotic detoxifying enzyme in human liver that metabolizes ester-containing substrates such as pesticides and chemotherapeutics.3−5 However, it also exhibits neutral cholesteryl ester hydrolase activity in both human macrophage cell lines and primary monocytes/macrophages.6 Interestingly, human macrophages express high levels of CES1, whereas mouse macrophages have minimal amounts of the orthologous murine isoform,7 which is termed Ces3 based on the nomenclature of Holmes et al.8 Ces3 has an important role in lipid mobilization from murine liver, but not murine macrophages, because of its triacylglycerol hydrolase activity,7 and Ces3–/–LdlR–/– double knockout mice placed on a high-fat diet were protected from atherosclerosis compared to LdlR–/– mice.9 However, mouse macrophages are not a good model of CES1-mediated cholesterol metabolism. Due to this important species difference, the use of cultured human cells is essential for studying the role of macrophage CES1 in atherogenesis.
CES1 has been proposed to play an important role in macrophage reverse cholesterol transport, viz., the hydrolysis of cholesteryl esters to yield free cholesterol for efflux, the initial step in the pathway by which cholesterol is removed from vessel walls and transported to the liver for disposal.6 This process is crucial for the regression of atherosclerotic plaques. We previously showed that treatment of human THP-1 foam cells with either a pharmacological inhibitor (diphenylethane-1,2-dione) or a toxicological inhibitor (paraoxon, the bioactive metabolite of the OP insecticide parathion) of CES1 resulted in increased levels of cholesteryl esters in foam cells compared to that in vehicle-treated foam cells.10 Thus, exposure to environmental chemicals that inhibit neutral cholesterol esterase activity might be an underappreciated means of inactivating reverse cholesterol transport, thereby increasing the risk of cardiovascular disease.
In addition to pollutants found in the environment, organisms produce and are exposed to a large number of endogenous toxins. For example, oxidized low-density lipoproteins (oxLDLs) are toxic molecules that accumulate in the subendothelial space of arterial walls.11 A large number of oxidants and electrophiles, including oxidized phospholipids, cholesteryl ester oxidation products, oxyanion radicals, and diffusible electrophilic α,β-unsaturated aldehydes (e.g., 4-hydroxynonenal or HNE), are found in oxLDLs. These endogenous toxins can activate vessel wall macrophages, thereby initiating a feed-forward mechanism in which additional inflammatory mediators and oxidants are released from the activated macrophages. This series of events leads to additional monocyte infiltration into the vessel wall, macrophage proliferation, and the development of chronic inflammation in the vessel wall.12 Cell-adaptive responses to these complex inflammatory and oxidative stimuli include the activation of the endocannabinoid system13 and the induction of antioxidant enzymes via Nrf2-mediated signaling.14 Therefore, monocytes and macrophages in the vascular system are exposed to a large number of xenobiotics and endogenous chemicals, which can exacerbate macrophage dysfunction during disease.15
Although many studies have focused on the roles of nutrients such as fatty acids and glucose during atherogenesis, little research has been aimed at the role of environmental toxicants in this context. In the earliest stages of atherosclerosis, most cholesterol is stored in macrophages as cholesteryl-fatty acid esters in lipid droplets, but as the atherosclerotic lesion progresses, the content of cholesteryl esters gradually decreases with reciprocal increases in unesterified or free cholesterol (FC) content.16 The resulting excess FC content in the macrophage causes endoplasmic reticulum-stress induced apoptotic events and subsequent secondary necrosis.17 In addition, FC accumulation in macrophages can trigger a marked synthesis and secretion of the pro-inflammatory cytokines TNFα and IL-6.16 Macrophage cholesterol efflux via the ATP-binding cassette transporters ABCA1 and ABCG1 is a vital mechanism of cholesterol homeostasis that helps to reduce cholesterol burden and inflammation. ABCA1 and ABCG1 promote the export of intracellular free cholesterol onto extracellular acceptors, such as lipid-free ApoA1 and high-density lipoprotein (HDL), respectively. With this in mind, studies that define how elements of macrophage cholesterol homeostasis are dysregulated by environmental and endogenous toxins are important to pursue. Therefore, the goal of this study was to examine the effects of toxicologically relevant xenobiotics (OP insecticide metabolites) and endogenous toxins (4-hydroxynonenal) on cholesterol efflux from human THP-1 macrophage foam cells, which had been preloaded with [3H]-cholesterol/acLDL. In addition, CES1 expression was knocked down to mimic the effects of CES1 inactivation by toxicants on macrophage cholesterol metabolism.
Experimental Procedures
Chemicals, Cells, and Reagents
Human THP-1 monocytes, COS-7 cells, RPMI-1640 medium containing high glucose and l-glutamine, Dulbecco’s modified Eagle’s medium (DMEM), gentamicin sulfate solution (50 mg/mL), and Hank’s balanced salt solution without calcium, magnesium, or phenol red were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Low-endotoxin containing fetal bovine serum (FBS) was purchased from Invitrogen (Carlsbad, CA). Acetylated low-density lipoprotein (acLDL) was from Intracel (Bethseda, MD), and [3H]-cholesterol, from PerkinElmer (Cambridge, MA). Acyl CoA:cholesterol acyltransferase (ACAT) inhibitor (Sandoz 58035) was purchased from Santa Cruz Biotechnology (Dallas, TX). THP-1 macrophages transduced with lentivirus containing either short-hairpin (sh)RNA that targets CES1 (CES1KD) or scrambled shRNA (control) were prepared as previously described.18 ABCA1, SR-A, GAPDH, and lysosomal acid lipase (LAL) antibodies were purchased from Santa Cruz Biotechnology or Abcam (Cambridge, MA). β-Actin antibody (cat. no. A5316) was from Sigma (St. Louis, MO). Total RNA isolation kits and quantitative real time (RT)-PCR reagents were purchased from Qiagen (Valencia, CA), and cDNA synthesis reagents were purchased from Bio-Rad Laboratories (Hercules, CA). Primers for RT-PCR consisted of both prevalidated Quantitect primer assays (Qiagen) and custom oligonucleotide primers purchased from Invitrogen (described in Table 1). Paraoxon (PO) and chlorpyrifos oxon (CPO) were gifts from Dr. Howard Chambers (Dept. of Biochemistry, MSU). HNE was from Cayman Chemical Company (Ann Arbor, MI). Phorbol 12-myristate 13-acetate (PMA), 4′,6-diamidino-2-phenylindole (DAPI), and T0901317 were from Sigma. Fluorophosphonate-biotin (FP-biotin) was from Toronto Research Chemicals (North York, Ontario). The expression vectors for human LAL and human CES1 were purchased from Origene (Rockville, MD).
Table 1. Primer Sequences Used for Quantitative Real-Time PCR.
| gene | forward sequence | reverse sequence |
|---|---|---|
| ABCA1 | 5′-GGGCCTCGTGAAGTATGGAG-3′ | 5′-GCCATCCTAGTGCAAAGAGC-3′ |
| ABCG1 | 5′-GACAGGGATGCGCATTTCAC-3′ | 5′-GCTGGCATTAGTAACTGTGTCC-3′ |
| CD36 | 5′-AGGACTTTCCTGCAGAATACCA-3′ | 5′-ACAAGCTCTGGTTCTTATTCACA-3′ |
| SR-A (MSR1) | 5′-CCTGTGCATTGATGAGAGTGC-3′ | 5′-TGCTCCATACTTCTTTCGTCCT-3′ |
| GAPDH | GAPDH_1_SG QuantiTect primer assay: QT00079247 | |
| CES1 | CES1_2_SG QuantiTect primer assay: QT01155581 | |
| CES3 | CES3_1_SG QuantiTect primer assay: QT00034692 | |
Macrophage Culture Conditions
THP-1 monocytes were grown in RPMI-1640 medium supplemented with 10% (v/v) FBS, 0.05 mM β-mercaptoethanol, and 50 μg/mL gentamicin (complete growth medium) at 37 °C in an atmosphere of 95% air/5% CO2. Cells were grown in suspension at a density between 0.2 × 106 and 1 × 106 cells/mL, as recommended by ATCC. THP-1 monocytes were differentiated into macrophages by the addition of PMA to the culture medium (final concentration 100 nM) for 48–72 h. The culture medium was replaced every 2 to 3 days with fresh growth medium containing PMA.
Cholesterol Mass in Macrophages Following Paraoxon Treatment
AcLDL is a modified form of LDL widely used in atherosclerosis studies to generate macrophage foam cells. It is handled by macrophages in the same manner as the more physiologically relevant oxidized (ox)LDL, i.e., acLDL is recognized by the same scavenger receptors (SR-A and CD36) as those for oxLDL.19 THP-1 macrophages were lipid-loaded by incubation with growth media containing 50 μg/mL acLDL and 1% (v/v) FBS for 24 h to produce foam cells. The cholesterol mass in foam cells was determined by a similar approach as that in our previous work10 but with some important differences. Foam cells were incubated overnight in serum-free growth medium containing 0.2% (w/v) BSA to allow equilibration of intracellular cholesterol pools.10 The cells were then treated simultaneously with an ACAT inhibitor (Sandoz 58035, 50 μM) and either paraoxon (10 μM) or vehicle (ethanol, 0.1% v/v) for 24 h in serum-free growth medium containing 0.2% BSA (no cholesterol acceptors are present at this stage). The ACAT inhibitor was used to prevent re-esterification of intracellular cholesterol during the treatment period. The intracellular cholesterol mass was determined before cholesterol efflux commenced to confirm foam cell formation (this was defined as 0 h) and after a 24 h cholesterol efflux period. Two extracellular cholesterol acceptors were used during efflux: lipid-free ApoA1 or FBS. At specified times (t = 0 and 24 h), the culture medium was removed, and macrophages were washed gently with PBS, scraped into ice-cold 50 mM Tris-HCl (pH 7.4) buffer, and sonicated. The whole-cell lysates were centrifuged at low speed to remove cellular debris (1000g, 5 min, 4 °C), the supernatant was collected, and aliquots were removed to measure the free cholesterol (unesterified) and total cholesterol (esterified plus unesterified) content using a commercial cholesterol assay kit (Invitrogen). Esterified cholesterol content was determined by subtracting the free cholesterol level from the total cholesterol level. In addition, aliquots of the lysate were removed to measure DNA content using DAPI dye for normalization. Cholesterol mass is reported as micrograms of cholesterol equivalents per microgram of DNA.
Cholesterol Efflux From Macrophages
Human THP-1 monocytes were seeded into a 6-well dish (2 × 106 cells/well), and PMA was added to the complete growth medium. Following 2–3 days of incubation to allow for cell differentiation, the resulting macrophages were cultured in complete growth medium supplemented with 50 μg/mL acetylated LDL, 1% (v/v) FBS, and [3H]-cholesterol (1 μCi/mL) for 24 h, followed by an overnight equilibration period in serum-free growth medium containing 0.2% BSA. Foam cells were treated with PO, CPO, or HNE (10 μM each) for 24 h without cholesterol acceptors, followed by a 0–48 h efflux period in the presence of either 10% (v/v) FBS, HDL (25 μg/mL), or ApoA1 (25 μg/mL). The toxicants were present in the culture media throughout the efflux period. At the specified time points, the growth medium from each well was removed and centrifuged briefly to remove cell debris and detached cells. In addition, the adherent cells were washed with PBS twice and lysed by addition of 1% (v/v) Triton X-100 in PBS (lysis buffer). Cells were allowed to stand for 15 min at room temperature in lysis buffer, followed by repetitive pipetting to ensure homogenization. The [3H]-cholesterol content in both the culture medium and whole-cell lysate was determined by radioassay of aliquots via liquid scintillation counting, and the percent efflux of [3H]-cholesterol was calculated as follows: % efflux = [cpm in medium/(cpm in cells + cpm in medium)] × 100. Data are presented as the mean ± SD of three experiments.
Inhibition of Overexpressed LAL Activity by Paraoxon
COS-7 cells were plated into 60 mm plates and transfected with either LAL or CES1 expression vectors, or mock transfected, using FuGene transfection reagent (Promega, Madison, WI) with the protocol outlined by the manufacturer. Forty-eight hours after transfection, the culture medium was removed, and the cells were washed with PBS. Cells were scraped into cold 50 mM Tris-HCl (pH 7.4) buffer and sonicated on ice. Overexpression of the desired protein was confirmed by immunoblotting analysis. Inhibition of LAL and CES1 following preincubation of the enzyme with paraoxon was determined by the approach described previously.20 The concentration of paraoxon that inhibits 50% of enzyme activity (IC50) was determined by incubating the cell lysate containing the overexpressed enzyme and paraoxon at 37 °C for either 30 or 15 min for LAL and CES1, respectively, followed by addition of ester substrates 4-methylumbelliferyl oleate (4-MUBO; for LAL) or p-nitrophenyl valerate (pNPV; for CES1). For LAL, the preincubation with paraoxon was carried out in 50 mM acetate buffer (pH 5.3) containing 0.01% Triton X-100. The enzymatic reaction was run in 50 mM acetate buffer (pH 5.3) containing 0.06% Triton X-100, 0.5% (v/v) ethanol, and 75 μM 4-MUBO in a final volume of 200 μL. At the end of the 30 min reaction, 100 μL of 100 mM Tris-HCl (pH 9.0) was added, and the amount of 4-methyumbelliferone generated in the reaction was quantified by fluorescence, λex 355 nm and λem 460 nm. For CES1, the reaction progress was monitored in a plate reader for 5 min, as previously described.21 The progress curves were linear over the reaction period, and slopes were calculated to determine the enzymatic activity. IC50 values were estimated by plotting the percent of enzyme activity versus paraoxon concentration.
Quantitative Real-Time PCR Analysis of mRNA Expression
Total RNA was isolated from both control and CES1KD THP-1 macrophages following 48–72 h differentiation and after loading with acLDL for an additional 24 h (post differentiation) using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. Recovered RNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA), and cDNA was synthesized with an iScript Select cDNA Synthesis Kit (BioRad) using oligo(dT) primers according to the manufacturer’s protocol. Real-time PCR of cDNA products was performed on a Stratagene Mx3005P thermal cycler with Quantifast SYBR Green PCR master mix (Qiagen) using the primers detailed in Table 1. The thermocycler program used for all target genes consisted of a 5 min hot start at 95 °C prior to 40 cycles of 10 s at 95 °C followed by 30 s at 60 °C, as recommended by the manufacturer. PCR product quality was assessed via dissociation curve analysis immediately following amplification. Differential expression of target genes was assessed by the ΔΔCt method using GAPDH as the reference gene, and results are presented as fold expression in CES1KD macrophages compared to control macrophages that were transduced with lentivirus containing a scrambled shRNA construct.
Immunoblotting Analysis of ABCA1, SR-A, CD36 and LAL Expression
Whole-cell lysates of acLDL-loaded control and CES1KD THP-1 macrophages (2 × 106 cells) were prepared in RIPA buffer containing protease inhibitors. Thirty micrograms of protein per sample, as determined by bicinchoninic acid assay (Thermo Scientific), was separated on 10% SDS-PAGE gels prior to semidry transfer (20 V for 30 min) onto PVDF membranes. Membranes were blocked in 5% (w/v) nonfat dry milk in Tris-buffered saline with Tween-20 (TBST: 10 mM Tris, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature and probed for GAPDH (Abcam 37168; final dilution 1:15 000), β-actin (Sigma A5316; final dilution 1:5000), ABCA1 (Abcam 18180; final dilution 1:1000), LAL (Abcam 36597; final dilution 1:500), SR-A (Abcam 151707; final dilution 1:1000), or CD36 [Abcam 133625 (rabbit monoclonal antibody); final dilution 1:1000 or Abcam 36977 (rabbit polyclonal antibody); final dilution 1:1000] overnight at 4 °C. After washing with TBST, blots were probed with either goat anti-rabbit IgG-HRP (Santa Cruz sc-2030; final dilution 1:15 000) or donkey anti-mouse IgG-HRP (Santa Cruz sc-2314; final dilution 1:10 000), as appropriate for the respective primary antibodies, for 1 h at room temperature. Following final washes, blots were visualized by enhanced chemiluminescence using Thermo Supersignal West Pico reagent. The resulting films were scanned, and densiometry analysis was performed using ImageJ v1.49a (NIH).
Activity-Based Protein Profiling (ABPP) of THP-1 Macrophage Lysates
Control and CES1KD THP-1 macrophages were loaded with acLDL (50 μg/mL) for 24 h, followed by overnight equilibration. Whole-cell lysates were prepared and treated with FP-biotin (5 μM, 1 h, room temperature). The treated proteomes were separated by SDS-PAGE, and biotin-labeled proteins were detected with avidin-peroxidase.18
Statistical Analysis
Results are presented as the mean ± SD. Statistical comparison between the means of two groups was done by Student’s t-test, and comparisons among multiple groups was done using one-way ANOVA followed by Dunnett’s test. SigmaStat or Excel was used for analysis.
Results
Cholesterol Mass in Macrophages Following Paraoxon Treatment
THP-1 macrophages were made foam cells by incubation with acLDL, and intracellular cholesterol pools were allowed to equilibrate in the absence of cholesterol acceptors (Supporting Information Figure S1). The foam cells were subsequently treated with an ACAT inhibitor and paraoxon (10 μM each) for 24 h in the absence of cholesterol acceptors, followed by an additional 24 h in the presence of the extracellular cholesterol acceptor ApoA1. The purpose for using an ACAT inhibitor (ACATi) was to remove the esterification arm from the macrophage cholesterol homeostasis model (Figure 1). Therefore, free cholesterol ferried into the cell via modified LDL particles or generated by macrophage cholesteryl ester hydrolase(s) cannot be esterified by ACAT; thus, ACAT will not compete with ABC transporters for free cholesterol. This serves to enhance the unidirectional transport of cholesterol out of the cell. Timing of the addition of ACATi is important, as shown in Figure 2A. Treatment of macrophages with Sandoz 58035 during the acLDL loading period prevents foam cell formation22 and enhanced [3H]-cholesterol efflux to ApoA1 (Figure 2A, left panel) because most of the [3H]-cholesterol pool is in the free form and poised for removal. On the other hand, addition of ACATi after the acLDL loading period was over resulted in a slight, but nonsignificant, increase in cholesterol efflux (Figure 2A, right panel). Thus, ACATi was added after acLDL loading in the subsequent experiments to allow foam cell formation and to isolate one limb (i.e., neutral cholesteryl ester hydrolysis) of the cholesterol esterification/de-esterification cycle for study. ACATi treatment during the 24 h efflux period caused a slight but significant increase in free cholesterol relative to the non-ACATi treatment when macrophage foam cells were incubated in serum-containing medium (Figure 2B), while redistribution of the two cholesterol pools [free cholesterol (FC) and cholesteryl ester (CE)] in macrophage foam cells following 24 h incubation with ACATi in serum-free medium is shown in Supporting Information Figure S2. CE and FC mass decreased and increased, respectively, while total cholesterol mass did not change.
Figure 1.

Macrophage cholesterol homeostasis/metabolism. Scheme showing the cholesteryl ester cycle in a macrophage foam cell, including mobilization of CEs from lipid droplets via neutral cholesteryl esterases and lipophagy involving lysosomal acid lipase (LAL). Inhibition of ACAT during cholesterol efflux prevents re-esterification of free cholesterol. ACAT, acyl CoA:cholesterol acyltransferase; nCEH, neutral cholesteryl ester hydrolase; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; ApoA1, extracellular cholesterol acceptor; CE, cholesteryl ester; FC, free cholesterol; HDL, high-density lipoprotein; LAL, lysosomal acid lipase.
Figure 2.

Paraoxon, the bioactive metabolite of parathion, increases cholesteryl ester content in macrophage foam cells. (A) Addition of ACATi during the acLDL loading period promotes [3H]-cholesterol efflux to ApoA1 (left panel), whereas ACATi treatment after acLDL loading period does not change efflux (right panel). (B) Free cholesterol mass in THP-1 macrophages after acLDL loading/equilibration (time = 0 h, black bar) and following 24 h efflux in the absence and presence of an ACAT inhibitor (ACATi) using FBS (10%, v/v) in the culture medium as a universal acceptor (gray bars). (C) Intracellular cholesterol levels (free cholesterol, FC; cholesteryl esters, CE) were quantified in macrophage foam cells after loading with acLDL (50 μg/mL), equilibration, and subsequent efflux using FBS (10%, v/v) in the culture medium as universal acceptor. Cells were treated with ACATi in the presence or absence of paraoxon (10 μM) during the 24 h efflux period. Data in each panel represent the mean ± SD of 3 dishes in a representative experiment; * p < 0.05, Student’s t-test; N.S., not significant.
When THP-1 macrophage foam cells were treated with ACATi in the presence or absence of paraoxon during the 24 h cholesterol efflux period, the amount of CE mass was significantly increased by the paraoxon treatment, whereas FC mass was unchanged (Figure 2C). This result suggested that paraoxon caused a buildup of cholesteryl ester-containing lipid droplets in the macrophages, which is consistent with our previous study10 showing that either paraoxon or pharmacological inhibition of CES1 by diphenylethane-1,2-dione increased the content of macrophage cholesteryl esters. However, in that study,10 we did not use an ACAT inhibitor as done here and thus cannot exclude the possibility that the increased cholesteryl ester content might have been caused by a paraoxon-mediated effect on ACAT. Use of the ACAT inhibitor disabled the esterification arm of the macrophage cholesteryl ester/free cholesterol cycle, thereby isolating the effects of paraoxon on macrophage cholesteryl ester hydrolase(s) (Figures 1 and 2C).
Effect of Oxons on Cholesterol Efflux From Macrophages
[3H]-Cholesterol-loaded THP-1 macrophage foam cells were treated with xenobiotics without cholesterol acceptors for 24 h, followed by a 24 h cholesterol efflux period using FBS, ApoA1, or HDL as acceptor (cumulative exposure time to xenobiotics was 48 h). JZL184, which can inhibit both CES1 and monoacylglycerol lipase,23 and paraoxon both significantly inhibited efflux of [3H]-cholesterol to ApoA1 (Figure 3A). On the other hand, treatment with T0901317, a synthetic liver X receptor (LXR) ligand, more than doubled [3H]-cholesterol efflux compared to that of the control (no xenobiotics). Figure 3B shows that paraoxon inhibited [3H]-cholesterol efflux to ApoA1 in a concentration-dependent manner, causing up to 50% reduction after 10 μM treatment. Addition of ACATi to the culture medium during the 24 h efflux period dampened the attenuating effects of paraoxon on cholesterol efflux (Figure 3C), which is presumably due to a small increase in the pool of FC available for efflux (Figure 2B). Importantly, the reduction in efflux caused by paraoxon was dependent on the cholesterol acceptor used, as only limited effects on cholesterol efflux were evident when HDL was the acceptor (Figure 3D), and paraoxon had no effect on efflux when using the universal acceptor fetal bovine serum (Figure 3F, left panel). This suggested that hydrolysis of cholesteryl esters might not be the rate-limiting step of cholesterol efflux because the efflux rate was dependent on the cholesterol acceptor used and dramatically increased with T0901317 treatment. Although the OPs paraoxon and chlorpyrifos oxon significantly inhibited [3H]-cholesterol efflux to HDL (Figure 3E), the lipid-derived electrophile HNE, which also can inhibit CES1 activity,21 had no effect on efflux. Furthermore, a time course for [3H]-cholesterol efflux from HNE-treated cells using fetal bovine serum as the extracellular acceptor indicated that there were no differences in efflux compared to vehicle-treated cells during the time period evaluated (Figure 3F, right panel). Toxicants were present for the entire 48 h efflux period shown in Figure 3F.
Figure 3.

Effect of xenobiotic and lipid electrophile HNE treatments on macrophage cholesterol efflux. (A) Macrophages loaded with acLDL/[3H]-cholesterol were treated with vehicle (ethanol), JZL184 (1 μM), paraoxon (1 μM), or synthetic LXR ligand T0901317 (10 μM) in the presence of ApoA1, and the extent of [3H]-cholesterol efflux after 24 h was determined. [3H]-Cholesterol efflux from macrophage foam cells to ApoA1 was determined as a function of paraoxon concentration in the absence (B) or presence (C) of ACAT inhibitor. (D) [3H]-Cholesterol efflux from macrophage foam cells to HDL was determined as a function of paraoxon concentration in the absence of ACAT inhibitor. (E) The extent of [3H]-cholesterol efflux from macrophages loaded with acLDL/[3H]-cholesterol and treated with either ethanol (control), paraoxon (PO), chlorpyrifos oxon (CPO), or 4-hydroxynonenal (HNE) in the presence of HDL was determined. (F) Time-course of [3H]-cholesterol efflux from acLDL/[3H]-cholesterol loaded macrophages treated with toxicants. Cells were treated with either PO or HNE (10 μM) for 24 h without cholesterol acceptors, followed by a 0–48 h efflux period with 10% v/v fetal bovine serum serving as the cholesterol acceptor. The toxicants were present in the culture media throughout the efflux period. Chemical structures for PO and HNE are indicated in graphs. Data in each panel represent the mean ± SD of 3 dishes; * p < 0.05, one-way ANOVA followed by Dunnett’s test.
Inhibition of Lysosomal Acid Lipase by Paraoxon
It was recently reported that lipophagy and lysosomal acid lipase (LAL) are responsible, in part, for the hydrolysis of cholesteryl esters that are contained in some cytoplasmic lipid droplets in macrophages.24 Free cholesterol generated in this manner is predominantly effluxed via ABCA1 to ApoA1. Immunoblotting of THP-1 cells indicated that LAL was detectable in macrophages but not in monocytes and could be expressed in COS7 cells following transient transfection of cDNA encoding human LAL (Figure 4A). It was also found that paraoxon could inhibit LAL activity in COS7 cell lysates following transient transfection, although it was not nearly as potent an inhibitor as it was for CES1 (Figure 4B). Indeed, on the basis of IC50 values, LAL was roughly 10 000 times less sensitive than CES1 toward paraoxon. Nevertheless, the concentrations of paraoxon necessary to partially inhibit LAL activity are similar to the concentrations required to reduce cholesterol efflux (1–10 μM, this study; 100 μM, Ouimet et al.24), whereas much lower concentrations of paraoxon are needed to inhibit CES1. Although it is difficult to determine from our data the relative amounts of CEs in foam cells that are mobilized for efflux by the neutral cholesteryl esterase pathway versus the lipophagic pathway, our findings are consistent with paraoxon partially affecting cholesterol efflux through its ability to inhibit LAL activity.
Figure 4.
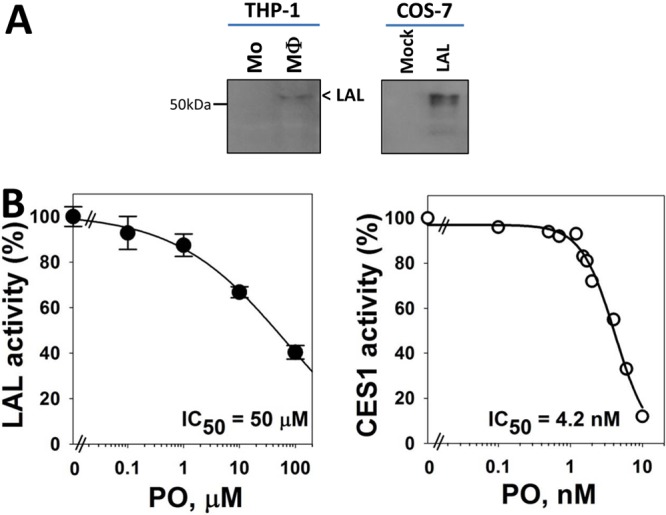
Inactivation of CES1 and lysosomal acid lipase by paraoxon. (A) Immunoblots of lysosomal acid lipase (LAL) in THP-1 monocytes (Mo) and macrophages (MΦ) (left). Human LAL was also overexpressed in COS-7 cells that were transiently transfected with LAL cDNA (right). (B) Recombinant human CES1 and LAL were overexpressed in COS7 cells and whole-cell lysates pretreated with the indicated concentrations of paraoxon for 15 and 30 min, respectively, before measuring enzymatic activity (see Experimental Procedures for details). IC50, concentration that inhibits 50% of enzymatic activity. Data in each panel represent the mean of duplicate or triplicate (±SD) measurements.
Effects of Paraoxon on ABCA1, ABCG1, and CES1 Expression
Because paraoxon’s ability to attenuate macrophage cholesterol efflux was dependent on the type of cholesterol acceptor used (Figure 3B,D,F), we speculated that paraoxon might reduce the expression of the cholesterol transporter ABCA1 that mediates cholesterol efflux to ApoA1 because efflux to this acceptor was the most reduced following paraoxon treatment of THP-1 foam cells. Exposure to paraoxon neither altered the expression of ABCA1 mRNA nor did it affect CES1 and ABCG1 mRNA levels (Figure 5A). On the other hand, treatment with T0901317 caused the expected increase in ABCA1 and ABCG1 mRNA expression (5- and 6-fold increase, respectively) (Figure 5A), whereas it had no effect CES1 mRNA levels. Nevertheless, although ABCA1 mRNA levels were unaltered by paraoxon, the quantity of ABCA1 protein in foam cells measured by immunoblotting appeared to decrease following paraoxon treatment relative to that in vehicle-treated cells (Figure 5B), which could account, in part, for the acceptor-dependent efflux effects observed following paraoxon treatments.
Figure 5.
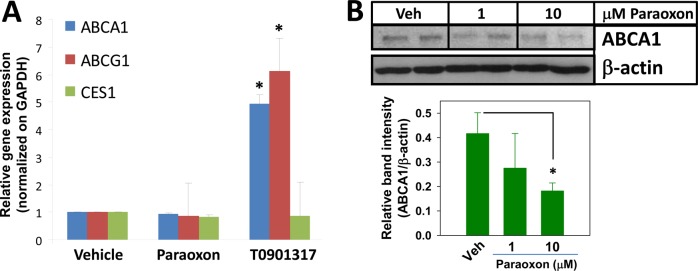
Effect of the organophosphate bioactive metabolite paraoxon on ABC transporter expression in THP-1 macrophages. (A) The effects of PO and LXR ligand T0901317 treatments (24 h) on the expression of the indicated genes in THP-1 macrophages were determined and compared to that from vehicle-treated cells. Data represent the mean ± SD of 3 dishes; * p < 0.05, Student’s t-test. (B) Immunoblotting analysis of ABCA1 protein in PO-treated THP-1 macrophage foam cells. Note that a 10% acrylamide gel was used for protein separation. Data represent the mean ± SD of 2–3 dishes; * p < 0.05, one-way ANOVA followed by Dunnett’s test.
Macrophage Cholesterol Efflux and Mass: Paradoxical Effects Following CES1 Silencing
The use of pharmacological and toxicological inhibitors of carboxylesterases inevitably leads to questions/concerns regarding their off-target effects. To address this issue, we stably knocked down the expression of CES1 in THP-1 macrophages using lentivirus harboring CES1 shRNA (denoted as CES1KD THP-1 cells) and examined the consequences on cholesterol efflux. We recently showed that silencing CES1 mRNA expression resulted in marked reductions in CES1 protein expression and enzymatic activity18 (Supporting Information Figure S3). On the basis of immunoblots of control and CES1KD macrophage lysates, CES1KD cells express only 12% of the CES1 protein present in control cells in Wang et al.18 However, despite efficient knockdown of CES1 protein, it was found that CES1KD macrophages showed no significant difference in the percent of cholesterol efflux to ApoA1 when compared to control macrophages that had been transduced with lentivirus containing scrambled shRNA (Figure 6A). Similar findings were observed when HDL was used as the acceptor instead of ApoA1 (data not shown). These experiments were repeated in the presence of T0901317 to ensure ABCA1 expression was maximal and cholesterol efflux via this transporter was not rate limiting. Although treatment with the LXR ligand caused the expected increase in cholesterol efflux in both cell types, the magnitude of the percent of cholesterol efflux to ApoA1 was no different for CES1KD macrophages compared to that for control macrophages (Figure 6A). These findings led us to compare the cholesterol mass in control macrophages and CES1KD macrophages after cholesterol loading using acLDL. Interestingly, CES1KD macrophages were found to contain significantly less cholesteryl ester than that in control macrophages immediately after acLDL loading and equilibration (t = 0 h) and following the efflux period (t = 24 h) using FBS as the universal cholesterol acceptor (Figure 6B, top panel). On the other hand, free cholesterol levels were unchanged (Figure 6B, bottom panel). Therefore, knocking down CES1 expression in THP-1 macrophages paradoxically resulted in cholesteryl ester levels being significantly lower than that in control THP-1 macrophages following acLDL loading, both before and after efflux.
Figure 6.

Effect of CES1 knockdown in THP-1 macrophages on cholesterol efflux and cholesteryl ester mass. (A) Efflux of [3H]-cholesterol to ApoA1 was unaltered in THP-1 macrophages transduced with lentivirus containing CES1 shRNA in either the presence or absence of LXR ligand T0901317. (B) Cholesteryl ester (CE) mass, but not free cholesterol (FC), was significantly lower in THP-1 macrophages transduced with lentivirus containing CES1 shRNA (CES1KD) compared to that in THP-1 macrophages transduced with lentivirus containing scrambled shRNA (Control). Intracellular cholesterol levels were determined at 0 h (following 24 h acLDL loading and an overnight equilibration period) and 24 h (allowing cholesterol to efflux in the presence of 10% fetal bovine serum). Data in each panel represent the mean ± SD of 3 wells; * p < 0.05, Student’s t-test.
Scavenger Receptor Expression in THP-1 Macrophages Following CES1 Silencing
The reduced amounts of total cholesterol mass and [3H]-cholesterol equivalents measured in CES1KD THP-1 macrophages relative to those in control THP-1 macrophages immediately following cholesterol loading (Figure 7A,B) suggested a deficiency in cholesterol uptake by these cells. Because SR-A and CD36 are responsible for the majority of acLDL uptake into foam cells, their transcription levels were determined by quantitative real-time PCR. RNA was isolated from control (cells transfected with a scrambled shRNA construct) and CES1KD THP-1 macrophages that had been loaded (or not) with acLDL for 24 h. For nonloaded CES1KD cells, we verified that CES1 mRNA expression was knocked down (10-fold) and that mRNA levels for scavenger receptors SR-A and CD36 were not significantly different when compared to that for nonloaded control cells (Figure 7C, left panel). On the other hand, the levels of SR-A and CD36 mRNA were significantly downregulated (3- and 4-fold, respectively) in the cholesterol-loaded CES1KD cells compared to that in cholesterol-loaded control cells (Figure 7C, right panel). As expected, CES1 mRNA was also downregulated (∼14.5-fold) in the cholesterol-loaded CES1KD THP-1 macrophages. Immunoblots of SR-A in cholesterol-loaded control and CES1KD cells indicated that SR-A protein expression was markedly decreased (∼4-fold, p < 0.01; Figure 8A). Immunoblots of CD36 in cholesterol-loaded control and CES1KD cells using a rabbit polyclonal CD36 antibody showed a slight reduction in immunoreactive antigen at 80 kDa (<2-fold, p < 0.05; Figure 8B), a mass that corresponded with the Mr of immunoreactive antigen in the 3T3L1 cell lysate used as a positive control (Figure 8B). CD36 is a glycosylated protein; therefore, it contains isoforms at higher apparent molecular weight than that of its nonglycosylated core protein (∼53 kDa).25
Figure 7.

Knockdown of CES1 in THP-1 macrophages affects cholesterol uptake: Effects of CES1 silencing on scavenger receptor mRNA levels. (A, B) Total cholesterol content (FC + CE mass, A) and radioactivity ([3H]-cholesterol equivalents, B) are reduced in CES1KD THP-1 macrophages (CES1 shRNA) compared to that in control THP-1 macrophages (scrambled shRNA) after cholesterol loading with acLDL/[3H]-cholesterol. scram, scrambled. (C) Real-time PCR analyses of select genes in non-acLDL loaded (left) or acLDL loaded (right) THP-1 macrophages. mRNA expression is normalized to GAPDH mRNA, and fold differences due to CES1 silencing are expressed relative to control conditions (i.e., non-CES1 silenced cells). Data represent the mean ± SD of 3 wells;* p < 0.05, Student’s t-test.
Figure 8.

CES1 silencing in THP-1 macrophage foam cells reduces the expression of SR-A and CD36. Immunoblotting analysis of SR-A protein (A) and CD36 protein (B) in control and CES1KD THP-1 macrophages that had been loaded with cholesterol (50 μg/mL acLDL for 24 h). 3T3L1 represents cell lysate used as positive control for polyclonal rabbit CD36 antibody. Quantitative densitometry is shown next to each immunoblot. Data in each panel represent the mean ± SD of 3 dishes; * p < 0.05, ** p < 0.01; Student’s t-test.
ABCA1 Expression in THP-1 Macrophages Following CES1 Silencing
Figure 9 shows the immunoblot of ABCA1 protein in control and CES1 KD cells that were either cholesterol-loaded or not loaded. A broad band at ∼250 kDa, which is consistent with the large molecular weight and extensive glycosylation of ABCA1, was detected (Figure 9A). On the basis of densitometry, non-acLDL-loaded cells had significantly less ABCA1 protein than that in acLDL-loaded cells (Figure 9B). There was no difference in ABCA1 levels between control and CES1 KD cells in nonloaded cells, whereas a small reduction was observed in CES1KD cells relative to that in control cells in the cholesterol-loaded state (Figure 9B).
Figure 9.

CES1 silencing slightly reduces ABCA1 protein expression in THP-1 foam cells but not in nonfoam cells. (A) Top, Immunoblotting analysis of ABCA1 in control and CES1KD THP-1 macrophages, which had been either loaded (+) or not loaded (−) with cholesterol (50 μg/mL acLDL, 24 h). Molecular weight (MW) markers on immunoblot are Magic Mark XP western MW standards. Bottom, PVDF membrane was stained with coomassie blue to demonstrate equal protein loading of lanes. Note that a 6% acrylamide gel was used for protein separation instead of a 10% gel; therefore, β-actin and GAPDH proteins are not detectable because proteins <50 kDa migrated through the gel. Molecular weight (MW) markers stained by coomassie blue are Fisher markers. (B) Quantitative densitometry is shown next to the immunoblot. Data represents the mean ± SD of 2 dishes; * p < 0.05, Student’s t-test.
CES3 Expression in THP-1 Macrophages Following CES1 Silencing
The amount of CES3 mRNA was upregulated 2.4-fold after silencing CES1 gene expression in cholesterol-loaded THP-1 macrophages (data not shown). This observation was originally reported before by Zhao et al.26 However, despite the apparent induction of CES3 mRNA, treatment of cholesterol-loaded control and CES1KD macrophage lysates with the ABPP probe, FP-biotin, which targets active serine hydrolases in native biological contexts,27 did not reveal a CES3 activity band in the CES1KD macrophage lysate, whereas a band representing CES1 was observed in control THP-1 macrophage lysate at 60 kDa (Figure 10 and Supporting Information Figure S3). Thus, although CES3 mRNA expression was apparently induced and might be a compensatory mechanism following knockdown of CES1 expression, it does not appear to be induced at the active enzyme level because CES3 also has a predicted molecular weight of 60 kDa. It is possible that CES3 does not react efficiently with FP-biotin, and we have previously shown that the serine hydrolase palmitoyl protein thioesterase 1 does not react strongly with this ABPP probe.18 However, this seems unlikely for CES3 given the promiscuous reactivity of the carboxylesterases.20,28 Thus, CES3 might not compensate for the loss of CES1 function in THP-1 macrophages.
Figure 10.
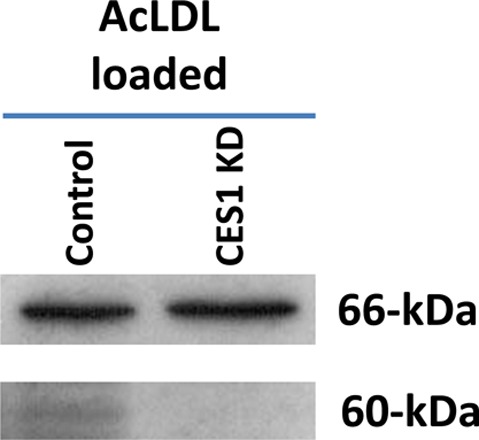
Activity-based protein profiling (ABPP) of control and CES1KD cell lysates. Control and CES1KD THP-1 macrophages were loaded with acLDL (50 μg/mL) for 24 h, followed by overnight equilibration. Whole-cell lysates were prepared and treated with FP-biotin (5 μM, 1 h, room temperature). The treated proteomes were separated by SDS-PAGE, and biotin-labeled proteins were detected with avidin-peroxidase. The 60 kDa region of the gel is where both CES1 and CES3 migrate, whereas the 66 kDa region represents an endogenous biotin-containing protein found in equal quantities in both control and CES1KD cells that serves as a gel-loading control. A full-length ABPP gel for control and CES1 KD THP-1 cells is shown in Supporting Information Figure S3B.
Discussion
CES1 is reported to have neutral cholesteryl ester hydrolase activity in macrophages6,29 and is highly sensitive to covalent modification and inactivation by OP poisons that react with the active-site serine residue.4,20 Consistent with this notion, we previously showed that pharmacological and toxicological inhibition of CES1 caused a significant buildup of cholesterol esters within THP-1 macrophages, which had been preloaded with acLDL, thus enhancing the foam cell phenotype.10 The purpose of this study was to confirm and extend these findings by examining the effects of toxicologically relevant molecules on macrophage cholesterol efflux and the expression of genes that encode proteins of importance to cholesterol homeostasis/metabolism. We hypothesized that treatments of cultured human macrophages preloaded with acLDL/[3H]-cholesterol with toxicants that are known to inhibit CES1 activity, as well as additional serine hydrolase activities, would disable cholesterol efflux. In most instances, our studies utilized an ACAT inhibitor to block the re-esterification arm of the cholesteryl ester cycle (Figure 1) so that one could determine the effect of the toxicants on hydrolysis of preformed cholesteryl esters (i.e., macrophages were loaded with acLDL before treatment with the toxicants). Indeed, we found that paraoxon effectively increased the cholesteryl ester content, but not free chlolesterol content, of treated cells (Figure 2C). This result was consistent with our hypothesis that inactivation of enzyme(s) responsible for cholesteryl ester hydrolase activity causes a buildup of intracellular cholesteryl esters in macrophages. Moreover, treatment of macrophage foam cells containing [3H]-cholesterol with paraoxon caused a concentration-dependent inhibition of efflux to ApoA1 and HDL (Figure 3). Both paraoxon and chlorpyrifos oxon inhibited macrophage cholesterol efflux to roughly the same extent (Figure 3E). On the other hand, the lipid electrophile HNE, even at a relatively high concentration of 10 μM in the culture medium, did not affect cholesterol efflux. We previously showed that exogenous HNE could inhibit CES1 activity in cultured THP-1 macrophages and that HNE covalently modifies a CES1 lysine residue not found in the active site.30 THP-1 cells are very efficient at metabolizing HNE, with glutathione conjugation of HNE, a soft electrophile, being a primary route of its detoxication.30 On the other hand, oxons are hard electrophiles and not detoxified by soft nucleophiles such as glutathione; oxons are instead removed by cellular scavengers such as carboxylesterases that possess a hard nucleophile (active-site catalytic serine residue) optimally positioned in catalytic triads.31 The efficient scavenging of oxons by carboxylesterases, however, might come with costs. We have already shown that paraoxon can efficiently inactivate CES1 in THP-1 macrophages, resulting in altered endocannabinoid metabolism.23 Interestingly, in the current study, we found that treatment of THP-1 macrophages with paraoxon neither altered the mRNA expression of CES1 nor did it affect ABCA1 and ABCG1 mRNA levels. However, ABCA1 protein levels did decline in foam cells following treatment with the highest concentration of paraoxon (Figure 5A,B), which could account, in part, for the reduced cholesterol efflux, although the mechanism for the downregulation is uncertain. Because ABCA1 levels in CES1 KD foam cells were also reduced to a small degree (∼20%) relative to that in control foam cells (Figure 9), this suggested that CES1 depletion (or inhibition) might directly lead to reduced ABCA1 expression. However, loading macrophages with cholesterol is known to induce ABCA1 levels relative to the nonloaded state,40 which is what our data also indicates (Figure 9A,B; compare acLDL-loaded cells to nonloaded cells). Thus, an alternative interpretation is that the reduced ABCA1 levels in CES1 KD foam cells compared to that in control foam cells is a consequence of the lower intracellular cholesterol content in the CES1 KD foam cells relative to that in control cells (Figures 6 and 7). Therefore, CES1 depletion (or inhibition) might have caused the reduction in ABCA1 levels by an indirect mechanism (see below). It was also notable that the synthetic LXR ligand T0901317 did not alter CES1 expression, yet it increased ABCA1 and ABCG1 mRNA levels in the expected manner. This result suggests that CES1 is not under the direct control of the nuclear receptor LXR (Figure 5A).
On the basis of activity-based serine hydrolase profiling, we previously showed that paraoxon and JZL184, at concentrations as low as 0.1 μM, can completely inhibit CES1 activity in THP-1 cells23 (Supporting Information Figure S3). Thus, the relatively modest effects of these xenobiotics on cholesterol efflux (Figure 3) suggested that mechanisms besides CES1-mediated hydrolysis of CEs were also important to consider in macrophage cholesterol efflux. This does not imply that CES1 and/or other cholesteryl esterases do not have a role. Rather, it suggests that the overall process is likely complex and that paraoxon may affect multiple components of the cholesterol efflux machinery, including several other candidates that catalyze cholesteryl ester hydrolysis that are known to exist.6 For example, LAL has been reported to participate in the lipophagy of cholesteryl ester-containing lipid droplets in macrophages.32 This mechanism involves fusion of lysosomes that contain LAL with autophagosomes that have engulfed cytosolic CE-containing lipid droplets. The resulting LAL-mediated hydrolysis of CEs produces a pool of free cholesterol available for efflux via ABCA1.24 Therefore, the partial inhibition of LAL activity by paraoxon (Figure 4) could partly explain the observed reduction in cholesterol efflux to apoA1 (Figure 3A,B). The concentrations of paraoxon used in our study would have likely inhibited several neutral cholesteryl ester hydrolase candidates, i.e., CES1, KIAA1363, and hormone-sensitive lipase, almost completely; however, as shown in Figure 3, cholesterol efflux is not completely inhibited. Thus, the correlation between the extent of LAL inhibition by paraoxon and the magnitude of cholesterol efflux reduction suggests another means by which oxons might impair macrophage cholesterol metabolism.
As already mentioned, the use of chemicals to inhibit enzyme function in cells is fraught with issues related to off-target effects. Because of their large catalytic sites, multiple chemicals have been shown to inhibit carboxylesterases.33 To avoid these issues, THP-1 macrophages were transduced with lentiviruses containing either CES1 shRNA or scrambled shRNA. Our previous study demonstrated that CES1 protein expression in cells was effectively knocked down by this approach;18 however, when we examined the impact of CES1 knockdown on cholesterol efflux (Figure 6A), we did not see a significant effect. Ghosh and colleagues have previously reported similar findings26 and attributed this to the compensatory upregulation of a carboxylesterase isoform, CES3, in macrophages that also exhibited CE hydrolase activity. We also observed a near 3-fold increase in CES3 mRNA in our CES1 silenced cells, although no increase in carboxylesterase activity was observed by ABPP of the cell lysates (Figure 10). Indeed, the observation that several different macrophage enzymes can exhibit neutral cholesteryl ester hydrolase activity, such as CES1, KIAA1363, and hormone-sensitive lipase, is consistent with the concept of enzyme redundancy or back-up systems in vital physiological processes.6 Thus, it was perhaps not surprising that knocking out CES1 activity did not result in changes in cholesterol efflux, as this finding could be attributed to the presence of other enzymes with redundant functions for CES1. Nevertheless, we also discovered that silencing CES1 caused significant reductions in CD36 and SR-A mRNA and protein levels. The reductions in scavenger receptor mRNA levels probably accounted for the markedly reduced cholesterol content found in CES1KD macrophages following acLDL loading as compared to that in control macrophages (Figures 6B and 7A). At present, it is unclear why silencing CES1 expression would reduce the levels of cholesterol scavenger receptor mRNA, but we are currently investigating this. Importantly, the reduction in CD36 and SR-A mRNA expression was found to be dependent on acLDL loading of the macrophages (Figure 7C). It is therefore tempting to speculate on possible CES1 substrates that might be present in the modified low-density lipoproteins taken up into the cells. These substrates might be pro-ligands for nuclear receptors that regulate the expression of genes encoding proteins involved in cholesterol homeostasis.
Our results demonstrated that bioactive oxon metabolites of OP pesticides can increase the cholesteryl ester content of cultured THP-1 macrophage foam cells and reduce the extent of cholesterol efflux. This effect was most pronounced at relatively high concentrations of oxon and when ApoA1 was used as cholesterol acceptor. Silencing of CES1 expression in THP-1 macrophages did not affect cholesterol efflux, even when ABCA1 and ABCG1 expression were induced with an LXR synthetic ligand. It is possible that other candidate neutral cholesteryl ester hydrolases that are sensitive to paraoxon, such as KIAA1363,34 might have also been inactivated along with CES1 by oxons in our study. We have previously shown that KIAA1363 is expressed in THP-1 macrophages, although in lower amounts compared to that of CES1,18 while it was recently shown that KIAA1363 is found in mouse peritoneal macrophages and cholesterol efflux from these cells was partially inhibited by paraoxon.35 Sakai et al.35 suggested that this effect was due to inhibition of the cholesteryl esterase activity of KIAA1363 (which they term nCEH1), although it should be noted that another study failed to observe a cholesteryl esterase activity with KIAA1363.36 However, which enzyme(s) are responsible for neutral cholesteryl esterase activity in macrophages and the methodology used to assay this activity are controversial issues. Although it was reported that THP-1 macrophages that are engineered to overexpress CES1 exhibited robust neutral cholesteryl esterase activity in cell homogenates and an enhanced cholesterol efflux capacity compared to that for control cells,37 it was also reported that homogenates of THP-1 macrophages infected with recombinant adenovirus encoding CES1 cDNA did not exhibit cholesteryl esterase activity.34 Furthermore, we could not detect cholesteryl esterase activity when using pure recombinant CES1 protein.10 That being said, it is important to stress the challenges often encountered when assaying lipase activity in vitro using water-insoluble substrates, such as cholesteryl esters, and broken cell preparations or purified proteins.38 Productive enzymatic reactions are dependent on the cellular activation state and/or the topology of the enzyme in question,7,39 which can be lost during cellular homogenization. The issue of subcellular location is particularly germane for CES1 (and nCEH1) because it is localized in the lumen of the endoplasmic reticulum.7 Thus, the mechanisms by which lumenal enzymes access cholesteryl esters, which are present in lipid droplets localized within the cytosol, are unclear. Cytosolic lipid droplets can form a continuum with the ER by fusing with the ER lipid bilayer enabling neutral lipid substrates to be transferred into the ER bilayer, thereby bringing lumenal hydrolases, which have active sites directed toward the ER lumen, into close proximity with its natural substrates.7 Alternatively, models that invoke the transfer of neutral lipids from cytosolic lipid droplets into the ER lumen and the subsequent formation of lumenal lipid droplets, which is similar to what is observed in hepatocytes, can also be envisioned.7 More work is needed to validate such models in macrophages.
Another potential mechanism for the oxon-mediated reduction in efflux includes an oxon-dependent downregulation of ABCA1, which is consistent with the more marked effects of paraoxon on efflux to ApoA1 compared to HDL (Figure 3B,D). In addition, our data indicated that an enzyme involved in the lipophagy of CE-containing lipid droplets, LAL,32 was partially inhibited by paraoxon, which suggested its inactivation might also contribute to paraoxon’s effects on efflux. On the basis of the differential sensitivity of LAL and CES1 to the inhibitory effects of paraoxon, and the relatively high concentrations of paraoxon required to attenuate cholesterol efflux, this possibility is compelling. Although silencing CES1 did not modulate the percent cholesterol efflux, it did significantly reduce cholesterol uptake by THP-1 macrophages. This finding could be attributed to the reduction in SR-A and CD36 levels, which recognize extracellular modified lipoproteins and facilitate their phagocytosis. Whether chronic CES1 inactivation by toxicants can lead to subsequent reductions in scavenger receptor levels and reduced cholesterol uptake by macrophages is currently under investigation. Together, our findings suggest that toxicants, such as oxons, can interfere with key steps in macrophage cholesterol homeostasis and might contribute to a pro-atherogenic phenotype. Due to the complex pathways that control cholesterol mobilization and efflux, and the redundancy of enzymes involved in these processes, it is difficult to identify one specific toxicological target that is responsible for the effects reported here.
Acknowledgments
We acknowledge Kim Pluta for her help with the CES1 overexpression experiments and IC50 determinations using paraoxon. In addition, we thank Drs. Barbara Kaplan and Edward Sharman for their careful review of the manuscript.
Glossary
Abbreviations
- ABPP
activity-based protein profiling
- ACATi
acyl CoA:cholesterol acyltransferase inhibitor
- nCEH
neutral cholesteryl ester hydrolase
- ABCA1
ATP-binding cassette transporter A1
- ABCG1
ATP-binding cassette transporter G1
- ApoA1
apolipoprotein A1
- CES1
carboxylesterase 1
- CE
cholesteryl ester
- CPO
chlorpyrifos oxon
- FC
free cholesterol
- HDL
high-density lipoprotein
- LAL
lysosomal acid lipase
- 4-MUBA
4-methylumbelliferyl acetate
- 4-MUBO
4-methylumbelliferyl oleate
- PO
paraoxon
- VLDL
very low-density lipoprotein
Supporting Information Available
Cholesteryl ester mass in THP-1 macrophages following acLDL loading and cholesterol efflux after 24 h incubation in serum-containing medium; cholesterol mass (free cholesterol and cholesteryl esters) in THP-1 macrophage foam cells in the presence and absence of ACATi following 24 h incubation in serum-free medium; evidence for knockdown of CES1 expression in THP-1 cells; and esterase activities of control and CES1 KD THP-1 monocytes following treatment with increasing amounts of paraoxon. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This study was supported by NIH 1R15ES015348-02 (M.K.R.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Casida J. E.; Quistad G. B. (2004) Organophosphate toxicology: safety aspects of nonacetylcholinesterase secondary targets. Chem. Res. Toxicol. 17, 983–998. [DOI] [PubMed] [Google Scholar]
- Crow J. A.; Borazjani A.; Potter P. M.; Ross M. K. (2007) Hydrolysis of pyrethroids by human and rat tissues: examination of intestinal, liver and serum carboxylesterases. Toxicol. Appl. Pharmacol. 221, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redinbo M. R.; Bencharit S.; Potter P. M. (2003) Human carboxylesterase 1: from drug metabolism to drug discovery. Biochem. Soc. Trans. 31, 620–624. [DOI] [PubMed] [Google Scholar]
- Ross M. K.; Borazjani A.; Wang R.; Crow J. A.; Xie S. (2012) Examination of the carboxylesterase phenotype in human liver. Arch. Biochem. Biophys. 522, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadkins R. M.; Hyatt J. L.; Wei X.; Yoon K. J.; Wierdl M.; Edwards C. C.; Morton C. L.; Obenauer J. C.; Damodaran K.; Beroza P.; Danks M. K.; Potter P. M. (2005) Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J. Med. Chem. 48, 2906–2915. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Zhao B.; Bie J.; Song J. (2010) Macrophage cholesteryl ester mobilization and atherosclerosis. Vasc. Pharmacol. 52, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga A. D.; Lehner R. (2011) Role of endoplasmic reticulum neutral lipid hydrolases. Trends. Endocrinol. Metab. 22, 218–225. [DOI] [PubMed] [Google Scholar]
- Holmes R. S.; Wright M. W.; Laulederkind S. J.; Cox L. A.; Hosokawa M.; Imai T.; Ishibashi S.; Lehner R.; Miyazaki M.; Perkins E. J.; Potter P. M.; Redinbo M. R.; Robert J.; Satoh T.; Yamashita T.; Yan B.; Yokoi T.; Zechner R.; Maltais L. J. (2010) Recommended nomenclature for five mammalian carboxylesterase gene families: human, mouse, and rat genes and proteins. Mamm. Genome 21, 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian J.; Quiroga A. D.; Li L.; Lehner R. (2012) Ces3/TGH deficiency improves dyslipidemia and reduces atherosclerosis in Ldlr–/– mice. Circ. Res. 111, 982–990. [DOI] [PubMed] [Google Scholar]
- Crow J. A.; Middleton B. L.; Borazjani A.; Hatfield M. J.; Potter P. M.; Ross M. K. (2008) Inhibition of carboxylesterase 1 is associated with cholesteryl ester retention in human THP-1 monocyte/macrophages. Biochim. Biophys. Acta 1781, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon R. G. (2012) Structural identification and cardiovascular activities of oxidized phospholipids. Circ. Res. 111, 930–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieur X.; Roszer T.; Ricote M. (2010) Lipotoxicity in macrophages: evidence from diseases associated with the metabolic syndrome. Biochim. Biophys. Acta 1801, 327–337. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Mechoulam R. (2011) Is lipid signaling through cannabinoid 2 receptors part of a protective system?. Prog. Lipid Res. 50, 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A.; Meher A. K.; Sharma P. R.; Lee M. Y.; Doran A. C.; Johnstone S. R.; Elliott M. R.; Gruber F.; Han J.; Chen W.; Kensler T.; Ravichandran K. S.; Isakson B. E.; Wamhoff B. R.; Leitinger N. (2010) Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 107, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M.; Matthews A.; Mangum L. (2014) Chemical atherogenesis: role of endogenous and exogenous poisons in disease development. Toxics 2, 17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Schwabe R. F.; DeVries-Seimon T.; Yao P. M.; Gerbod-Giannone M. C.; Tall A. R.; Davis R. J.; Flavell R.; Brenner D. A.; Tabas I. (2005) Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 280, 21763–21772. [DOI] [PubMed] [Google Scholar]
- Feng B.; Yao P. M.; Li Y.; Devlin C. M.; Zhang D.; Harding H. P.; Sweeney M.; Rong J. X.; Kuriakose G.; Fisher E. A.; Marks A. R.; Ron D.; Tabas I. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792. [DOI] [PubMed] [Google Scholar]
- Wang R.; Borazjani A.; Matthews A. T.; Mangum L. C.; Edelmann M. J.; Ross M. K. (2013) Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry 52, 7559–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon R. G.; Gu X. (2011) Critical insights into cardiovascular disease from basic research on the oxidation of phospholipids: the gamma-hydroxyalkenal phospholipid hypothesis. Chem. Res. Toxicol. 24, 1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. A.; Bittles V.; Herring K. L.; Borazjani A.; Potter P. M.; Ross M. K. (2012) Inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by chlorpyrifos oxon, paraoxon and methyl paraoxon. Toxicol. Appl. Pharmacol. 258, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borazjani A.; Edelmann M. J.; Hardin K. L.; Herring K. L.; Crow J. A.; Ross M. K. (2011) Catabolism of 4-hydroxy-2-trans-nonenal by THP1 monocytes/macrophages and inactivation of carboxylesterases by this lipid electrophile. Chem. Biol. Interact. 194, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritharides L.; Christian A.; Stoudt G.; Morel D.; Rothblat G. H. (1998) Cholesterol metabolism and efflux in human THP-1 macrophages. Arterioscler., Thromb., Vasc. Biol. 18, 1589–1599. [DOI] [PubMed] [Google Scholar]
- Xie S.; Borazjani A.; Hatfield M. J.; Edwards C. C.; Potter P. M.; Ross M. K. (2010) Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: role of carboxylesterases 1 and 2. Chem. Res. Toxicol. 23, 1890–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M.; Marcel Y. L. (2012) Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler., Thromb., Vasc. Biol. 32, 575–581. [DOI] [PubMed] [Google Scholar]
- Alessio M.; De Monte L.; Scirea A.; Gruarin P.; Tandon N. N.; Sitia R. (1996) Synthesis, processing, and intracellular transport of CD36 during monocytic differentiation. J. Biol. Chem. 271, 1770–1775. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Bie J.; Wang J.; Marqueen S. A.; Ghosh S. (2012) Identification of a novel intracellular cholesteryl ester hydrolase (carboxylesterase 3) in human macrophages: compensatory increase in its expression after carboxylesterase 1 silencing. Am. J. Physiol.: Cell Physiol. 303, C427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman J. L.; Cravatt B. F. (2013) Chemical probes of endocannabinoid metabolism. Pharmacol. Rev. 65, 849–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. A.; Bittles V.; Borazjani A.; Potter P. M.; Ross M. K. (2012) Covalent inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by the carbamates JZL184 and URB597. Biochem. Pharmacol. 84, 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S. (2000) Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol. Genomics 2, 1–8. [DOI] [PubMed] [Google Scholar]
- Borazjani A.; Edelmann M. J.; Hardin K. L.; Herring K. L.; Crow J. A.; Ross M. K. (2011) Catabolism of 4-hydroxy-2-trans-nonenal by THP1 monocytes/macrophages and inactivation of carboxylesterases by this lipid electrophile. Chem.–Biol. Interact. 194, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S.; Wang C.; Yu K.; Kuzin A. P.; Richter F.; Lew S.; Miklos A. E.; Matthews M. L.; Seetharaman J.; Su M.; Hunt J. F.; Cravatt B. F.; Baker D. (2014) Design of activated serine-containing catalytic triads with atomic-level accuracy. Nat. Chem. Biol. 10, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M.; Franklin V.; Mak E.; Liao X.; Tabas I.; Marcel Y. L. (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 13, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M. K.; Crow J. A. (2007) Human carboxylesterases and their role in xenobiotic and endobiotic metabolism. J. Biochem. Mol. Toxicol. 21, 187–196. [DOI] [PubMed] [Google Scholar]
- Igarashi M.; Osuga J.; Uozaki H.; Sekiya M.; Nagashima S.; Takahashi M.; Takase S.; Takanashi M.; Li Y.; Ohta K.; Kumagai M.; Nishi M.; Hosokawa M.; Fledelius C.; Jacobsen P.; Yagyu H.; Fukayama M.; Nagai R.; Kadowaki T.; Ohashi K.; Ishibashi S. (2010) The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ. Res. 107, 1387–1395. [DOI] [PubMed] [Google Scholar]
- Sakai K.; Igarashi M.; Yamamuro D.; Ohshiro T.; Nagashima S.; Takahashi M.; Enkhtuvshin B.; Sekiya M.; Okazak H.; Osuga J. I.; Ishibashi S. (2014) Critical role of neutral cholesterol ester hydrolase 1 in cholesterol ester hydrolysis in murine macrophages. J. Lipid Res. 55, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchebner M.; Pfeifer T.; Rathke N.; Chandak P. G.; Lass A.; Schreiber R.; Kratzer A.; Zimmermann R.; Sattler W.; Koefeler H.; Frohlich E.; Kostner G. M.; Birner-Gruenberger R.; Chiang K. P.; Haemmerle G.; Zechner R.; Levak-Frank S.; Cravatt B.; Kratky D. (2010) Cholesteryl ester hydrolase activity is abolished in HSL–/– macrophages but unchanged in macrophages lacking KIAA1363. J. Lipid Res. 51, 2896–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Song J.; St Clair R. W.; Ghosh S. (2007) Stable overexpression of human macrophage cholesteryl ester hydrolase results in enhanced free cholesterol efflux from human THP1 macrophages. Am. J. Physiol.: Cell Physiol. 292, C405–412. [DOI] [PubMed] [Google Scholar]
- Gilham D.; Lehner R. (2005) Techniques to measure lipase and esterase activity in vitro. Methods 36, 139–147. [DOI] [PubMed] [Google Scholar]
- Bencharit S.; Edwards C. C.; Morton C. L.; Howard-Williams E. L.; Kuhn P.; Potter P. M.; Redinbo M. R. (2006) Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J. Mol. Biol. 363, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K. J.; Tabas I. (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.