

Abstract

Base propenals are products of the reaction of DNA with oxidants such as peroxynitrite and bleomycin. The most reactive base propenal, adenine propenal, is mutagenic in Escherichia coli and reacts with DNA to form covalent adducts; however, the reaction of adenine propenal with protein has not yet been investigated. A survey of the reaction of adenine propenal with amino acids revealed that lysine and cysteine form adducts, whereas histidine and arginine do not. Nε-Oxopropenyllysine, a lysine–lysine cross-link, and S-oxopropenyl cysteine are the major products. Comprehensive profiling of the reaction of adenine propenal with human serum albumin and the DNA repair protein, XPA, revealed that the only stable adduct is Nε-oxopropenyllysine. The most reactive sites for modification in human albumin are K190 and K351. Three sites of modification of XPA are in the DNA-binding domain, and two sites are subject to regulatory acetylation. Modification by adenine propenal dramatically reduces XPA’s ability to bind to a DNA substrate.
Introduction
The major reactive aldehyde produced during peroxidation of polyunsaturated fatty acids, malondialdehyde (MDA), reacts with nucleophilic sites of DNA and protein to form oxopropenyl adducts. Reaction of MDA with DNA produces primarily 3-(2-deoxy-β-d-erythro-pentofuranosyl)pyrimido[1,2-α]purine-10(3H)-one (M1dG) and lesser amounts of 2′-oxopropenyl-deoxyadenosine (Figure 1).1 M1dG is mutagenic in vitro and in vivo, and 2′-oxopropenyl-deoxyadenosine alters DNA polymerase activity.2−4
Figure 1.

Schematic of base propenal and MDA production. Both species can react with dA or dG, resulting in oxypropenylation of the base. M1dG is subsequently formed by ring closure of the oxypropenylated dG.
The reaction of MDA with protein occurs predominantly at lysine residues under physiological conditions.5−9 Our understanding of the detailed chemistry and physiological consequences of MDA–protein adduct formation is rudimentary, as the exact targets and sites of adduction are largely unknown. However, a particularly interesting effect of MDA exposure in human cells is the inhibition of nucleotide excision repair (NER), which is associated with increased sensitivity to the mutagenicity of ultraviolet light and benzo[a]pyrene dihydrodiolepoxide.10 NER is the primary process for repair of bulky DNA adducts, and in humans, NER deficiencies result in xeroderma pigmentosum, a spectrum of disorders characterized by hypersensitivity to sunlight, neurological degeneration, and a greatly increased incidence of cancer.11 Although not verified experimentally, inhibition of NER by MDA was proposed to arise from loss of function of MDA-modified NER proteins.10 A potential MDA target is the NER scaffold protein, xeroderma pigmentosum complementation group A (XPA), which binds to the damaged DNA substrate via a C-terminal basic cleft comprising multiple lysine residues.12,13 XPA coordinates recruitment and assembly of the enzymes that repair the damaged DNA, and mutations in its DNA-binding domain are associated with the most severe clinical XP phenotypes.11
Oxopropenyl adducts to DNA and protein can also be formed through reaction with base propenal, a reactive aldehyde produced by oxidant-mediated hydrogen abstraction from the 4′-position of DNA bases (Figure 1).14−17 Base propenals are approximately 100-fold more reactive to DNA than MDA, and experiments in which Escherichia coli strains of varying membrane composition were exposed to oxidants demonstrated that base propenals are more important than MDA as a source of M1dG.18 Currently, very little is known about potential protein targets of base propenal adduction; however, as a highly reactive form of DNA damage, base propenals are ideally situated to oxopropenylate DNA-binding proteins such as XPA. This has led us to hypothesize that base propenal-mediated protein damage contributes to toxicity and mutagenicity associated with oxidative or electrophile stress. To test this hypothesis, a comprehensive evaluation of base propenal’s ability to directly damage proteins is required. Adenine propenal is known to react with glutathione via Michael addition to form glutathionylpropenal and a glutathione–adenine propenal cross-link,19 but, to date, no detailed investigation of the reaction of any base propenal with amino acids or proteins has been reported.
One approach to evaluate the protein adduction potential of an electrophile is to identify all sites of modification and the structures of those modifications using a model protein. A frequently used target for this purpose is human serum albumin (HSA).20,21 Albumin is highly abundant and has affinity for a broad range of ligands, a relatively long half-life, and the potential for noninvasive sampling. Thus, it is an ideal candidate for the global qualitative and quantitative evaluation of exposure to electrophiles of endogenous or exogenous origin (the exposome). In fact, in a large multilaboratory comparison of biomarkers of in vivo oxidant generation, the level of carbonylated plasma protein (i.e., albumin) was one of only two markers that correlated with oxidant exposure and tissue pathology, and it was the only marker that was effective longer than 6 h after exposure.22 To exploit albumin’s potential as a biomarker, however, requires a more complete understanding of its major sites of adduction and determinants of their reactivity to a range of electrophiles. For example, whereas much attention has been directed to HSA’s highly reactive Cys-34 as a sensor of oxidant damage,23 the major site of oxopropenylation by MDA is Lys-525.7
Here, we report the application of a recently described mass spectrometric approach24 to identify adenine propenal modifications of HSA and XPA. Our data demonstrate that, like MDA, adenine propenal primarily reacts at protein lysine residues; however, the major site of attack on HSA is distinct from that of MDA. We also report that adenine propenal modifies lysine residues of XPA, including residues within its DNA-binding domain and residues reported to be sites of regulation via SIRT1-mediated deacetylation.12,13,25 Biophysical analysis confirms that modification of XPA by adenine propenal decreases its affinity for damaged DNA.
Experimental Procedures
Materials
HSA was obtained from Abcam (Cambridge, MA). N-α-Acetyllysine, trichloroacetic acid (TCA), dithiothreitol, iodoacetate (Sigma Ultra), and NaCNBH3 were obtained from Sigma-Aldrich (St. Louis, MO). Trypsin (gold) was obtained from Promega (Madison, WI). 2,2,2-Trifluoroethanol was obtained from Acros Organics (Pittsburgh, PA).
Characterization of Compounds
NMR experiments were acquired using a 14.0 T Bruker magnet equipped with a Bruker AV-III console operating at 600.13 MHz. All spectra were acquired in 3 mm NMR tubes using a Bruker 5 mm TCI cryogenically cooled NMR probe. Chemical shifts were referenced internally to D2O (4.70 ppm), which also served as the 2H lock solvent. For 1D 1H NMR, experiments were acquired with presaturation in order to suppress the residual H2O signal, and typical experimental conditions included 32K data points, 13 ppm sweep width, a recycle delay of 1.5 s, and 32 scans. For 2D 1H–1H COSY, presaturation was also performed, and experimental conditions included a 2048 × 1024 data matrix, 13 ppm sweep width, recycle delay of 1.5 s, and 8 scans per increment. The data were processed using squared sinebell window function, symmetrized, and displayed in magnitude mode. Multiplicity-edited 1H–13C HSQC experiments were acquired using a 1024 × 128 data matrix, a J(C–H) value of 145 Hz, which resulted in a multiplicity selection delay of 34 ms, a recycle delay of 1.5 s, and 80 scans per increment along with GARP decoupling on 13C during the acquisition time (150 ms). The data were processed using a p/2 shifted squared sine window function and displayed with CH/CH3 signals phased positive and CH2 signals phased negative. J1(C–H) filtered 1H–13C HMBC experiments were acquired using a 2048 × 128 data matrix, a J(C–H) value of 9 Hz for detection of long-range couplings resulting in an evolution delay of 55 ms, J1(C–H) filter delay of 145 Hz (34 ms) for the suppression of one-bond couplings, a recycle delay of 1.5 s, and 144 scans per increment. The HMBC data were processed using a p/2 shifted squared sine window function and displayed in magnitude mode. Compounds were greater than 90% pure, and mass spectral analysis was accomplished on an Applied Biosystems 3200 Q trap mass spectrometer (MDS Sciex). The mass spectrometer was equipped with an electrospray source and operated in positive ion mode.
Synthesis of Adenine Propenal
Adenine propenal was synthesized as previously described.23,26 NMR data were consistent with literature values. 1H NMR (DMSO-d6): δ (ppm) 7.17 (dd, J1 = 7.88 Hz, J2 = 14.4 Hz, 1H), 7.55 (br s, 2H), 8.27 (s, 1H), 8.35 (d, J = 14.4 Hz, 1H), 8.63 (s, 1H), 9.67 (d, J = 7.88 Hz, 1H). 13C NMR (DMSO-d6): δ (ppm) 119.2, 120.5, 136.4, 142.8, 147.6, 151.4, 158.2, 190.8.
Preparation of the Sodium Salt of Malondialdehyde (MDA)
Sodium malondialdehyde was synthesized as previously described.28 NMR data were consistent with literature values. 1H NMR (D2O): δ (ppm) 9.15 (d, J = 10.0 Hz, 2H), 5.73 (t, J = 10.0 Hz, 1H). 13C NMR (D2O): δ (ppm) 193.3, 110.7.
Preparation of Nε-Oxopropenyl-N-α-acetyllysine (3)
The sodium salt of malondialdehyde (MDA) (100 mg, 1.06 mmol) was added to a solution of N-α-acetyllysine (200 mg, 1.06 mmol) in 5 mL of ammonium acetate buffer (pH 4.5). The reaction was then stirred at room temperature for 15 h. The reaction mixture was lyophilized, and the residue was purified by column chromatography (EtOAc/MeOH, 5:1) on silica gel (60 Å porosity, 40–63 μM particle size). 1H NMR (D2O): δ (ppm) 1.33 (m, 2H), 1.56–1.61 (m, 3H), 1.71 (m, 1H), 1.93 (s, 3H), 2.91 (t, J = 7.3 Hz, 2H), 4.05 (m, 1H), 5.31 (m, 1H), 7.44 (d, J = 13.0 Hz, 1H), 8.59 (d, J = 8.5 Hz, 1H). 13C NMR (D2O): δ (ppm) 22.0, 23.4, 27.2, 32.0, 39.3, 55.3, 86.6, 163.7, 173.7, 179.1, 191.2. ESI-MS: [MH]+, m/z 243.4. (See Supporting Information Figures S5–S7.)
Preparation of the Lysine–Lysine Cross-Link (4)
The sodium salt of MDA (60 mg, 0.319 mmol) was added to a solution of N-α-acetyllysine (15 mg, 0.159 mmol) in 2 mL of anhydrous methanol. The mixture was heated at reflux, and the progress of the reaction was monitored by UV–vis spectroscopy. The product was then purified by column chromatography (EtOAc/MeOH, 5:1) on silica gel (60 Å porosity, 40–63 μM particle size) and HPLC. HPLC chromatography was carried out on a Luna C18 column (250 × 4.60 mm, 5 μm) at a flow rate of 1.25 mL/min. The eluting solvents included solvent A (10 mM ammonium acetate) and solvent B (acetonitrile). The following solvent gradient was used: 0–10.5 min linear gradient from 5% B to 20% B, followed by a 2 min linear gradient to 80% B, hold for 6.50 min at 80% B, followed by a 1 min linear gradient to the initial conditions of 5% B. The product was observed with a retention time of 6.1 min. 1H NMR (D2O): δ (ppm) 1.30 (m, 4H), 1.59–1.61 (m, 6H), 1.73 (m, 2H), 1.88 (s, 6H), 3.22 (m, 2H), 3.30 (t, J = 7.3 Hz, 2H), 4.08 (m, 2H), 5.45 (m, 1H), 7.49 (d, J = 11.5 Hz, 1H), 7.56 (d, J = 11.5 Hz, 1H). 13C NMR (D2O): δ (ppm) 21.3, 21.8, 25.7, 30.2, 42.0, 53.7, 88.5, 158.9, 160.5, 177.5, 178.9. ESI-MS: [MH]+, m/z 413.3. (See Supporting Information Figures S8–S10.)
HPLC Analysis of N-α-Acetyllysine and Adenine Propenal Reactions
N-α-Acetyllysine (various concentrations) was mixed with 10 mM adenine propenal in 50 μL reaction mixtures containing 10 mM NaCl and 10 mM 4-morpholinepropanesulfonic acid (MOPS) (pH 6.5). At 1 h intervals, aliquots of each reaction mixture were injected onto a Waters HPLC system equipped with a 717 Plus autosampler, 1525 Binary HPLC pump, and 2996 photodiode array detector. Analytes were separated using a Phenomenex Polar-RP Synergi HPLC column, 75 cm × 2 mm with 4 μm particle size, at a flow rate of 0.6 mL/min. The mobile phase solvents were H2O + 0.05% acetic acid (v/v, solvent A) and 3:1 MeOH/ACN + 0.05% acetic acid (v/v, solvent B) delivered in a 19 min gradient consisting of the following: 0–10.5 min, 5–20% B; 10.5–12.5 min, 20–80% B; 12.5–16 min, 80% B; 16–19 min, 5% B. Data were analyzed using Empower software, and peak areas were manually integrated. Eluting peaks were collected and analyzed by MS (see below).
MS Analysis of the Reaction Products of Adenine Propenal and N-α-Acetyllysine
Reactions were performed as described above and analyzed using a Thermo Accela HPLC pump (Thermo Fisher Scientific) and HTC PAL autosampler (LEAP Technologies) in-line with a Thermo Quantum triple quadrupole mass spectrometer (Thermo Fisher Scientific). The Quantum was equipped with an electrospray source operated in Q1 full scan, positive ion mode. Q1 was scanned from 100–600 amu in 0.45 s. Analytes were chromatographically separated on a Synergi Polar-RP HPLC column (Phenomenex, 7.5 × 0.2 cm, 4 μm particle size) using the following gradient: 0–10.5 min, 5–20% B; 10.5–12.5 min, 20–80% B; 12.5–16 min, 80% B; 16–17 min, 5% B. The mobile phase solvents were H2O + 0.05% acetic acid (v/v, solvent A) and acetonitrile + 0.05% acetic acid (v/v, solvent B), and they were delivered at a flow rate of 0.6 mL/min. All data were acquired and analyzed using Thermo Xcalibur software.
Spectrophotometric Quantification of the Lysine–Lysine Cross-Link (4) in Reaction Mixtures of N-α-Acetyllysine and Adenine Propenal
Solutions containing known concentrations of synthetic 3 and 4 were used to measure extinction coefficients of each compound. The value obtained for both compounds (33 600 ± 200 M–1cm–1) was the same within experimental error; however, the wavelengths of maximal absorbance were different (280 nm for 3 and 300 nm for 4) (Figure S1).
Adenine propenal (10 mM) was incubated with increasing concentrations of N-α-acetyllysine (1, 2, 5, 10, 50, and 100 mM) in reaction mixtures containing 10 mM NaCl and 10 mM MOPS (pH 6.5). The mixtures were incubated for 2 h at room temperature and then diluted 1:1000 in H2O. The absorbance at 300 nm of each dilution was then obtained using a DU800 spectrophotometer (Beckman Coulter), and the experimental extinction coefficient of 4 was used to estimate its concentration in the reaction mixture. Note that, for reaction mixtures containing >10 mM N-α-acetyllysine, complete consumption of adenine propenal eliminates concerns of its contribution to the absorbance of the final reaction mixture. The adenine product does not absorb significantly at 300 nm (Figure S1). On the basis of its full UV spectrum (Figure S1), the extinction coefficient of 3 at 300 nm is estimated to be 7.8 × 103 M–1 cm–1. The presence of this product at a concentration equal to approximately 10% of the concentration of 4, as found in the reaction mixtures, indicates that its contribution to the absorbance at 300 nm will be no more than 3%. Consequently, the absorbance at 300 nm can be used to estimate the concentration of 4 with reasonable accuracy.
Reaction of N-α-Acetyllysine with Adenine Propenal under Reducing Conditions
N-α-Acetyllysine (10 mM) was incubated with adenine propenal (10 mM) in 10 mM MOPS (pH 6.5) containing 10 mM NaCl in a 200 μL reaction mixture for 30 min. After incubation, 50 mM NaCNBH3 (in H2O) (Sigma-Aldrich) was added for an additional 30 min, and samples were then analyzed using LC-MS as described below.
Analysis of Reduced Intermediate (6) by LC-MS
LC-MS analysis was performed on the Thermo Quantum triple quadrupole mass spectrometer. Here, the instrument was operated in Q1 full-scan negative ion mode using electrospray ionization. Q1 was scanned over the range 100–400 amu over 0.5 s. Chromatography was carried out on a Synergi Max-RP column (Phenomenex, 7.5 × 0.2 cm, 5 μm particle size) using the following gradient: 0–5 min linear gradient from 2% B to 50% B, 5–7 min at 50% B, 7–10 min 50% B to 2% B. The mobile phase solvents were H2O + 0.05% formic acid (v/v, solvent A) and 3:1 CH3CN/MeOH + 0.05% formic acid (v/v, solvent B), and they were delivered at a flow rate of 400 μL/min. All data were acquired and analyzed using Thermo Xcalibur software. Using this chromatographic system, the reduced intermediate eluted at approximately 2.9 min.
Analysis of the Reaction of N-Acetylcysteine with Adenine Propenal
N-Acetylcysteine (200 μM) was combined with 50 μM adenine propenal in 50 mM sodium phosphate buffer, pH 7.4, and incubated for 6 h at 37 °C. Unreduced sample was immediately frozen. NaBH4 (50 mM) (Sigma-Aldrich) was used to reduce samples as indicated. Analytes were separated on a Supelco Ascentis C18 column (50 × 2.1 mm, 3 μm) with buffers A and B comprising 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The gradient was as follows: 1–3 min, 1% B; 3–7 min, 98% B; 7–11 min, 98% B; 11–17 min, 1% B. Samples were analyzed on a ThermoFinnigan TSQ Quantum mass spectrometer with ESI source interfaced to a ThermoFinnigan MS pump plus and autosampler plus (Thermo, San Jose, CA). N-Acetylcysteine-adducted species were analyzed with data-dependent scanning enabled in negative ion mode, scanning from m/z 150–650 over 0.5 s. The top two m/z peaks from each MS scan were fragmented with 10 eV collision energy for 0.5 s.
LC-MS/MS Analysis of HSA Modification by Adenine Propenal
HSA (15 μM) (Abcam, Cambridge, MA) was incubated with increasing amounts of adenine propenal (0.15, 0.38, 1.5, 3, and 7 mM), representing a molar excess of adenine propenal to HSA of 10, 25, 100, 250, and 500, respectively. Reactions were incubated for 6 h at room temperature in a buffer containing 10 mM NaCl and 10 mM MOPS (pH 6.5). Following incubation, samples were precipitated with 25% trichloroacetic acid (Sigma-Aldrich) on ice for 1 h, washed with cold acetone, dried, and reconstituted in 50 mM Tris (pH 8.0) containing 50% 2,2,2-trifluoroethanol (Acros Organics, Pittsburgh, PA). Samples were reduced with DTT (Sigma-Aldrich), carbamidomethylated with iodoacetamide (Sigma-Alrich), and diluted 5-fold with 100 mM Tris (to obtain a final solution containing 10% 2,2,2-trifluoroethanol). Following dilution, samples were digested with sequencing-grade trypsin (Promega, Madison, WI) overnight, acidified, and diluted in 0.1% formic acid. The resulting solutions of trypsin-generated peptides were loaded onto a capillary reverse-phase analytical column (360 μm o.d. × 100 μm i.d.) using an Eksigent NanoLC Ultra HPLC and autosampler. The 20 cm analytical column was directly packed into a laser-pulled emitter tip using Jupiter C18 reverse-phase medium (3 μm beads, 300 Å pore size, Phenomenex). Mobile phase solvents consisting of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B) at a flow rate of 500 nL/min were used to elute the peptides. The 90 min gradient consisted of the following: 0–10 min, 2% B; 10–50 min, 2–35% B; 50–60 min, 35–95% B; 60–65 min, 95% B; 65–70 min 95–2% B; 70–90 min, 2% B. Mass analysis of eluting peptides was performed on an LTQ Orbitrap XL mass spectrometer (Thermo Scientific), equipped with a nanoelectrospray ionization source with detection in positive ion mode. The LTQ Orbitrap was operated using a data-dependent method, enabling dynamic exclusion. Full scan (m/z 400–2000) spectra were acquired with the Orbitrap (resolution 60 000), and the five most abundant ions in each MS scan were selected for fragmentation in the LTQ. An isolation width of 2 m/z, 30 ms activation time, and collision energy normalized to 35% were used to generate MS2 spectra. Dynamic exclusion settings allowed for a repeat count of 2 within a 10 s duration, with exclusion duration time set to 15 s. For identification of modified peptides, tandem mass spectra were searched with SEQUEST (Thermo Fisher Scientific) against a human subset database created from the UniProtKB protein database (www.uniprot.org). Variable modifications of +57.0214 on Cys (carbamidomethylation), +15.9949 on Met (oxidation), and +54.0105 on Lys or Cys (oxypropenylation) were included for database searching. Search results were assembled with Scaffold 3.0 (Proteome Software) using threshold filtering criteria consisting of 95% peptide probability to achieve a peptide false discovery rate estimate of ≤0.2%. Sites of modification were validated by manual interrogation of tandem mass spectra using Xcalibur 2.1 Qual Browser software (Thermo Scientific). Calculations of mass errors (in ppm) of precursor ions mass analyzed in the Orbitrap were performed using the following equation: ((theoretical mass – observed mass)/theoretical mass) × 106. To obtain theoretical masses, the Protein Prospector MS-Product tool was used (v.5.10.4. UCSF). Analysis of cross-links was performed in a similar manner with inclusion of +36 on Lys for database searching.
Proteolytic Digestion of HSA Treated with Adenine Propenal for Lysine–Lysine Cross-Link (4) Identification
Purified HSA (0.75 μM) was incubated with increasing concentrations of adenine propenal in reaction mixtures containing 10 mM NaCl and 10 mM MOPS (pH 6.5) for 6 h at room temperature. Following incubation, the protein was heated at 95 °C for 10 min, cooled to room temperature, and then treated with 1 mg of Pronase (Calbiochem, Gibbstown, NJ) per mg of protein at 37 °C for 24 h. Pronase was inactivated by heating at 95 °C for 10 min, followed by cooling to room temperature, and treating with 1 μL of aminopeptidase M (Calbiochem, Gibbstown, NJ) at 37 °C for 24 h. A final incubation at 95 °C for 10 min inactivated the aminopeptidase M, and the samples were then cooled to room temperature in preparation for adduct purification.
Purification of Lysine–Lysine Cross-Links (4) and Analysis by MS
For use as an internal standard, [3H]-[14C]-4 was prepared by reaction of MDA with [3H]-lysine containing [14C]-lysine as a tracer and quantified using a method similar to that described for the [3H]-[14C]-lysine-lactam internal standard as described.29 An Oasis HLB cartridge (1 cm3) (Waters, Milford, MA) was equilibrated with 2 mL of methanol followed by 2 mL of H2O. Following addition of [3H]-[13C]-4, the sample was loaded onto the cartridge using gravity flow. The cartridge was washed with 1 mL of H2O, and 4 was eluted with 3 mL 1:1 methanol/ethyl acetate and dried to 100 μL under N2 gas. The sample was then diluted in 0.1% formic acid solution to a final volume of 1 mL and filtered using a 0.22 μm nylon Spin-x centrifuge tube spun at 6000 rpm for 5 min.
Samples were purified using a Thermo Scientific Aquasil C18 5 μm reverse-phase column using liquid scintillation counting to monitor elution of the radioactive [3H]-[13C]-lysine–lysine cross-link standard. The internal standard [3H]-[14C]-lysine–lysine cross-link was prepared by reaction of MDA with [3H]-lysine containing [14C]-lysine as a tracer and quantified using a method similar to that described for the [3H]-[14C]-lysine-lactam internal standard.28 For quantitation, the transition between m/z 341 and m/z 306 was monitored; it corresponds to the loss of one molecule of water and one molecule of ammonia. Portions of samples containing the standard were then concentrated using 1 cc Oasis HLB columns and dried under nitrogen. The purified samples were analyzed using a Thermo Scientific TSQ Vantage quadrupole mass spectrometer equipped with Xcalibur software for data collection and manipulation. All measured MDA-adduct signal in each sample was divided by the signal for the [3H]-[13C]-lysine–lysine cross-link standard of known concentration to quantify experimental results. These methods will be published in detail elsewhere.
Proteomics Analysis of XPA Modification by Adenine Propenal
XPA was expressed and purified as described.30 To modify XPA, 3.2 μM of purified protein was incubated with 500× (1.6 mM) of adenine propenal for 6 h at room temperature in a buffer containing 10 mM NaCl and 10 mM MOPS (pH 6.5). Modifications were identified as described above for HSA.
Fluorescence Anisotropy DNA Binding Assay
A high-throughput fluorescence anisotropy assay was used to measure the DNA-binding activity of XPA.31,32 The substrate for this assay was a fluorescein-labeled Y-shaped ssDNA–dsDNA junction substrate containing 8 base pairs of duplex and 12 nucleotide noncomplementary ssDNA overhangs at both the 3′ and 5′ ends. XPA was modified with NHS–biotin as described previously.33 Modified or mock-treated XPA protein and the DNA substrate were diluted in binding buffer (20 mM HEPES, pH 7.9, 75 M KCl, 5 mM MgCl2, 5% glycerol, 1 mM DTT). DNA substrate was added to a final concentration of 50 nM. Fluorescence anisotropy was measured using a Synergy H1 plate reader (λEx = 485 nm, λEm = 528 nm).30 Binding measurements were acquired in triplicate for each DNA substrate. Apparent dissociation constants (Kd) were determined for each individual titration by plotting fluorescence anisotropy against protein concentration and fitting to a simple two-state binding model with KaleidaGraph (v4.03) software.
Results
Adenine Propenal Modifies N-α-Acetyllysine, Forming Nε-Oxopropenyl-N-α-acetyllysine (3) and Lysine–Lysine Cross-Links (4)
Adenine propenal was reacted with N-α-acetyllysine, N-α-acetylhistidine, N-α-acetylarginine, or N-α-acetylcysteine in 10 mM NaCl buffered with 10 mM MOPS (pH 6.5). Reaction progress was monitored hourly by HPLC using a C18 reverse-phase column for product separation. Adenine propenal eluted at ∼8 min and displayed a maximum absorbance of 260 nm (Figures 2 and S1). Reaction with N-α-acetyllysine resulted in the appearance of three new chromatographic peaks at 1.0, 4.8, and 5.3 min, which were analyzed by UV/vis, NMR, and MS (Figures 2 and S1). The peak eluting at 1 min displayed a UV absorbance maximum at 260 nm and an m/z of 136, corresponding to protonated adenine ([M + H]+) (1). The 4.8 min peak displayed a UV maximum at 280 nm. On the basis of NMR analysis, this product was identified as 3, and LC-MS analysis gave an m/z of 243, which corresponds to the protonated species ([M + H]+). The 5.3 min peak, with a maximum absorbance at 300 nm and an m/z of 413, which corresponds to the protonated species ([M + H]+), was identified as 4 by NMR analysis. The total amount of product formed was unaffected by variations in pH over a range of 6.5 to 9.5.
Figure 2.

HPLC analysis of an N-α-acetyllysine and adenine propenal reaction mixture. N-α-Acetyllysine (50 mM) was incubated with adenine propenal (10 mM) in a 50 μL reaction mixture for 2 h at room temperature. Following incubation, 4 μL of the reaction mixture was analyzed by HPLC. The column eluate was monitored at 260 nm (A) or 300 nm (B). Analytes present in each peak were subjected to MS, UV, and NMR spectrometry. Numbers correspond to the identified products, represented in (C).
Chemical synthesis of 3 and 4 yielded standards that verified the product structure assignments. The molar extinction coefficients of the pure compounds (ε = 33.6 × 103 M–1 cm–1 at 280 nm for 3 and ε = 33.6 × 103 M–1 cm–1 at 300 nm for 4) were similar to that of adenine propenal (ε = 32.5 × 103 M–1 cm–1 at 260 nm). HPLC analysis of reaction mixtures of N-α-acetyllysine and adenine propenal using diode-array detection enabled direct comparison of peak areas for each compound measured at its optimal wavelength (Figure 3). The essentially identical absorbance coefficients of 3 and 4 allowed peak areas to be used to estimate relative product quantity. These comparisons indicated that the major product formed from the reaction was 4, with much lower quantities of 3.
Figure 3.

HPLC analysis of product formation in the reaction of adenine propenal and N-α-acetyllysine. The indicated amounts of N-α-acetyllysine (NAL) were incubated with 10 mM adenine propenal, and reactions were monitored hourly for 9–10 h. Peaks corresponding to 3 and 4 were monitored at 280 and 300 nm, respectively, by diode-array detection. Data show the mean ± SD of three determinations.
Variation of the amount of N-α-acetyllysine (from 2 to 100 mM) in reaction mixtures while holding the amount of adenine propenal (10 mM) fixed demonstrated that the amount of 3 formed increased with N-α-acetyllysine concentrations from 2 to 10 mM and then decreased somewhat at the higher concentrations. In contrast, the amount of 4 produced increased with N-α-acetyllysine concentrations up to 50 mM. On the basis of the relative peak area, the ratio of 4 to 3 in most reaction mixtures was approximately 10:1. Estimation of the concentration of 4 using absorbance at 300 nm indicated a maximum of ∼9 mM formed in reaction mixtures containing 50–100 mM N-α-acetyllysine (Figure S2). These results are consistent with complete consumption of adenine propenal under those conditions.
Our initial hypothesis for the production of 4 was that it arose by reaction of N-α-acetyllysine with 3, but this reaction did not occur under conditions comparable to those used for the reaction of adenine propenal with N-α-acetyllysine. An alternative reaction scheme, in which an enaminoimine cross-link between adenine propenal and N-α-acetyllysine is first formed and then reacts with H2O to produce 3 or another equivalent of N-α-acetyllysine to form 4, is shown in Figure 4. If this scheme is correct, then our data indicate that, at the concentrations of N-α-acetyllysine and adenine propenal used here, reaction of the proposed intermediate with another molecule of N-α-acetyllysine is more favorable than its reaction with water.
Figure 4.

Proposed scheme for the reaction of adenine propenal with N-α-acetyllysine.
HPLC analysis with UV or in-line NMR was unable to detect the putative intermediate 5 (Figure 4). Therefore, we attempted to trap it by reduction with NaCNBH3. Adenine propenal was incubated with N-α-acetyllysine in 10 mM NaCl/10 mM MOPS (pH 6.5) in the presence of NaCNBH3. We utilized LC-MS analysis of the reaction mixture to detect ions with m/z 360, which corresponds to the partially reduced intermediate (6). A peak corresponding to this mass eluted at 2.9 min, consistent with the transient formation of the adenine propenal adduct to N-α-acetyllysine (Figure 5).
Figure 5.

LC-MS analysis of the reduced reaction intermediate (6). (A) Representative LC-MS chromatogram of 6. In this example, 5 mM N-α-acetyllysine was reacted with 10 mM adenine propenal for 30 min followed by reduction with 50 mM NaCNBH3. (B) Mass spectrum of the parent ion of the reduced intermediate displaying m/z 360 with detection in negative ion mode.
Reactions of adenine propenal with N-α-acetylhistidine and N-α-acetylarginine did not result in the formation of new chromatographic peaks. Reaction with N-α-acetylcysteine resulted in the formation of an oxopropenylated product, which displayed m/z 216, corresponding to the deprotonated molecular ion, and its major fragmentation product, m/z 86.9. Treatment of the reaction mixture with NaBH4 yielded the partially reduced product (m/z 218) and its major fragmentation product (m/z 89) (Figure S3).
Adenine Propenal Reacts Predominantly with Lys-214 and Lys-375 in HSA
To ascertain adenine propenal’s ability to react with protein, we incubated increasing amounts of the electrophile with a fixed amount of purified HSA for 6 h, and the samples were prepared for LC-MS/MS by trichloroacetic acid precipitation, resolubilization of the precipitated protein pellet, reduction and carbamidomethylation of cysteine residues, and proteolytic digestion with sequencing-grade trypsin. The resultant proteolytic peptides were analyzed via LC-MS/MS using an LTQ-Orbitrap Velos mass spectrometer with collision-induced dissociation. Sequence coverage for unmodified HSA was found to be 90%, but exposure to increasing concentrations of adenine propenal reduced coverage to a minimum value of 62% (Table S1). HSA peptides were interrogated for the presence of a +54.0105 mass increase, as would be expected from the addition of an oxopropenyl group. To verify positively identified residues, data were manually examined using Xcalibur software. Extracted ion chromatograms were generated from precursor peptide masses, and theoretical masses calculated from Protein Prospector were compared to observed masses to calculate ppm mass error. For modified precursor peptides displaying a ppm mass error less than 10, MS/MS spectra were interrogated with Xcalibur software. At least 40% of each theoretical b- and y-product ion series was present in the observed product ion spectra of positively identified peptides, and 50% sequence coverage was obtained for each peptide validated. To validate the site of adduction, at least two ions within a product ion series with a +54.0105 adduction mass shift were required. Multiple lysine residues were found to be modified in adenine propenal-exposed HSA, including Lys26, Lys36, Lys161, Lys183, Lys214, Lys219, Lys236, Lys249, Lys257, Lys347, Lys375, Lys402, Lys413, Lys438, Lys549, Lys569, and Lys598. Residues verified to have a +54.0105 adduction mass were mapped to the crystal structure of HSA (PDB ID: 1AO6) and were found to be largely confined to the protein surface (Figure 6A). The 17 modified lysine residues represented 28% of the total lysines within the protein (Table 1). Although we determined that adenine propenal can also form oxopropenylated cysteine, searching for adducts on cysteine residues within HSA did not result in the identification of any modified sites. S-Oxopropenal-N-acetylcysteine was stable to treatment with TCA as well a DTT, which are reagents used for proteomic analysis, so any oxopropenylated cysteine residues on HSA-derived peptides should have survived the workup prior to mass spectrometry.48 Therefore, adenine propenal is acting as a lysine-specific modifier of HSA.
Figure 6.
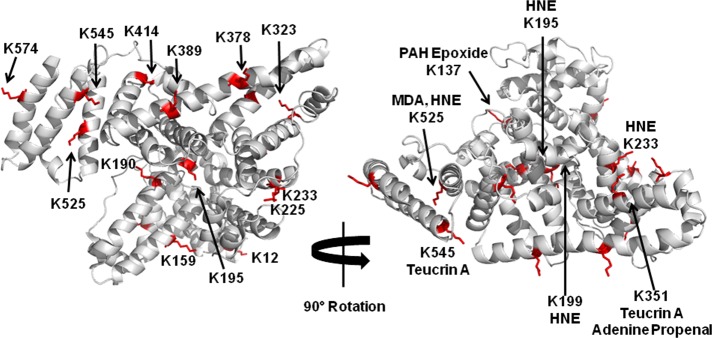
Human serum albumin modification by adenine propenal. (A) Modified lysine residues identified in Table 1 were mapped to the crystal structure of HSA (1AO6). Lysine residues with an additional mass of +54.0105 are indicated in red. (B) The most highly modified residues by individual electrophiles are highlighted with the modifying electrophile noted.
Table 1. Modified Lysine Residues within HSAa.
| molar
ratio adenine propenal:HSA |
||||||
|---|---|---|---|---|---|---|
| residue | peptide | 10:1 | 25:1 | 100:1 | 250:1 | 500:1 |
| Lys-28 (N/A) | DAHK*SEVAHR | ● | ● | ● | ● | |
| Lys-36 (12) | FK*DLGEENFK | ● | ● | ● | ||
| Lys-161 (137) | K*YLYEIAR | ● | ● | ● | ● | |
| Lys-183 (159) | HPYFYAPELLFFAK*R | ● | ● | ● | ● | |
| Lys-214 (190) | LDELRDEGK*ASSAK | ● | ● | ● | ● | ● |
| Lys-219 (195) | LDELRDEGKASSAK*QR | ● | ||||
| Lys-236 (212) | AFK*AWAVAR | ● | ● | ● | ||
| Lys-249 (225) | FPK*AEFAEVSK | ● | ● | ● | ||
| Lys-257 (233) | AEFAEVSK*LVTDLTK | ● | ● | ● | ||
| Lys-347 (323) | NYAEAK*DVFLGMFLYEYAR | ● | ● | |||
| Lys-375 (351) | LAK*TYETTLEK | ● | ● | ● | ● | ● |
| Lys-402 (378) | VFDEFK*PLVEEPQNLIK | ● | ● | ● | ● | |
| Lys-413 (389) | VFDEFKPLVEEPQNLIK* | ● | ||||
| Lys-438 (414) | K*VPQVSTPTLVEVSR | ● | ● | |||
| Lys-549 (525) | K*QTALVELVK | ● | ● | ● | ● | |
| Lys-569 (545) | EQLK*AVMDDFAAFVEK | ● | ● | |||
| Lys-598 (574) | K*LVAASQAALGL | ● | ● | ● | ||
HSA was treated at the indicated molar ratios of adenine propenal and analyzed for lysine residues modified with +54.0105. The presence of each modified lysine in a sample is indicated with ●. The modified lysine in each peptide sequence is indicated with an asterisk. Equivalent lysine residues within the crystal structure of HSA are provided in parentheses.
The use of HSA as a biomarker has primarily been described in the context of electrophilic modifications at Cys34.23 Adenine propenal did not modify Cys34, so we sought to identify the most reactive lysine residue(s) that might be used as a biomarker for modification by adenine propenal. We reduced the levels of adenine propenal relative to protein to a ratio of 10:1, which was the lowest ratio at which modification was observed. Using the interrogation methods described above to verify adduction sites, two lysine residues, K190 and K351, displayed the highest reactivity (Table 1). Mapping of those residues to the crystal structure of HSA revealed that both are located within domain II, a recognized binding region for many compounds (Figure 6A).34
Investigation of Cross-Link Formation in the Reaction of Adenine Propenal with HSA
Decreased HSA sequence coverage observed with increasing concentrations of adenine propenal suggested that adducts impede protein digestion and/or fragmentation required for peptide identification. As lysine–lysine cross-links are formed from the reaction of adenine propenal with N-α-acetyllysine, we hypothesized that at high concentrations of adenine propenal, lysine–lysine cross-links may be induced in HSA. To investigate the presence of cross-links, we interrogated previously acquired proteomics data for a +36 adduct mass, corresponding to an unreduced 3-carbon linkage between lysine residues.31 This analysis did not result in the identification of any lysine–lysine cross-links, which was not surprising considering the decreased sequence coverage and potential hindrance of peptide fragmentation by cross-links. Although cross-linked sites were not identified with these algorithm-based data searching methods, we manually interrogated the data to look for the presence of precursor peptides containing cross-linked residues. To predict lysine residues likely to participate in cross-link formation, we performed in silico analysis of the HSA crystal structure (1AO6). As a typical carbon–carbon bond is approximately 1.5 Å in length, we hypothesized that the 3-carbon cross-link would be ∼5–7 Å long. Flexible, dynamic protein movements in solution may result in cross-link formation between lysines at a distance greater than 7 Å. We therefore examined the crystal structure to identify lysine residue pairs separated by a distance of less than or equal to 20 Å as potential sites of cross-link formation. Following identification of these residues, tryptic peptide sequences were determined from the primary sequence, and theoretical peptide masses containing the cross-link mass of +36 were calculated using Protein Prospector. We then thoroughly interrogated the data manually for the presence of ions of the theoretical precursor peptide masses. No evidence for the presence of cross-link-containing peptides was obtained.
As proteomics-based mass spectrometric analyses were unsuccessful, we employed a different analytical approach to search for lysine–lysine cross-links, in which modified HSA was degraded to the level of amino acids, and the resultant hydrolysate was analyzed by MS. This methodology employed selected reaction monitoring to identify analytes exhibiting a mass transition from m/z 329 to m/z 294, as exhibited by analysis of the lysine–lysine cross-link standard. Addition of a [3H]-[13C]-4 internal standard (m/z 341 to m/z 306 transition) enabled identification of analyte-containing fractions during purification by scintillation counting and quantification by stable isotope dilution. Application of this approach to HSA that had been incubated with adenine propenal resulted in the identification of 30 ± 12 ng cross-link/mg protein in vehicle-treated HSA and 57 ± 17 ng cross-link/mg protein in adenine propenal-treated HSA. These differences were not statistically significant, supporting the conclusion that adenine propenal did not induce intramolecular cross-links in HSA.
Adenine Propenal Oxopropenylates XPA
Having confirmed adenine propenal’s capacity to modify lysine residues in HSA, we investigated its reaction with XPA. Adenine propenal was incubated with XPA for 6 h at room temperature, and the samples were prepared for and analyzed by LC-MS/MS using the same methods as described for HSA. As for HSA, peptides were interrogated for the presence of a +54.0105 mass addition, and rigorous criteria were applied to peptide and site of adduction identification. The data revealed six modified residues, listed in Table 2. Of note, three of these, K145, K183, and K204, are located in the proposed DNA-binding domain of XPA (Figure 7A),12,13 and two, K63 and K67, are known sites of acetylation that reduces XPA’s interaction with replication protein A.25
Table 2. XPA Lysine Modifications Induced by Adenine Propenala.
| residue | peptide |
|---|---|
| Lys-63 | PYSATAAAATGGMANVK*AAPK |
| Lys-67 | AAPK*IIDTGGGFILEEEEEEEQK |
| Lys-86 | IIDTGGGFILEEEEEEEQK*IGK |
| Lys-145 | TEAK*QEYLLK |
| Lys-183 | NPHHSQWGDMK*LYLK |
| Lys-204 | SLEVWGSQEALEEAK*EVR |
Purified XPA was incubated with a 500-fold molar excess of adenine propenal and analyzed for lysine residues modified with +54.0105. The modified lysine in each peptide sequence is indicated with an asterisk.
Figure 7.
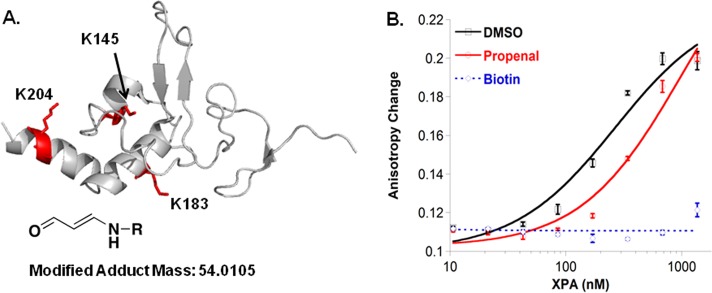
Adenine propenal modifies XPA and reduces its DNA-binding activity. (A) Lysine residues modified with an adduct mass of +54.0105 were mapped to the XPA NMR solution structure (1D4U). (B) Plots of the change in fluorescence anisotropy for a Y-shaped 8/12 ssDNA–dsDNA junction substrate (50 nM) versus added protein treated with DMSO, adenine propenal, or NHS-biotin. Each data point represents the mean ± SD of three titrations.
Modification of residues within the XPA DNA-binding domain has the potential to affect DNA-binding activity. Using a plate-based fluoresence anisotropy assay, the effect of adenine propenal-mediated oxopropenylation on XPA’s affinity for a Y-shaped ssDNA–dsDNA junction substrate was evaluated.32 Fitting of the data for binding of unmodified XPA to the fluorescein-labeled 20-nucleotide substrate provided a dissociation constant (Kd) of 300 ± 30 nM (Figure S4). Binding affinities for the 20-nucleotide substrate were measured for purified XPA incubated with vehicle (DMSO), adenine propenal, or NHS–biotin (biotin N-hydroxysuccinimide ester) (Table 3).33 As expected, incubation of XPA with DMSO had no effect on its DNA-binding activity. In contrast, exposure to NHS–biotin, which efficiently modifies lysine residues and disrupts the DNA-binding activity of many proteins,33,35,36 resulted in a nearly complete loss of XPA’s DNA-binding affinity. XPA modified with adenine propenal bound to the substrate, but with a greater than 3-fold reduction in affinity as compared to that of the unmodified protein (Figure 7B). In fact, oxopropenylation of the protein reduced the affinity to such an extent that it could not be accurately measured; thus, the Kd value reported in Table 3 (1 ± 0.1 μM) is an estimate.
Table 3. Dissociation Constants for XPA Binding to Y-Shaped ssDNA–dsDNA Junction Substratea.
| treatment | Kd (μM) |
|---|---|
| unmodified | 0.30 ± 0.03 |
| DMSO | 0.31 ± 0.03 |
| adenine propenal | 1 ± 0.1 |
| NHS–biotin | n.d. |
Dissociation constants (Kd) were determined by fitting fluorescence anisotropy data to a simple two-state binding model. Kd values represent the mean ± SD of at least three titrations. The biotin-modification of XPA causes such a large loss of DNA-binding affinity that there is no significant change in fluorescence anisotropy; hence, a Kd value could not be determined.
Discussion
Covalent modifications of DNA and proteins by electrophiles produced during oxidative stress have multiple consequences on cellular function, including mutagenesis, stress responses, cellular dysfunction, and ultimately cell death. All of these may contribute to pathological processes such as carcinogenesis, neurodegeneration, atherosclerosis, and chronic inflammation. However, elucidation of the exact mechanisms by which specific macromolecular damage leads to pathology requires a better understanding of the sites and nature of that damage.
The most frequently used biomarker of electrophile damage to protein during oxidative stress is the change in global protein carbonylation.37 Although useful as a general indicator that stress has occurred, this approach does not identify the exact type of modification or the modifying electrophile. Recent developments in highly sensitive MS-based proteomics techniques have allowed for expansion of this approach to the investigation of the specific types of modifications induced and the impact of these modifications on protein activity. The resultant increased ability to identify specific protein modifications induced by a single electrophile enables the search for biomarkers that distinguish between different mechanisms of oxidant damage.
The generation of adenine propenal occurs predominantly in intact DNA, suggesting that most of its targets will likely be DNA-associated proteins in the nucleus or mitochondria. Nevertheless, we initiated our investigation of adenine propenal’s protein-modifying profile, using HSA as the model protein. HSA’s reactivity with electrophilic agents has been studied extensively in the past,7,38−44 allowing us to place adenine propenal-dependent modifications in the context of those existing data. For example, teucrin A was found to preferentially modify Lys351 and Lys545, while 4-hydroxynonenal modifies Lys195, Lys199, Lys233, and Lys525, and polycyclic aromatic hydrocarbon epoxides modify Lys137.43−45 MDA has been shown to modify multiple lysine residues including Lys136, Lys174, Lys240, Lys281, Lys525, and Lys541.7 As MDA and adenine propenal both produce oxopropenyl adducts on lysine residues, it is interesting to note that the major sites of adduction for the two electrophiles are different: Lys190 and Lys351 for adenine propenal versus Lys525 for MDA. These findings suggest that distinct HSA modification patterns may be used as markers for different electrophiles (Figure 6B). Clearly, the use of HSA as a biomarker of electrophile damage is attractive, given its high concentration in the blood and ease of sampling. However, as noted above, adenine propenal generation is largely confined to intact DNA, so it remains to be seen if enough of the electrophile reaches the circulation to allow the modification of HSA to serve as a biomarker of exposure.
The impact of electrophiles has long been studied in the context of covalent modification of DNA and its potential to lead directly to mutations. However, adenine propenal produced in the nucleus also has the potential to modify DNA-associated proteins, including those involved in DNA repair. We report here that adenine propenal covalently modifies the NER protein XPA in vitro and that this modification leads to a reduction in XPA’s DNA-binding activity. Oxopropenylation of residues known to be acetylated further suggests that adenine propenal-dependent modification could result in failure of regulation of XPA’s activity through SIRT-1-dependent deacetylation.25 XPA is a critical component of the NER machinery, without which repair activity ceases.46 Mutations in XPA, particularly in the DNA-binding domain, are associated with severe XP clinical phenotypes.47 Thus, it is possible that decreased XPA-binding activity resulting from adenine propenal-dependent modification could result in reduced NER activity in cells and increased potential for mutagenesis and carcinogenesis. It should be noted that, in this model, the modification of repair proteins may have a synergistic effect on mutagenesis by preventing the repair of the original DNA lesion. In this case, even moderate reductions in the activity of the repair proteins have the potential to result in significant increases in genomic instability. Further analysis will be required to determine whether reactive base propenal species modify DNA repair proteins in cells under oxidative stress and to characterize the effects of modification on DNA repair activity in vivo.
Glossary
Abbreviations
- MDA
malondialdehyde
- M1dG
3-(2′-deoxy-β-d-erythro-pentofuranosyl)-pyrimido[1,2-α]-purin-10(3H)-one
- NER
nucleotide excision repair
- XPA
xeroderma pigmentosum complementation group A
- HSA
human serum albumin
- MOPS
4-morpholinepropanesulfonic acid
Supporting Information Available
UV/vis spectra of adenine propenal reactant and products with N-α-acetyllysine; quantification of lysine cross-link formation; MS spectrum of adenine propenal–cysteine adduct; XPA-junction substrate-binding data; COSY spectrum, HMBC, and HSQC of 3 and 4; and sequence coverage of HSA. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by research and training grants from the National Institutes of Health [R37 CA087819 (L.J.M), R01 ES1065561 (W.J.C.), DK020593 (J.A.O.), GM15431 (J.A.O.), and F32 CA159701-01 (S.C.S.)]. S.M.S is supported by postdoctoral fellowship 119569-PF-11-271-01-DMC from the American Cancer Society. Partial support for the NMR and MS was provided by P30 ES000267. Funds for the 600 MHz spectrometer was provided from the National Institutes of Health (S10 RR019022), and we thank Dr. Don Stec for providing this service.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Marnett L. J.; Riggins J. N.; West J. D. (2003) Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J. Clin. Invest. 111, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink S. P.; Reddy G. R.; Marnett L. J. (1997) Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc. Natl. Acad. Sci. U.S.A. 94, 8652–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddukuri L.; Eoff R. L.; Choi J. Y.; Rizzo C. J.; Guengerich F. P.; Marnett L. J. (2010) In vitro bypass of the major malondialdehyde- and base propenal-derived DNA adduct by human Y-family DNA polymerases kappa, iota, and Rev1. Biochemistry 49, 8415–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddukuri L.; Shuck S. C.; Eoff R. L.; Zhao L.; Rizzo C. J.; Guengerich F. P.; Marnett L. J. (2013) Replication, repair, and translesion polymerase bypass of N6-oxopropenyl-2′-deoxyadenosine. Biochemistry 52, 8766–8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley D. P.; Kolaja K. L.; Reichard J.; Petersen D. R. (1999) 4-Hydroxynonenal and malondialdehyde hepatic protein adducts in rats treated with carbon tetrachloride: immunochemical detection and lobular localization. Toxicol. Appl. Pharmacol. 161, 23–33. [DOI] [PubMed] [Google Scholar]
- Hartley D. P.; Kroll D. J.; Petersen D. R. (1997) Prooxidant-initiated lipid peroxidation in isolated rat hepatocytes: detection of 4-hydroxynonenal– and malondialdehyde–protein adducts. Chem. Res. Toxicol. 10, 895–905. [DOI] [PubMed] [Google Scholar]
- Ishii T.; Ito S.; Kumazawa S.; Sakurai T.; Yamaguchi S.; Mori T.; Nakayama T.; Uchida K. (2008) Site-specific modification of positively-charged surfaces on human serum albumin by malondialdehyde. Biochem. Biophys. Res. Commun. 371, 28–32. [DOI] [PubMed] [Google Scholar]
- Ishii T.; Kumazawa S.; Sakurai T.; Nakayama T.; Uchida K. (2006) Mass spectroscopic characterization of protein modification by malondialdehyde. Chem. Res. Toxicol. 19, 122–129. [DOI] [PubMed] [Google Scholar]
- Uchida K.; Sakai K.; Itakura K.; Osawa T.; Toyokuni S. (1997) Protein modification by lipid peroxidation products: formation of malondialdehyde-derived Nε-(2-propenol)lysine in proteins. Arch. Biochem. Biophys. 346, 45–52. [DOI] [PubMed] [Google Scholar]
- Feng Z.; Hu W.; Marnett L. J.; Tang M. S. (2006) Malondialdehyde, a major endogenous lipid peroxidation product, sensitizes human cells to UV- and BPDE-induced killing and mutagenesis through inhibition of nucleotide excision repair. Mutat. Res. 601, 125–136. [DOI] [PubMed] [Google Scholar]
- Cleaver J. E. (2005) Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 5, 564–573. [DOI] [PubMed] [Google Scholar]
- Buchko G. W.; Tung C. S.; McAteer K.; Isern N. G.; Spicer L. D.; Kennedy M. A. (2001) DNA–XPA interactions: a 31P NMR and molecular modeling study of dCCAATAACC association with the minimal DNA-binding domain (M98–F219) of the nucleotide excision repair protein XPA. Nucleic Acids Res. 29, 2635–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T.; Kuraoka I.; Saijo M.; Kodo N.; Kyogoku Y.; Morikawa K.; Tanaka K.; Shirakawa M. (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat. Struct. Biol. 5, 701–706. [DOI] [PubMed] [Google Scholar]
- Dedon P. C.; Plastaras J. P.; Rouzer C. A.; Marnett L. J. (1998) Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc. Natl. Acad. Sci. U.S.A. 95, 11113–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Zhou X.; Taghizadeh K.; Chen J.; Stubbe J.; Dedon P. C. (2007) GC/MS methods to quantify the 2-deoxypentos-4-ulose and 3′-phosphoglycolate pathways of 4′ oxidation of 2-deoxyribose in DNA: application to DNA damage produced by gamma radiation and bleomycin. Chem. Res. Toxicol. 20, 1701–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Stubbe J. (2005) Bleomycins: towards better therapeutics. Nat. Rev. Cancer 5, 102–112. [DOI] [PubMed] [Google Scholar]
- Dedon P. C.; Tannenbaum S. R. (2004) Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 423, 12–22. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Taghizadeh K.; Dedon P. C. (2005) Chemical and biological evidence for base propenals as the major source of the endogenous M1dG adduct in cellular DNA. J. Biol. Chem. 280, 25377–25382. [DOI] [PubMed] [Google Scholar]
- Berhane K.; Widersten M.; Engstrom A.; Kozarich J. W.; Mannervik B. (1994) Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. U.S.A. 91, 1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I.; Scaloni A.; Giustarini D.; Cavarra E.; Tell G.; Lungarella G.; Colombo R.; Rossi R.; Milzani A. (2005) Proteins as biomarkers of oxidative/nitrosative stress in diseases: the contribution of redox proteomics. Mass Spectrom. Rev. 24, 55–99. [DOI] [PubMed] [Google Scholar]
- Fanali G.; di Masi A.; Trezza V.; Marino M.; Fasano M.; Ascenzi P. (2012) Human serum albumin: from bench to bedside. Mol. Aspects Med. 33, 209–290. [DOI] [PubMed] [Google Scholar]
- Kadiiska M. B.; Gladen B. C.; Baird D. D.; Germolec D.; Graham L. B.; Parker C. E.; Nyska A.; Wachsman J. T.; Ames B. N.; Basu S.; Brot N.; Fitzgerald G. A.; Floyd R. A.; George M.; Heinecke J. W.; Hatch G. E.; Hensley K.; Lawson J. A.; Marnett L. J.; Morrow J. D.; Murray D. M.; Plastaras J.; Roberts L. J. II; Rokach J.; Shigenaga M. K.; Sohal R. S.; Sun J.; Tice R. R.; Van Thiel D. H.; Wellner D.; Walter P. B.; Tomer K. B.; Mason R. P.; Barrett J. C. (2005) Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning?. Free Radical Biol. Med. 38, 698–710. [DOI] [PubMed] [Google Scholar]
- Nagumo K.; Tanaka M.; Chuang V. T.; Setoyama H.; Watanabe H.; Yamada N.; Kubota K.; Tanaka M.; Matsushita K.; Yoshida A.; Jinnouchi H.; Anraku M.; Kadowaki D.; Ishima Y.; Sasaki Y.; Otagiri M.; Maruyama T. (2014) Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS One 9, e85216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck S. C.; Rose K. L.; Marnett L. J. (2014) Mass spectrometric methods for the analysis of nucleoside-protein cross-links: application to oxopropenyl-deoxyadenosine. Chem. Res. Toxicol. 27, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W.; Luo J. (2010) SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 39, 247–258. [DOI] [PubMed] [Google Scholar]
- Johnson F.; Pillai K. M.; Grollman A. P.; Tseng L.; Takeshita M. (1984) Synthesis and biological activity of a new class of cytotoxic agents: N-(3-oxoprop-1-enyl)-substituted pyrimidines and purines. J. Med. Chem. 27, 954–958. [DOI] [PubMed] [Google Scholar]
- Nair V.; Vietti D. E.; Cooper C. S. (1981) Degenerative chemistry of malondialdehyde. Structure, stereochemistry, and kinetics of formation of enaminals from reaction with amino acids. J. Am. Chem. Soc. 103, 3030–3036. [Google Scholar]
- Marnett L. J.; Bienkowski M. J.; Raban M.; Tuttle M. A. (1979) Studies of the hydrolysis of 14C-labeled tetraethoxypropane to malondialdehyde. Anal. Biochem. 99, 458–463. [DOI] [PubMed] [Google Scholar]
- Bernoud-Hubac N.; Davies S. S.; Boutaud O.; Montine T. J.; Roberts L. J. II. (2001) Formation of highly reactive gamma-ketoaldehydes (neuroketals) as products of the neuroprostane pathway. J. Biol. Chem. 276, 30964–30970. [DOI] [PubMed] [Google Scholar]
- Sugitani N.; Shell S. M.; Soss S. E.; Chazin W. J. (2014) Redefining the DNA-binding domain of human XPA. J. Am. Chem. Soc. 136, 10830–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao B.; Pennathur S.; Pagani I.; Oda M. N.; Witztum J. L.; Oram J. F.; Heinecke J. W. (2010) Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 285, 18473–18484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S. M.; Hawkins E. K.; Tsai M. S.; Hlaing A. S.; Rizzo C. J.; Chazin W. J. (2013) Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair 12, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S. M.; Li Z.; Shkriabai N.; Kvaratskhelia M.; Brosey C.; Serrano M. A.; Chazin W. J.; Musich P. R.; Zou Y. (2009) Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J. Biol. Chem. 284, 24213–24222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan J. Unpublished results.
- Sugio S.; Kashima A.; Mochizuki S.; Noda M.; Kobayashi K. (1999) Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 12, 439–446. [DOI] [PubMed] [Google Scholar]
- Kvaratskhelia M.; Miller J. T.; Budihas S. R.; Pannell L. K.; Le Grice S. F. (2002) Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc. Natl. Acad. Sci. U.S.A. 99, 15988–15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S. M.; Hess S.; Kvaratskhelia M.; Zou Y. (2005) Mass spectrometric identification of lysines involved in the interaction of human replication protein a with single-stranded DNA. Biochemistry 44, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I.; Rossi R.; Giustarini D.; Milzani A.; Colombo R. (2003) Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 329, 23–38. [DOI] [PubMed] [Google Scholar]
- Antunes A. M.; Godinho A. L.; Martins I. L.; Oliveira M. C.; Gomes R. A.; Coelho A. V.; Beland F. A.; Marques M. M. (2010) Protein adducts as prospective biomarkers of nevirapine toxicity. Chem. Res. Toxicol. 23, 1714–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y.; Mitsuda Y.; Taguchi H.; Planque S.; Hara M.; Karle S.; Hanson C. V.; Uda T.; Paul S. (2005) Broadly distributed nucleophilic reactivity of proteins coordinated with specific ligand binding activity. J. Mol. Recognit. 18, 295–306. [DOI] [PubMed] [Google Scholar]
- Burcham P. C.; Fontaine F. R.; Kaminskas L. M.; Petersen D. R.; Pyke S. M. (2004) Protein adduct-trapping by hydrazinophthalazine drugs: mechanisms of cytoprotection against acrolein-mediated toxicity. Mol. Pharmacol. 65, 655–664. [DOI] [PubMed] [Google Scholar]
- Gibson J. D.; Pumford N. R.; Samokyszyn V. M.; Hinson J. A. (1996) Mechanism of acetaminophen-induced hepatotoxicity: covalent binding versus oxidative stress. Chem. Res. Toxicol. 9, 580–585. [DOI] [PubMed] [Google Scholar]
- Switzar L.; Kwast L. M.; Lingeman H.; Giera M.; Pieters R. H.; Niessen W. M. (2013) Identification and quantification of drug-albumin adducts in serum samples from a drug exposure study in mice. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 917–918, 53–61. [DOI] [PubMed] [Google Scholar]
- Aldini G.; Gamberoni L.; Orioli M.; Beretta G.; Regazzoni L.; Maffei Facino R.; Carini M. (2006) Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. J. Mass Spectrom. 41, 1149–1161. [DOI] [PubMed] [Google Scholar]
- Szapacs M. E.; Riggins J. N.; Zimmerman L. J.; Liebler D. C. (2006) Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: kinetic analysis of competing alkylation reactions. Biochemistry 45, 10521–10528. [DOI] [PubMed] [Google Scholar]
- Druckova A.; Mernaugh R. L.; Ham A. J.; Marnett L. J. (2007) Identification of the protein targets of the reactive metabolite of teucrin A in vivo in the rat. Chem. Res. Toxicol. 20, 1393–1408. [DOI] [PubMed] [Google Scholar]
- Camenisch U.; Nageli H. (2008) XPA gene, its product and biological roles. Adv. Exp. Med. Biol. 637, 28–38. [DOI] [PubMed] [Google Scholar]
- Hengge U. R.; Emmert S. (2008) Clinical features of xeroderma pigmentosum. Adv. Exp. Med. Biol. 637, 10–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.