

Abstract
DNA polymerase (pol) ι is the most error-prone among the Y-family polymerases that participate in translesion synthesis (TLS). Pol ι can bypass various DNA lesions, e.g., N2-ethyl(Et)G, O6-methyl(Me)G, 8-oxo-7,8-dihydroguanine (8-oxoG), and an abasic site, though frequently with low fidelity. We assessed the biochemical effects of six reported genetic variations of human pol ι on its TLS properties, using the recombinant pol ι (residues 1–445) proteins and DNA templates containing a G, N2-EtG, O6-MeG, 8-oxoG, or abasic site. The Δ1–25 variant, which is the N-terminal truncation of 25 residues resulting from an initiation codon variant (c.3G > A) and also is the formerly misassigned wild-type, exhibited considerably higher polymerase activity than wild-type with Mg2+ (but not with Mn2+), coinciding with its steady-state kinetic data showing a ∼10-fold increase in kcat/Km for nucleotide incorporation opposite templates (only with Mg2+). The R96G variant, which lacks a R96 residue known to interact with the incoming nucleotide, lost much of its polymerase activity, consistent with the kinetic data displaying 5- to 72-fold decreases in kcat/Km for nucleotide incorporation opposite templates either with Mg2+ or Mn2+, except for that opposite N2-EtG with Mn2+ (showing a 9-fold increase for dCTP incorporation). The Δ1–25 variant bound DNA 20- to 29-fold more tightly than wild-type (with Mg2+), but the R96G variant bound DNA 2-fold less tightly than wild-type. The DNA-binding affinity of wild-type, but not of the Δ1–25 variant, was ∼7-fold stronger with 0.15 mM Mn2+ than with Mg2+. The results indicate that the R96G variation severely impairs most of the Mg2+- and Mn2+-dependent TLS abilities of pol ι, whereas the Δ1–25 variation selectively and substantially enhances the Mg2+-dependent TLS capability of pol ι, emphasizing the potential translational importance of these pol ι genetic variations, e.g., individual differences in TLS, mutation, and cancer susceptibility to genotoxic carcinogens.
Introduction
DNA damage is constantly generated from endogenous and exogenous sources in cells and poses a major obstacle to vital cellular processes of replication and transcription, possibly leading to mutation and cell death. To cope with the constant threat of DNA damage, cells are equipped with a sophisticated network of DNA damage response systems, including DNA repair mechanisms, damage tolerance processes, and cell-cycle checkpoints.1,2 Such systems should ideally have high fidelity, efficiency, and coordination with each other to preserve genome integrity, but these properties are not perfect nor the same for all lesions. Sometimes the attempts at repair can result in genomic errors and cell apoptosis. Inherited defects in human DNA damage response machineries (e.g., XPC, POLH, ATM) cause the faulty repair, damage tolerance, and checkpoints and commonly result in severe cancer predisposition disorders along with other different disease phenotypes.3,4 Reduced DNA repair capacity and related genetic variations have been shown to be associated with enhanced cancer risks in human individuals.5−8 Along the same line, it can be speculated that the differential cellular capacity for DNA damage tolerance influences the final biological outcomes from residual genomic lesions and thus could be a determining factor for mutation and cancer predisposition in individuals.
Persistent unrepaired DNA lesions can interfere with DNA replication, which can lead to replication fork stalling and copying errors. As a prompt response to the lesion-blocked replication fork, cells are able to utilize a DNA damage tolerance system involving specialized translesion polymerases, mostly belonging to the Y-Family, which overcomes DNA lesions and performs translesion synthesis (TLS). TLS is a potentially mutagenic process due to the low fidelity of TLS polymerases with lesions in many cases, while serving to avoid the permanent cell cycle arrest and cell death. Indeed, each Y-Family polymerase can carry out its unique TLS, varying in both efficiency and fidelity depending on the type, size, and location of the lesion.9 Therefore, individual TLS polymerases may play distinctive roles, i.e., protective (error-free), provocative (error-prone), or neutral, in mutagenesis induced by each specific DNA lesion in cells. For instance, both pol κ and REV1 can perform error-free and efficient TLS at bulky N2-guanine (G) lesions, such as benzo[a]pyrene-derived N2-G adducts but pol η and pol ι perform relatively error-prone TLS (albeit yielding different types of errors) at those adducts,10−13 suggesting error-free roles for the former two pols but error-prone roles for the latter two pols in TLS (at least regarding bulky carcinogen-bound N2-G adducts). In these circumstances, it can be postulated that the overall cellular TLS capacity, comprising behaviors of multiple individual polymerases employed against carcinogen-specific DNA lesions, will determine the levels of lesion-derived mutations in the newly synthesized genome and thus play a role in preventing or facilitating mutagenesis resulting from genotoxic carcinogens in cells, which could further relate to cancer susceptibility in individuals.
DNA polymerase (pol) ι, a member of human Y-family DNA polymerases, has been known to perform TLS at various DNA lesions although it has the lowest fidelity in DNA synthesis among polymerases. Pol ι is inherently very error-prone in nucleotide insertion, particularly opposite undamaged template bases G and T, respectively, yielding misinsertion of either dTTP or dGTP at a frequency of about 0.1 and 1 (relative to the correct nucleotide insertion), which is ascribed to its unique active site and related non-Watson–Crick base pairing.14−16 Pol ι is able to mediate relatively mutagenic but occasionally accurate replicative bypass with a variety of DNA lesions, including minor-groove N2-G adducts, major-groove O6-G adducts, 8-oxo-7,8-dihydroG (8-oxoG), pyrimidine dimers, and abasic lesions, with different nucleotide selectivities according to lesion types. Pol ι inserts both dCTP and dTTP opposite N2-G and O6-G adducts, with a slight preference of either dCTP or dTTP, respectively.17 Pol ι slightly prefers dCTP over dGTP for insertion opposite 8-oxoG, prefers dATP opposite the 3′ T (but both dGTP and dTTP opposite 5′ T) of (6–4) TT photoproducts, and slightly prefers to insert dTTP and dGTP opposite abasic lesions.18,19 Pol ι is distinctively known to prefer Mn2+ over Mg2+ ions as a metal in polymerase catalysis and is maximally active at low concentrations (0.05–0.25 mM) of Mn2+.20
Substantial evidence suggests a possible implication of pol ι in cancer in mammals. Pol ι deficiency results in a higher susceptibility to UV-induced skin cancers in mice under pol η-null conditions,21,22 and the loss of pol ι increases urethane-induced lung mutagenesis and tumorigenesis in C57BL/6J pol ι-knockout mice.23 Dysregulation of pol ι is also found in many types of cancers. Pol ι is overexpressed in various types of cancerous tissues or cells including breast, prostate, uterus, stomach, rectal, esophageal, and bladder cancers,24−27 which might hypothetically lead to a mutator phenotype due to an elevated error-prone DNA replication. Two germline POLI single nucleotide variations (SNVs), which might result in a missense change at codon 532 or 731 located in the ubiquitin-binding motifs, have been associated with a significantly higher risk of some subsets of lung and prostate cancers, respectively, although their specific mechanisms have not been elucidated yet.28,29 On the basis of these circumstances, we can infer that pol ι would serve a protective (or sometimes facilitative) role in genomic mutagenesis induced by genotoxic agents in cells, and the altered status of pol ι function by genetic variation might affect individual risks of mutation and cancer from exposure to certain genotoxic carcinogens.
The human POLI gene encodes the pol ι protein consisting of 740 amino acids according to NCBI GenBank database (http://www.ncbi.nlm.nih.gov/genbank/), which has the additional N-terminal 25 residues to the formerly erroneously designated open-reading frame (ORF) amino acid sequence. For such a reason, most of the previous biochemical and structural experiments on pol ι were performed using the sequence information on the N-terminal truncated (25-amino acids-shorter) ORF. The catalytic core of pol ι is positioned in the N-terminal region (amino acids 26–445), and its ternary complex crystal structure has been determined.14 However, biochemical properties of the new wild-type pol ι, as well as the effect of the extra 25 amino acids (containing 12 acidic residues) added at the N-terminus, on pol ι function, have not been reported. Until the present time, a total of ∼122 germline variations in POLI gene have been described for human individuals in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), but the functional impacts of these genetic variations have not been biochemically evaluated yet. Biochemical approaches to evaluate the effects of germline genetic variations on pol ι function are indispensable for understanding and predicting their mechanistic basis and biological outcomes either before or after studying their clinical associations. In this study, we focused on the nonsynonymous coding POLI gene variations, which are located in the polymerase core domains and the N-terminal 25 amino acids region, and predicted to have damaging effects by in silico prediction analysis tools, e.g., SIFT and PolyPhen,30−32 because they would be likely to affect the catalytic function of pol ι and change its TLS function. In order to characterize the putatively functional genetic variations of human pol ι, we investigated the biochemical effects of six selected missense or deletion genetic variations on the enzymatic properties of human pol ι regarding both normal DNA synthesis and bypass of various DNA lesions. We performed the experiments with “standing-start” full-length primer extensions, steady-state kinetics, and pol-DNA binding in the presence of either Mg2+ or Mn2+ ions, using wild-type recombinant human pol ι (1–445 amino acids) and six variants with primer-annealed oligonucleotide DNA templates containing an undamaged G, N2-ethyl(Et)G, O6-methyl(Me)G, 8-oxoG, or abasic site. Here we describe two germline genetic variations that can alter in vitro enzyme function of pol ι in nucleotide incorporation with normal and lesion DNA substrates, as well as DNA substrate binding. These observations are discussed in the context of understanding the possible mechanistic and functional aspects of altered TLS with pol ι variants.
Experimental Procedures
Materials
T4 polynucleotide kinase, restriction endonucleases, and dNTPs were purchased from New England Biolabs (Ipswich, MA). [γ-32P]ATP (specific activity 3 × 103 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). Biospin columns were purchased from Bio-Rad (Hercules, CA). A protease inhibitor cocktail was obtained from Roche Applied Science (Indianapolis, IN). The vector pBG101 was gratefully obtained from the Center for Structural Biology, Vanderbilt University. The pCR2.1-TOPO TA cloning kit was from Invitrogen (Carlsbad, CA), and the QuickChange mutagenesis kit was from Stratagene (La Jolla, CA). FPLC columns were purchased from GE Healthcare (Uppsala, Sweden).
DNA Substrates
24-Mer (5′-GCC TCG AGC CAG CCG CAG ACG CAG-3′) and 36-mer (3′-CGG AGC TCG GTC GGC GTC TGC GTC XCT CCT GCG GCT-5′; X = G, O6-MeG, or tetrahydrofuran (abasic site analogue)) oligonucleotides containing a G, O6-MeG, or abasic site (stable tetrahydrofuran derivative) were obtained from Midland Certified Reagent Co. (Midland, TX). A 36-mer (X = N2-EtG) containing N2-EtG was prepared as previously described.33 A 36-mer (X = 8-oxoG) containing an 8-oxoG, and an 18-FAM-mer (5′-(FAM)-AGC CAG CCG CAG ACG CAG-3′; FAM = 6-carboxyfluorescein) were obtained from Bioneer (Daejeon, Korea). Primers (24-mers) were 5′ end-labeled using T4 polynucleotide kinase with [γ-32P]ATP and annealed with 36-mer templates to make duplex primer-template DNA substrates for use in polymerase activity assays. 18-FAM-mer primers were annealed with 36-G-mer templates for use in DNA binding assays.
Selection of Human POLI Gene Variations Having Potentially Functional Impact
We searched for human POLI gene variations that are highly likely to alter enzyme function. First, we screened the naturally occurring germ-line genetic variations in the protein-coding sequence of the POLI gene from the public database dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP). We selected four candidate variations to be likely dysfunctional based on three criteria: (i) nonsynonymous coding variations that cause a missense or nonsense change, (ii) variations located in the polymerase core domain (amino acid residues 1 to 445), and (iii) missense variations predicted to be deleterious or damaging on protein function by SIFT and Polyphen.30−32 We also included two more candidate variations, which cause the initiation codon change or an amino acid deletion and thus might exert functional changes. Thus, we selected six variants (i.e., a deletion, an initiator codon, and four missense variants) and performed detailed biochemical analyses using the corresponding recombinant protein pol ι1–445 proteins purified from Escherichia coli. Current information for the six POLI gene variations included in this study is summarized in Table 1, based on public databases, e.g., dbSNP, 1000 genomes (http://browser.1000genomes.org).
Table 1. POLI Gene Variations Studied.
| predictionb |
||||||
|---|---|---|---|---|---|---|
| rs IDa | nucleotide change | amino acid change | protein domain | minor allele frequency | SIFT | PolyPhen-2 |
| rs199757163 | c.3G > A | M1_A25del (Δ1–25)c | 0.001d | N/Ae | N/A | |
| rs10584411 | c.51_53del | D17del (ΔD17) | 0.747d | N/A | N/A | |
| rs3218778 | c.286A > G | R96G | finger | 0.006f | damaging | probably damaging |
| rs3218784 | c.783A > G | I261M | thumb | 0.011d | damaging | possibly damaging |
| rs3218783 | c.826G > A | E276K | thumb | 0.0005d | damaging | possibly damaging |
| rs11558769 | c.1120T > A | Y374N | PAD | N/A | damaging | benign |
A reference SNP identification number provided by dbSNP.
Possible functional effects of genetic variations are predicted in silico using SIFT and PolyPhen-2.30−32
The Greek symbol Δ denotes a deletion.
From 1000 Genomes project.
Not available.
From NIHPDR and PDR90 resources described in dbSNP.
Construction of Expression Vectors for Core Proteins of Wild-type Pol ι and Six Variants
The gene fragments covering the core proteins (amino acids 1–445) of wild-type pol ι and the Δ1–25 variant were obtained by PCR amplification from human testis cDNA (Clontech, Mountain View, CA) as template using AccuTaq LA DNA polymerase (Sigma, St. Louis, MO) with a forward primer (5′-GAA TCC ATG GAG AAG CTG GGG GTG G-3′ for wild-type or 5′-GAA TCC ATG GAG TCG GCA GAG GGT GTG-3′ for Δ1–25) and a reverse primer (5′-CTA CTT AGC AGT ATT TAG TGC-3′). Each of the resulting PCR products of 1.3 kb POLI core was cloned into the vector pCR2.1-TOPO, and the nucleotide sequences of pol ι gene inserts for wild-type and Δ1–25 were confirmed. From the nucleotide sequencing of wild-type gene inserts, we obtained two kinds of vectors encoding the wild-type or ΔD17 variant pol ι(1–445), indicating that ΔD17 variation is very common in the human population. Each of the POLI gene fragments were then cloned into the BamHI and EcoRI sites of the vector pBG101, which can generate the pBG101-wtPOLI1–445, and two vectors encoding the Δ1–25 or ΔD17 pol ι(1–445) variant. Each of the four different mutations in the POLI gene was created by a QuickChange mutagenesis kit with the pBG101-wtPOLI1–445 vector as template. The oligonucleotide primers for introducing the point mutations in POLI were 5′-GGT TAC CTG CAA CTA TGA AGC TGG GAA ACT TGG AG-3′ for R96G, 5′-CTT ATT CAT AGT TTG AAT CAC ATG AAG GAA ATA CCT GGT ATT GGC-3′ for I261M, 5′-CCA AAT GTC TTA AAG CAC TGG GTA TCA ATA GTG TGC G-3′ for E276K, 5′-GTG AGA TTA ATA ATC CGT CGG AAT TCC TCT GAG AAG C-3′ for Y374N, and the corresponding antiparallel primer for each mutation. All four substitutions were confirmed by nucleotide sequence analyses of the constructed vectors.
Expression and Purification of Recombinant Proteins
The wild-type and variant forms of recombinant pol ι core proteins were expressed in E. coli strain BL21(DE3) cells. E. coli harboring each vector for the recombinant protein were grown in Luria–Bertani broth supplemented with kanamycin (50 μg mL–1) at 37 °C, with aeration, to an OD600 of 0.6. Isopropyl-β-d-thiogalactopyranoside was added to 0.2 mM, and incubation was continued for 14 h at 16 °C. The cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, containing 300 mM NaCl, 10% glycerol (v/v), 5 mM β-mercaptoethanol, 1 mg lysozyme mL–1, and protease inhibitor cocktail), cooled on ice for 30 min, and then lysed by sonication (12 × 10 s duration with a Branson digital sonifier microtip, (VWR, West Chester, PA), 45% amplitude, with intermittent cooling time). The cell lysate was clarified by centrifugation at 4 × 104 × g for 60 min at 4 °C. The resulting supernatant was loaded onto a 1 mL GSTrap 4B column, and the column was washed with 20 mL of Buffer A (50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 10% glycerol (v/v), and 5 mM β-mercaptoethanol). GST-tagged pol ι core bound on the column was cleaved by Prescission Protease for 14 h at 4 °C. Cleaved pol ι core fractions (eluted with Buffer A) were collected and diluted 6-fold with buffer B (50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 10% glycerol (v/v), and 5 mM β-mercaptoethanol). Pol ι core was further purified to near homogeneity with the use of a Mono S column and a 50 mM to 2 M NaCl gradient in buffer B. Pol ι core was eluted at ∼250 mM NaCl. Protein concentrations were estimated using a Bradford protein assay, and the quality of purified proteins was assessed by SDS-polyacrylamide gel electrophoresis and Coomassie Brilliant Blue staining (Figure S1, Supporting Information).
DNA Polymerization Assays and Steady-State Kinetic Analysis
Standard DNA polymerase reactions of 8 μL were performed in 50 mM Tris-HCl (pH 7.5) buffer containing 50 mM NaCl, 5 mM dithiothreitol, 100 μg mL–1 bovine serum albumin (BSA) (w/v), and 10% glycerol (v/v) with 100 nM primer-template substrate at 37 °C. Reactions were initiated by the addition of dNTPs with MgCl2 (5 mM final concentration) or MnCl2 (0.15 mM final concentration) to preincubated enzyme/DNA mixtures and terminated with six volumes of a solution of 20 mM EDTA in 95% formamide (v/v). For steady-state kinetic analysis, the primer-template was extended in the presence of 0.1–33 nM pol ι enzyme (or up to 400 nM enzyme for R96G) with increasing concentrations of individual dNTPs for 10 min, when the maximum amount of extension products was ≤20% of total DNA substrates. Products were resolved using a 16% polyacrylamide (w/v) gel electrophoresis system containing 8 M urea and visualized using a Bio-Rad Personal Molecular Imager and Quantity One software (Bio-Rad). The product formation rates (as a function of dNTP concentration) were plotted to estimate the kinetic parameters Km and kcat by nonlinear regression fitting to the Michaelis–Menten equation using Graph Pad Prism 5.0 software (GraphPad, San Diego, CA). Misinsertion frequency (fins) opposite G or G adducts was calculated as fins = (kcat/Km)dNTP/(kcat/Km)dCTP.34
Fluorescence Polarization Experiments
The 2 nM 18-FAM-mer primer annealed with unmodified 36-mer template was incubated with varying concentrations of pol ι, and fluorescence polarization (FP) was measured with a Biotek Synergy NEO plate reader (Winooski, VT) using excitation and emission wavelengths of 485 and 528 nm, respectively. The polymerase-DNA binding reaction was done in the presence of 50 mM HEPES buffer (pH 7.5) containing 10 mM potassium acetate, 10 mM KCl, 0.1 mM EDTA, 2 mM β-mercaptoethanol, and 0.1 mg mL–1 BSA in the presence of MgCl2 or MnCl2 (final 0.15 or 1 mM concentration), as modified from a previous study.35 FP data were plotted vs enzyme concentration and fit to a quadratic equation to estimate Kd,DNA using the equation: P = P0 + (Pmax – P0)((Dt + Et + Kd,DNA) – ((Dt + Et + Kd,DNA)2 – (4DtEt))1/2)/(2Dt), where P is the measured change in polarization (in units of millipolarization (mP)), P0 is initial polarization (DNA alone), Pmax is maximum polarization, Dt is DNA concentration, Et is enzyme concentration, and Kd,DNA is the equilibrium dissociation constant for enzyme binding to DNA.
Results
Overall Study Approach
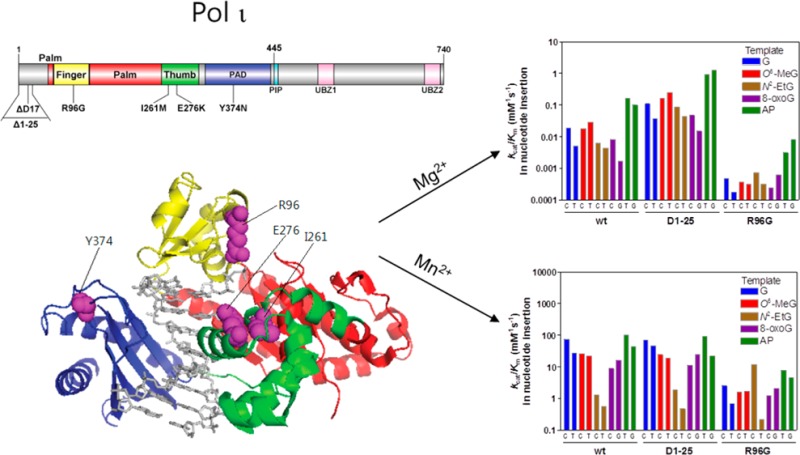
The aim of this study was to analyze the potentially dysfunctional germ-line genetic variants of human pol ι and identify functional pol ι variants. To achieve this, we first screened for human POLI genetic variations likely to alter the enzymatic function of pol ι from the dbSNP database. We utilized the new annotated open reading frame (ORF) sequence of the POLI gene as described in GenBank accession number NM_007195 (http://www.ncbi.nlm.nih.gov/genbank/), which encodes a pol ι protein of 740 amino acids with an additional 25 amino acids at the N-terminus compared to the previous wild-type, in that the previous ORF of pol ι was erroneously annotated to start at a site 75 bases downstream of the actual translational initiation site.36 We picked four candidate variations by searching for nonsynonymous coding variations that are located in the polymerase core domains (finger, palm, thumb, and little finger) and are also predicted as damaging with prediction tools (SIFT and PolyPhen). The SIFT algorithm utilizes a sequence homology-based approach to classify amino acid substitutions, which is based on the evolutionary conservation of the amino acids within protein families,30 while the PolyPhen algorithm uses both sequence- and structure-based prediction.31,32 The R96G variation (close to the incoming nucleotide at the finger domain) and the I261M and E276K variations (in the thumb domain) were predicted to be damaging by both SIFT and PolyPhen, while the Y374N variation in the little finger domain was predicted to be damaging only by SIFT (Table 1 and Figure 1). We also selected two more candidate variations that would result in an altered translation initiation or an amino acid deletion at the N-terminal extension of 25 amino acids (Table 1 and Figure 1), one of which is the initiator codon variation (c.3G > A) to mutate the start codon (ATG) to a nonstart codon (ATA) and thus theoretically yield the variant protein deleted of the first 25 N-terminal residues (Δ1–25) that was previously referred to the wild-type enzyme. After that, we investigated the biochemical impact of six genetic variations on the enzymatic features of pol ι in both normal and translesion DNA synthesis at G and various DNA lesions. To effectively observe the alterations of polymerase function in six selected pol ι variants of the polymerase core domains, we utilized the core protein (amino acids 1–445) of pol ι, which contains all the polymerase core domains critical to polymerase activity. The previous core protein (26–445) was also shown to have the similar polymerase activity to the previous full-length pol ι (26–740).37 A set of experiments, including “standing-start” full-length primer extensions, steady-state kinetics of nucleotide incorporation opposite the lesions, and pol ι-DNA binding assays, was carried out successively using the recombinant core (1–445 amino acids) of pol ι enzymes and oligonucleotides containing a normal G or each of four bypassable DNA lesions, i.e., N2-EtG, O6-MeG, 8-oxoG, or abasic site at a defined site. We also compared the effects of two metals—Mn2+ and Mg2+—in these experiments because pol ι is known to prefer Mn2+ to Mg2+ in polymerase catalysis.20
Figure 1.

Locations of genetic pol ι variations. Structure of human pol ι(26–445) (PDB code, 2FLL) bound to primer/template DNA and incoming nucleotide is shown using Pymol. Pol ι(26–445) is shown in cartoon ribbons, and the primer/template DNA and nucleotide are shown in gray sticks. The finger, palm, thumb, and PAD domains are colored yellow, red, green, and blue, respectively. The amino acid residues (in purple spheres) of genetic pol ι variants are indicated. The structural domains of pol ι are shown in the upper schematic diagram using DOG (version 2.0),60 where positions of amino acids related to six studied variations are indicated.
Primer Extension across G and DNA Lesions with All Four dNTPs by Wild-Type and Variant Pol ι Enzymes in the Presence of MgCl2
To evaluate the possible changes in Mg2+-dependent DNA polymerase activities of six human pol ι variants at undamaged and damaged DNA templates, we performed “standing-start” primer extensions with the wild-type and variant pol ι proteins using 24-mer/36-mer duplexes containing a G, N2-EtG, O6-MeG, 8-oxoG, or abasic site at position 25 of the template in the presence of all four dNTPs and 5 mM MgCl2 (Figure 2). Those four DNA adducts, which were previously found to be bypassed relatively efficiently by human pol ι,11,17,19,38 were selected as the favored substrate lesions of pol ι. Wild-type pol ι extended about half of the primers past G and yielded mainly one-base extended 25-mer products with some traces of 26- and 27-mers (with a 50 nM enzyme concentration), and this pattern was also observed with the ΔD17, I261M, E276K, and Y374N variants. However, the Δ1–25 variant readily synthesized products mainly up to 27- and 28-mers and seemed much more effective than the wild-type enzyme. In contrast, the R96G variant generated almost no extension at G even with a 50 nM enzyme concentration, indicating severe impairment of polymerase activity due to this amino acid substitution. For translesion synthesis at N2-EtG, O6-MeG, 8-oxoG, or an abasic lesion, those six variants showed a similar trend of results as that observed with an undamaged G template, although the extents of bypass synthesis across each lesions differed from one another. With each of those four lesions, the Δ1–25 variant yielded considerably more one-base or up to four-base extended products than the wild-type, while the R96G variant yielded only a trace of one-base or almost no extension at the lesions. Similar patterns were also observed in the presence of 1 mM MgCl2 (at which pol ι was observed to be maximally active with normal DNA substrates tested, results not shown). These results indicate that R96G and Δ1–25 pol ι variants might have a defective and hyperactive Mg2+-dependent TLS ability, respectively.
Figure 2.

Extension of 32P-labeled primers opposite G, O6-MeG, N2-EtG, 8-oxoG, and an abasic site by human wild-type pol ι (1–445) and variants in the presence of Mg2+. The primer (24-mer) was annealed with each of the five different 36-mer templates containing an unmodified G, O6-MeG, N2-EtG, 8-oxoG, or abasic site placed at the 25th position from the 3′-end. Reactions were done in the presence of 5 mM MgCl2 for 15 min with DNA substrate (100 nM primer/template), all four dNTPs (50 μM each), and increasing concentrations of pol ι (0–50 nM) as indicated. The extension products were separated by denaturing gel electrophoresis and imaged using a phosphorimager.
Primer Extension across G and DNA Lesions with All Four dNTPs by Wild-Type and Variant Pol ι Enzymes in the Presence of MnCl2
To examine the effect of Mn2+ as a prosthetic group (instead of Mg2+) on normal and translesion polymerase activities by six variants, standing-start primer extension experiments were done with the wild-type and variant pol ι proteins using 24-mer primers annealed to 36-mer templates containing G, N2-EtG, O6-MeG, 8-oxoG, or an abasic site in the presence of all four dNTPs and 0.15 mM MnCl2 (Figure 3). Wild-type pol ι extended most 24-mer primers across G and four lesions and yielded 25- or up to 28-mer products (10 nM enzyme concentration with 0.15 mM Mn2+, Figure 3A), more effectively than with 5 mM Mg2+ (Figure 2A), indicating a catalytic preference of pol ι for Mn2+ compared to Mg2+, as expected from the previous literature.20 The primer extension results for each pol ι protein across G and four lesions in the presence of Mn2+ were almost similar to those with Mg2+ except for the case of the Δ1–25 variant. Opposite G and four DNA lesions, the R96G variant generated extension products considerably lower in extent than the wild-type in the presence of Mn2+ (Figure 3A), as similarly observed with Mg2+ (Figure 2A), although the decrease of R96G bypass extent was less opposite template N2-EtG than the other templates. However, the Δ1–25 variant extended the primers across G and four lesions to a similar extent as the wild-type pol ι in the presence of Mn2+ (Figure 3), in contrast to the increase in Mg2+-dependent polymerase activity of this variant (Figure 2).
Figure 3.

Extension of 32P-labeled primers opposite G, O6-MeG, N2-EtG, 8-oxoG, and an abasic site by human wild-type pol ι (1–445) and variants in the presence of Mn2+. Primer (24-mer) was annealed with each of the five different 36-mer templates containing an unmodified G, O6-MeG, N2-EtG, 8-oxoG, or abasic site placed at the 25th position from the 3′-end. Reactions were done in the presence of 0.15 mM MnCl2 for 15 min with DNA substrate (100 nM primer/template), all four dNTPs (50 μM each), and increasing concentrations of pol ι (0–10 nM) as indicated. The extension products were separated by denaturing gel electrophoresis and imaged using a phosphorimager.
Steady-State Kinetics of Nucleotide Incorporation Opposite DNA Lesions by the Wild-Type and Variant Pol ι Enzymes in the Presence of MgCl2
To analyze the efficiency and fidelity of six pol ι variants for Mg2+-dependent nucleotide insertion opposite G and four DNA lesions, we determined steady-state kinetic parameters for incorporation of single nucleotides into 24-mer/36-mer duplexes opposite a G or each of four lesions in the presence of 5 mM MgCl2 by six variants in comparison to wild-type pol ι (Tables 2–4). The values of kcat/Km and misinsertion frequency (f = (kcat/Km)incorrect dNTP/(kcat/Km)correct dNTP) were employed as semiquantitative measures for the nucleotide insertion efficiency and fidelity of a distributive pol ι, as applied in previous work.19 Wild-type pol ι inserted single nucleotides opposite each template with the efficiency order (based on the maximum kcat/Km) abasic lesion > O6-MeG > G > 8-oxoG > N2-EtG. Wild-type pol ι inserted the correct dCTP in slight preference to dTTP opposite G and N2-EtG but misinserted dTTP and dGTP (in preference to dCTP) opposite O6-MeG and 8-oxoG, respectively, while inserting nucleoside triphosphates opposite an abasic site in the preferential order of dGTP > dTTP > dATP > dCTP. The ΔD17, I261M, E276K, and Y374N variants inserted dCTP in preference to dTTP opposite undamaged G, with the values of kcat/Km similar to those of wild-type pol ι. However, the R96G variant showed ∼40-fold reductions in kcat/Km values for dCTP and dTTP insertion opposite G compared to wild-type pol ι, while the Δ1–25 variant showed 6- to 7-fold increases in those values. Similar trends of results were observed with four DNA lesion templates, with some alterations in nucleotide preference for some cases. The R96G variant displayed about 8-, 33-, 50-, and 72-fold reductions in kcat/Km values for dCTP insertion, respectively, opposite N2-EtG, 8-oxoG, O6-MeG, and an abasic site compared to wild-type pol ι, with 5- and 10-fold increases in misinsertion frequencies for A and T opposite 8-oxoG but a 3-fold decrease in misinsertion frequency for T opposite O6-MeG. However, the Δ1–25 variant displayed 6- to 14-fold increases in kcat/Km for correct dCTP insertion opposite four lesions compared to wild-type pol ι, with misinsertion frequencies similar to the wild-type protein. Interestingly, both the Δ1–25 and R96G variants slightly preferred dTTP over dGTP for insertion opposite an abasic site, as opposed to the case of the wild-type pol ι, which prefers to insert dGTP (Table 4).
Table 2. Steady-State Kinetic Parameters for dNTP Incorporation Opposite G, O6-MeG, and N2-EtG by Wild-Type and Variant hPols ι(1-445) in the Presence of 5 mM Mg2+.
| template | pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|---|
| G | wild-type | C | 1100 ± 300 | 0.021 ± 0.002 | 0.019 | 1 | 1 |
| T | 1800 ± 400 | 0.0093 ± 0.0007 | 0.0052 | 0.27 | |||
| Δ1–25 | C | 370 ± 47 | 0.042 ± 0.002 | 0.11 | 1 | 5.8 | |
| T | 1000 ± 100 | 0.037 ± 0.002 | 0.037 | 0.34 | |||
| ΔD17 | C | 1900 ± 300 | 0.046 ± 0.002 | 0.024 | 1 | 1.3 | |
| T | 2900 ± 1000 | 0.013 ± 0.002 | 0.0045 | 0.19 | |||
| R96G | C | 3100 ± 800 | 0.0015 ± 0.0001 | 0.00048 | 1 | 0.025 | |
| T | 820 ± 230c | 0.00015 ± 0.00001 | 0.00018 | 0.38 | |||
| I261M | C | 1500 ± 100 | 0.034 ± 0.0008 | 0.023 | 1 | 1.2 | |
| T | 2400 ± 600 | 0.0078 ± 0.0007 | 0.0033 | 0.14 | |||
| E276K | C | 1700 ± 200 | 0.03 ± 0.001 | 0.018 | 1 | 0.95 | |
| T | 4700 ± 1600 | 0.014 ± 0.002 | 0.0030 | 0.17 | |||
| Y374N | C | 1900 ± 200 | 0.022 ± 0.001 | 0.012 | 1 | 0.63 | |
| T | 3400 ± 900 | 0.0079 ± 0.0008 | 0.0023 | 0.19 | |||
| O6-MeG | wild-type | C | 1800 ± 500 | 0.033 ± 0.004 | 0.018 | 1 | 1 |
| T | 940 ± 170 | 0.027 ± 0.002 | 0.029 | 1.6 | |||
| Δ1–25 | C | 620 ± 180 | 0.10 ± 0.01 | 0.16 | 1 | 8.9 | |
| T | 170 ± 50 | 0.043 ± 0.003 | 0.25 | 1.6 | |||
| ΔD17 | C | 2800 ± 600 | 0.055 ± 0.005 | 0.020 | 1 | 1.1 | |
| T | 1400 ± 300 | 0.088 ± 0.006 | 0.063 | 3.2 | |||
| R96G | C | 3000 ± 800 | 0.0011 ± 0.0001 | 0.00036 | 1 | 0.02 | |
| T | 3200 ± 600 | 0.0010 ± 0.0001 | 0.00032 | 0.89 | |||
| I261M | C | 2200 ± 800 | 0.034 ± 0.004 | 0.018 | 1 | 1.0 | |
| T | 1100 ± 200 | 0.039 ± 0.002 | 0.035 | 1.9 | |||
| E276K | C | 1800 ± 400 | 0.029 ± 0.002 | 0.016 | 1 | 0.89 | |
| T | 1600 ± 300 | 0.042 ± 0.003 | 0.026 | 1.6 | |||
| Y374N | C | 2200 ± 500 | 0.048 ± 0.005 | 0.022 | 1 | 1.2 | |
| T | 1500 ± 400 | 0.043 ± 0.004 | 0.029 | 1.3 | |||
| N2-EtG | wild-type | C | 2800 ± 400 | 0.018 ± 0.001 | 0.0064 | 1 | 1 |
| T | 2700 ± 200 | 0.012 ± 0.0003 | 0.0044 | 0.69 | |||
| Δ1–25 | C | 550 ± 60 | 0.048 ± 0.001 | 0.087 | 1 | 14 | |
| T | 770 ± 110 | 0.033 ± 0.001 | 0.043 | 0.49 | |||
| ΔD17 | C | 3000 ± 300 | 0.028 ± 0.0001 | 0.0093 | 1 | 1.5 | |
| T | 2800 ± 200 | 0.020 ± 0.001 | 0.0071 | 0.76 | |||
| R96G | C | 5400 ± 1600 | 0.0040 ± 0.0005 | 0.00074 | 1 | 0.12 | |
| T | 1600 ± 100 | 0.00049 ± 0.00009 | 0.00031 | 0.41 | |||
| I261M | C | 2000 ± 300 | 0.015 ± 0.001 | 0.0075 | 1 | 1.2 | |
| T | 1900 ± 700 | 0.012 ± 0.002 | 0.0063 | 0.84 | |||
| E276K | C | 3700 ± 500 | 0.026 ± 0.002 | 0.0070 | 1 | 1.1 | |
| T | 3700 ± 400 | 0.021 ± 0.001 | 0.0057 | 0.81 | |||
| Y374N | C | 1400 ± 200 | 0.017 ± 0.001 | 0.012 | 1 | 1.9 | |
| T | 2100 ± 700 | 0.013 ± 0.002 | 0.0062 | 0.52 |
Misinsertion frequency, calculated by dividing kcat/Km for dNTP incorporation by the kcat/Km for dCTP incorporation opposite template base. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dCTP incorporation opposite template base by kcat/Km of wild-type pol ι(1–445) for dCTP incorporation opposite template base.
The apparent Km value, determined under the condition where the amount of enzyme is greater than the amount of DNA and thus is not strictly steady-state.
Table 4. Steady-State Kinetic Parameters for dNTP Incorporation Opposite Abasic Site by Wild-Type and Variant hPols ι(1-445) in the Presence of 5 mM Mg2+.
| pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | dNTP selectivity ratioa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 1100 ± 30 | 0.081 ± 0.007 | 0.074 | 0.46 | |
| T | 600 ± 90 | 0.058 ± 0.002 | 0.097 | 0.61 | ||
| G | 770 ± 110 | 0.12 ± 0.01 | 0.16 | 1 | 1 | |
| C | 830 ± 550 | 0.027 ± 0.005 | 0.033 | 0.21 | ||
| Δ1–25 | A | 160 ± 10 | 0.15 ± 0.002 | 0.94 | 0.72 | |
| T | 140 ± 20 | 0.18 ± 0.01 | 1.3 | 1 | ||
| G | 230 ± 50 | 0.21 ± 0.01 | 0.91 | 0.70 | 5.7 | |
| C | 680 ± 100 | 0.14 ± 0.01 | 0.21 | 0.16 | ||
| ΔD17 | A | 770 ± 200 | 0.081 ± 0.006 | 0.11 | 0.61 | |
| T | 760 ± 50 | 0.10 ± 0.002 | 0.13 | 0.72 | ||
| G | 910 ± 90 | 0.17 ± 0.01 | 0.18 | 1 | 1.1 | |
| C | 2100 ± 800 | 0.074 ± 0.010 | 0.035 | 0.19 | ||
| R96G | A | 1700 ± 500 | 0.0035 ± 0.0003 | 0.0021 | 0.26 | |
| T | 820 ± 64 | 0.0066 ± 0.0002 | 0.0080 | 1 | ||
| G | 1600 ± 400 | 0.0048 ± 0.0008 | 0.0030 | 0.38 | 0.019 | |
| C | 1800 ± 400 | 0.0012 ± 0.0001 | 0.00067 | 0.084 | ||
| I261M | A | 690 ± 60 | 0.053 ± 0.002 | 0.077 | 0.64 | |
| T | 480 ± 90 | 0.051 ± 0.003 | 0.11 | 0.92 | ||
| G | 670 ± 80 | 0.080 ± 0.003 | 0.12 | 1 | 0.75 | |
| C | 860 ± 250 | 0.031 ± 0.002 | 0.036 | 0.30 | ||
| E276K | A | 1400 ± 100 | 0.086 ± 0.004 | 0.064 | 0.49 | |
| T | 1000 ± 100 | 0.079 ± 0.002 | 0.079 | 0.61 | ||
| G | 950 ± 110 | 0.13 ± 0.01 | 0.13 | 1 | 0.81 | |
| C | 2000 ± 300 | 0.054 ± 0.002 | 0.027 | 0.21 | ||
| Y374N | A | 850 ± 210 | 0.047 ± 0.004 | 0.056 | 0.58 | |
| T | 600 ± 90 | 0.056 ± 0.003 | 0.093 | 0.97 | ||
| G | 1000 ± 200 | 0.096 ± 0.007 | 0.096 | 1 | 0.60 | |
| C | 1200 ± 100 | 0.032 ± 0.003 | 0.027 | 0.28 |
dNTP selectivity ratio, calculated by dividing kcat/Km of each dNTP incorporation by the highest kcat/Km for dNTP incorporation opposite the abasic site. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dGTP incorporation opposite the abasic site by kcat/Km of wild-type pol ι(1–445) for dGTP incorporation opposite the abasic site.
Table 3. Steady-State Kinetic Parameters for dNTP Incorporation Opposite 8-oxoG by Wild-Type and Variant hPols ι(1-445) in the Presence of 5 mM Mg2+.
| pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 1300 ± 200 | 0.00081 ± 0.00005 | 0.00062 | 0.077 | |
| T | 1600 ± 300 | 0.0027 ± 0.0002 | 0.0017 | 0.27 | ||
| G | 230 ± 40 | 0.0032 ± 0.0001 | 0.014 | 1.7 | ||
| C | 1600 ± 300 | 0.013 ± 0.001 | 0.0081 | 1 | 1 | |
| Δ1–25 | A | 530 ± 80 | 0.0034 ± 0.00017 | 0.0064 | 0.13 | |
| T | 510 ± 70 | 0.0074 ± 0.0003 | 0.015 | 0.31 | ||
| G | 120 ± 10 | 0.0061 ± 0.00014 | 0.051 | 1.1 | ||
| C | 1200 ± 100 | 0.057 ± 0.002 | 0.048 | 1 | 5.9 | |
| ΔD17 | A | 1000 ± 200 | 0.00088 ± 0.00005 | 0.00088 | 0.14 | |
| T | 2400 ± 200 | 0.0046 ± 0.0001 | 0.0019 | 0.30 | ||
| G | 530 ± 50 | 0.0062 ± 0.0002 | 0.012 | 1.9 | ||
| C | 4000 ± 900 | 0.025 ± 0.003 | 0.0063 | 1 | 0.78 | |
| R96G | A | 840 ± 30c | 0.000072 ± 0.000008 | 0.000086 | 0.36 | |
| T | 290 ± 70 | 0.00018 ± 0.00001 | 0.00062 | 2.6 | ||
| G | 160 ± 20c | 0.00010 ± 0.000003 | 0.00063 | 2.6 | ||
| C | 1900 ± 400 | 0.00045 ± 0.00004 | 0.00024 | 1 | 0.030 | |
| I261M | A | 820 ± 110 | 0.00075 ± 0.00003 | 0.00092 | 0.21 | |
| T | 1400 ± 300 | 0.0028 ± 0.0002 | 0.0020 | 0.47 | ||
| G | 320 ± 20 | 0.0044 ± 0.0001 | 0.014 | 3.3 | ||
| C | 2100 ± 300 | 0.0090 ± 0.0005 | 0.0043 | 1 | 0.53 | |
| E276K | A | 1500 ± 100 | 0.0010 ± 0.00002 | 0.00066 | 0.11 | |
| T | 2200 ± 200 | 0.0055 ± 0.0002 | 0.0025 | 0.43 | ||
| G | 920 ± 30 | 0.012 ± 0.0001 | 0.013 | 2.2 | ||
| C | 3800 ± 200 | 0.022 ± 0.001 | 0.0058 | 1 | 0.72 | |
| Y374N | A | 1000 ± 100 | 0.00061 ± 0.00003 | 0.00061 | 0.086 | |
| T | 1200 ± 200 | 0.0023 ± 0.0001 | 0.0019 | 0.27 | ||
| G | 260 ± 20 | 0.0030 ± 0.0001 | 0.013 | 1.8 | ||
| C | 1700 ± 800 | 0.012 ± 0.002 | 0.0071 | 1 | 0.88 |
Misinsertion frequency, calculated by dividing kcat/Km for dNTP incorporation by the kcat/Km for dCTP incorporation opposite 8-oxoG. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dCTP incorporation opposite 8-oxoG by kcat/Km of wild-type pol ι(1–445) for dCTP incorporation opposite 8-oxoG.
The apparent Km value, determined under the condition where the amount of enzyme is greater than the amount of DNA and thus is not strictly steady-state.
Steady-State Kinetics of Nucleotide Incorporation Opposite DNA Lesions by the Wild-Type and Variant Pol ι Enzymes in the Presence of MnCl2
To evaluate the efficiency and fidelity in Mn2+-dependent nucleotide insertion opposite G and DNA lesions by six pol ι variants, we determined steady-state kinetic parameters for nucleotide incorporation opposite a G or each of four lesions in the presence of 0.15 mM MnCl2 by six variants in comparison to the wild-type (Tables 5–7). The kcat/Km values of wild-type pol ι for Mn2+-dependent dCTP and dTTP insertion opposite G were 3 orders of magnitude higher than those of the wild-type protein for Mg2+-dependent insertion. For Mn2+-dependent nucleotide insertion, most of the variants (including the Δ1–25 variant) showed the kcat/Km values similar to those of wild-type, whereas the R96G variant displayed 29- to 41-fold reduction in kcat/Km compared to wild-type (Table 5). Similar trends of results were observed with four DNA lesion templates, except for the case with the template N2-EtG and the R96G variant. The R96G variant showed about 16-, 8-, and 5-fold decreases in kcat/Km values for dCTP insertion, respectively, opposite O6-MeG, 8-oxoG, and the abasic site compared to those of the wild-type protein, while showing a 9-fold increase in activity opposite N2-EtG compared to wild-type, indicating that the R96G variation might substantially impair Mn2+-dependent TLS opposite O6-MeG, 8-oxoG, and an abasic site but facilitate that only opposite N2-EtG. These steady-state kinetic data might in large part explain the relatively proficient Mn2+-dependent bypass of the R96G variant opposite N2-EtG compared to the other lesions (Figure 3).
Table 5. Steady-State Kinetic Parameters for dNTP Incorporation Opposite G, O6-MeG, and N2-EtG by Wild-Type and Variant hPols ι(1-445) in the Presence of 0.15 mM Mn2+.
| template | pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|---|
| G | wild-type | C | 1.5 ± 0.1 | 0.11 ± 0.002 | 73 | 1 | 1 |
| T | 3.3 ± 0.2 | 0.094 ± 0.002 | 28 | 0.38 | |||
| Δ1–25 | C | 0.52 ± 0.05 | 0.036 ± 0.0007 | 72 | 1 | 0.99 | |
| T | 1.2 ± 0.1 | 0.055 ± 0.001 | 46 | 0.64 | |||
| ΔD17 | C | 1.4 ± 0.003 | 0.12 ± 0.003 | 86 | 1 | 1.2 | |
| T | 3.6 ± 0.2 | 0.12 ± 0.002 | 33 | 0.38 | |||
| R96G | C | 11 ± 1 | 0.028 ± 0.001 | 2.5 | 1 | 0.034 | |
| T | 11 ± 1 | 0.0075 ± 0.0003 | 0.68 | 0.27 | |||
| I261M | C | 1.3 ± 0.08 | 0.068 ± 0.0009 | 52 | 1 | 0.71 | |
| T | 3.0 ± 0.2 | 0.068 ± 0.001 | 23 | 0.44 | |||
| E276K | C | 1.4 ± 0.08 | 0.051 ± 0.0006 | 36 | 1 | 0.49 | |
| T | 3.4 ± 0.1 | 0.047 ± 0.0005 | 14 | 0.39 | |||
| Y374N | C | 2.6 ± 0.2 | 0.14 ± 0.003 | 54 | 1 | 0.74 | |
| T | 3.8 ± 0.2 | 0.095 ± 0.001 | 25 | 0.46 | |||
| O6-MeG | wild-type | C | 3.9 ± 0.1 | 0.10 ± 0.001 | 26 | 1 | 1 |
| T | 3.2 ± 0.2 | 0.069 ± 0.001 | 22 | 0.85 | |||
| Δ1–25 | C | 1.6 ± 0.09 | 0.039 ± 0.0005 | 24 | 1 | 0.93 | |
| T | 1.4 ± 0.2 | 0.027 ± 0.001 | 19 | 0.79 | |||
| ΔD17 | C | 3.8 ± 0.2 | 0.18 ± 0.003 | 47 | 1 | 1.8 | |
| T | 2.5 ± 0.4 | 0.087 ± 0.003 | 35 | 0.74 | |||
| R96G | C | 12 ± 3 | 0.019 ± 0.002 | 1.6 | 1 | 0.062 | |
| T | 4.3 ± 0.6 | 0.0073 ± 0.0003 | 1.7 | 1.1 | |||
| I261M | C | 2.3 ± 0.1 | 0.075 ± 0.001 | 33 | 1 | 1.3 | |
| T | 4.0 ± 0.5 | 0.036 ± 0.001 | 9.0 | 0.27 | |||
| E276K | C | 2.5 ± 0.1 | 0.044 ± 0.0006 | 18 | 1 | 0.69 | |
| T | 3.2 ± 0.4 | 0.038 ± 0.001 | 12 | 0.67 | |||
| Y374N | C | 3.7 ± 0.3 | 0.077 ± 0.001 | 21 | 1 | 0.81 | |
| T | 2.0 ± 0.3 | 0.050 ± 0.002 | 25 | 1.2 | |||
| N2-EtG | wild-type | C | 27 ± 2 | 0.034 ± 0.001 | 1.3 | 1 | 1 |
| T | 80 ± 5 | 0.045 ± 0.001 | 0.56 | 0.44 | |||
| Δ1–25 | C | 7.5 ± 1.1 | 0.011 ± 0.001 | 1.5 | 1 | 1.2 | |
| T | 29 ± 2 | 0.018 ± 0.0005 | 0.62 | 0.42 | |||
| ΔD17 | C | 13 ± 1 | 0.025 ± 0.001 | 1.9 | 1 | 1.5 | |
| T | 93 ± 12 | 0.044 ± 0.003 | 0.47 | 0.24 | |||
| R96G | C | 3.7 ± 0.1 | 0.045 ± 0.0004 | 12 | 1 | 9.2 | |
| T | 26 ± 9 | 0.0058 ± 0.0008 | 0.22 | 0.018 | |||
| I261M | C | 16 ± 2 | 0.023 ± 0.001 | 1.4 | 1 | 1.1 | |
| T | 87 ± 11 | 0.042 ± 0.002 | 0.48 | 0.34 | |||
| E276K | C | 20 ± 5 | 0.018 ± 0.002 | 0.90 | 1 | 0.69 | |
| T | 92 ± 13 | 0.042 ± 0.003 | 0.46 | 0.51 | |||
| Y374N | C | 33 ± 3 | 0.039 ± 0.02 | 1.2 | 1 | 0.92 | |
| T | 82 ± 4 | 0.035 ± 0.001 | 0.43 | 0.36 |
Misinsertion frequency, calculated by dividing kcat/Km for each dNTP incorporation by the kcat/Km for dCTP incorporation opposite template base. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dCTP incorporation opposite template base by kcat/Km of wild-type pol ι(1–445) for dCTP incorporation opposite template base.
Table 7. Steady-State Kinetic Parameters for dNTP Incorporation Opposite Abasic Site by Wild-Type and Variant hPols ι(1-445) in the Presence of 0.15 mM Mn2+.
| pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | dNTP selectivity ratioa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 0.68 ± 0.04 | 0.012 ± 0.0001 | 18 | 0.18 | |
| T | 1.5 ± 0.1 | 0.064 ± 0.001 | 43 | 0.43 | ||
| G | 0.29 ± 0.06 | 0.029 ± 0.001 | 100 | 1 | 1 | |
| C | 2.8 ± 0.1 | 0.066 ± 0.001 | 24 | 0.24 | ||
| Δ1–25 | A | 0.50 ± 0.06 | 0.010 ± 0.003 | 20 | 0.22 | |
| T | 1.1 ± 0.1 | 0.024 ± 0.001 | 22 | 0.24 | ||
| G | 0.099 ± 0.005 | 0.0091 ± 0.0001 | 92 | 1 | 0.92 | |
| C | 0.98 ± 0.08 | 0.043 ± 0.001 | 44 | 0.48 | ||
| ΔD17 | A | 0.83 ± 0.13 | 0.016 ± 0.001 | 19 | 0.31 | |
| T | 1.8 ± 0.2 | 0.048 ± 0.001 | 27 | 0.44 | ||
| G | 0.34 ± 0.04 | 0.021 ± 0.0004 | 62 | 1 | 0.62 | |
| C | 1.8 ± 0.3 | 0.11 ± 0.004 | 61 | 0.98 | ||
| R96G | A | 1.9 ± 0.3 | 0.011 ± 0.0004 | 5.8 | 0.76 | |
| T | 1.9 ± 0.2 | 0.0088 ± 0.0002 | 4.6 | 0.61 | ||
| G | 0.49 ± 0.06 | 0.0037 ± 0.0001 | 7.6 | 1 | 0.076 | |
| C | 3.4 ± 0.3 | 0.015 ± 0.0004 | 4.4 | 0.58 | ||
| I261M | A | 0.35 ± 0.05 | 0.021 ± 0.001 | 60 | 0.50 | |
| T | 1.3 ± 0.09 | 0.026 ± 0.0004 | 20 | 0.17 | ||
| G | 0.27 ± 0.02 | 0.032 ± 0.001 | 120 | 1 | 1.2 | |
| C | 2.5 ± 0.4 | 0.050 ± 0.002 | 20 | 0.17 | ||
| E276K | A | 0.72 ± 0.11 | 0.017 ± 0.001 | 24 | 0.39 | |
| T | 2.7 ± 0.3 | 0.028 ± 0.0007 | 10 | 0.16 | ||
| G | 0.34 ± 0.02 | 0.021 ± 0.0002 | 62 | 1 | 0.62 | |
| C | 2.2 ± 0.1 | 0.041 ± 0.0005 | 19 | 0.31 | ||
| Y374N | A | 0.71 ± 0.09 | 0.026 ± 0.001 | 37 | 0.38 | |
| T | 2.0 ± 0.2 | 0.082 ± 0.002 | 41 | 0.42 | ||
| G | 0.39 ± 0.03 | 0.038 ± 0.001 | 97 | 1 | 0.97 | |
| C | 3.3 ± 0.3 | 0.047 ± 0.001 | 14 | 0.14 |
dNTP selectivity ratio, calculated by dividing kcat/Km for each dNTP incorporation by the highest kcat/Km for dNTP incorporation opposite the abasic site. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dGTP incorporation opposite the abasic site by kcat/Km of wild-type pol ι(1–445) for dGTP incorporation opposite the abasic site.
Table 6. Steady-State Kinetic Parameters for dNTP Incorporation Opposite 8-oxoG by Wild-Type and Variant hPols ι(1-445) in the Presence of 0.15 mM Mn2+.
| pol ι(1–445) | dNTP | Km (μM) | kcat (s–1) | kcat/Km (s–1 mM–1) | finsa | relative efficiencyb |
|---|---|---|---|---|---|---|
| wild-type | A | 2.0 ± 0.3 | 0.015 ± 0.001 | 7.5 | 0.83 | |
| T | 30 ± 2 | 0.046 ± 0.002 | 1.5 | 0.17 | ||
| G | 0.90 ± 0.07 | 0.014 ± 0.0002 | 16 | 1.8 | ||
| C | 4.2 ± 0.2 | 0.038 ± 0.001 | 9.0 | 1 | 1 | |
| Δ1–25 | A | 17 ± 1 | 0.021 ± 0.0003 | 1.2 | 0.092 | |
| T | 5.2 ± 0.3 | 0.028 ± 0.001 | 5.4 | 0.42 | ||
| G | 0.21 ± 0.1 | 0.0082 ± 0.0004 | 39 | 3.0 | ||
| C | 1.8 ± 0.2 | 0.023 ± 0.001 | 13 | 1 | 1.4 | |
| ΔD17 | A | 2.0 ± 0.2 | 0.017 ± 0.0003 | 8.5 | 0.77 | |
| T | 29 ± 1 | 0.055 ± 0.001 | 5.3 | 0.48 | ||
| G | 0.53 ± 0.06 | 0.013 ± 0.0003 | 25 | 2.3 | ||
| C | 3.5 ± 0.2 | 0.040 ± 0.0006 | 11 | 1 | 1.2 | |
| R96G | A | 3.3 ± 0.3 | 0.0017 ± 0.00004 | 0.52 | 0.43 | |
| T | 13 ± 1 | 0.0046 ± 0.0001 | 0.35 | 0.29 | ||
| G | 1.8 ± 0.2 | 0.0038 ± 0.0001 | 2.1 | 1.8 | ||
| C | 8.5 ± 0.5 | 0.010 ± 0.0002 | 1.2 | 1 | 0.13 | |
| I261M | A | 1.4 ± 0.2 | 0.015 ± 0.0004 | 11 | 1.0 | |
| T | 25 ± 3 | 0.026 ± 0.001 | 1.0 | 0.091 | ||
| G | 0.66 ± 0.07 | 0.012 ± 0.0003 | 18 | 1.6 | ||
| C | 3.1 ± 0.1 | 0.034 ± 1.0003 | 11 | 1 | 1.2 | |
| E276K | A | 2.2 ± 0.3 | 0.022 ± 0.001 | 10 | 1.4 | |
| T | 24 ± 4 | 0.044 ± 0.003 | 1.8 | 0.26 | ||
| G | 0.60 ± 0.04 | 0.016 ± 0.0002 | 27 | 3.9 | ||
| C | 3.2 ± 0.2 | 0.022 ± 0.0004 | 6.9 | 1 | 0.77 | |
| Y374N | A | 1.4 ± 0.1 | 0.015 ± 0.0003 | 11 | 1.0 | |
| T | 29 ± 6 | 0.025 ± 0.003 | 0.86 | 0.078 | ||
| G | 0.72 ± 0.06 | 0.018 ± 0.0003 | 25 | 2.3 | ||
| C | 3.6 ± 0.1 | 0.040 ± 0.0003 | 11 | 1 | 1.2 |
Misinsertion frequency, calculated by dividing kcat/Km for each dNTP incorporation by the kcat/Km for dCTP incorporation opposite 8-oxoG. All values are presented to two significant digits.
Relative efficiency, calculated by dividing kcat/Km of each pol ι(1–445) for dCTP incorporation opposite 8-oxoG by kcat/Km of wild-type pol ι(1–445) for dCTP incorporation opposite 8-oxoG.
Binding of the Wild-Type Pol ι(1–445) and the Variants Δ1–25 and R96G to DNA Substrate
To analyze the binding affinities of two dysfunctional pol ι variants, Δ1–25 and R96G, for the primer-template DNA substrate in the presence of either Mg2+ or Mn2+, we performed fluorescence polarization experiments. The equilibrium dissociation constants (Kd,DNA) of wild-type pol ι and the Δ1–25 and R96G variants were estimated by fitting the fluorescence polarization values of fluorescein-labeled DNA substrates (18-FAM-mer primers annealed to unmodified 36-G-mer templates) as a function of the pol ι concentration to a quadratic equation (Table 8). Wild-type pol ι bound weakly to DNA with a Kd,DNA of 490 and 840 nM, respectively, at 0.15 and 1 mM Mg2+concentrations, while binding relatively tightly to DNA with a Kd,DNA of 68 nM at 0.15 mM Mn2+ (but not at 1 mM Mn2+). The R96G variant had Kd,DNA values 2- to 3-fold higher than wild-type in the presence of either metal, indicating a slight decrease in DNA binding affinity of pol ι by this variant. In contrast, the Δ1–25 variant bound DNA much more tightly (20- to 29-fold) than the wild-type protein in the presence of 0.15 or 1 mM Mg2+. Similarly, the Δ1–25 variant also had a DNA-binding affinity (Kd,DNA = 140 nM) ∼10-fold lower than that of wild-type in the presence of 5 mM Mg2+ (results not shown). This large difference in the DNA-binding affinities was reduced to only 4-fold in the presence of 0.15 mM of Mn2+. These data indicate that the N-terminal region (residues 1–25), rich in negatively charged amino acids, might severely interfere with DNA substrate binding of wild-type pol ι, but this interference could be totally ablated by the deletion of N-terminal 25 amino acids or could be substantially overcome by the low level of Mn2+. These features might at least in part explain the substantial increase in polymerase activity of the Δ1–25 variant versus the wild-type pol ι seen in the presence of Mg2+ (Figure 2, Tables 2–4) but not with Mn2+ (Figure 3, Tables 5–7). Here we also note that the Kd,DNA value (68 nM) of wild-type pol ι with Mn2+ would yield a relatively high dissociation rate, koff (∼0.68 s–1), which might make pol ι very distributive.
Table 8. Kd Values of Wild-Type hPol ι (1-445), and the Δ1–25 and R96G Variants for 18-FAM-mer/36-G-mer DNA Substrate in the Presence of MnCl2 or MgCl2.
|
Kd (nM) of pol ι(1–445) |
|||
|---|---|---|---|
| MnCl2 or MgCl2 | wild-type | Δ1–25 | R96G |
| 0.15 mM MnCl2 | 68 ± 11 | 17 ± 3 | 220 ± 50 |
| 0.15 mM MgCl2 | 490 ± 70 | 17 ± 3 | 900 ± 200 |
| 1 mM MnCl2 | 840 ± 250 | 88 ± 16 | 1800 ± 400 |
| 1 mM MgCl2 | 840 ± 320 | 41 ± 7 | 1800 ± 600 |
Discussion
In this study we examined the biochemical properties of six nonsynonymous coding variants of human pol ι in comparison with the wild-type, based on the newly annotated ORF sequence information. Four missense and two deletion variations were selected for this study because they were expected to cause functional alterations on pol ι on the basis of the nature of the changes, positions in the pol ι catalytic core, and predicted effects. Our biochemical data revealed that the R96G variation severely impairs the efficiency of pol ι for both normal and translesion syntheses at G, N2-EtG, 8-oxoG, O6-MeG, and an abasic site regardless of the presence of Mn2+ or Mg2+ (except for the case of a Mn2+-facilitated C insertion opposite N2-EtG), whereas the Δ1–25 deletion variation, which is equivalent to the previously misannotated wild-type, considerably increases pol ι efficiency for both normal and translesion syntheses in the presence of Mg2+ (but not with Mn2+). The Δ1–25 variation greatly (20- to 29-fold) increased the DNA-binding affinity of pol ι in the presence of Mg2+, which was much less prominent (only about ∼4-fold) in the presence of a low concentration of Mn2+. We also note that the poor DNA-binding affinity of the wild-type pol ι was considerably improved by the addition of a low level of Mn2+ when compared to that of Mg2+. In this study we report that two rare, nonsynonymous POLI genetic variations can affect the TLS activities of human pol ι in opposite directions in vitro and that wild-type pol ι is substantially more sluggish in activity than expected from analysis of the previous wild-type protein (Δ1–25) in the presence of Mg2+, unlike in the presence of Mn2+.
To the best of our knowledge, this is the first study analyzing biochemical alterations in germline genetic variants and the newly denoted wild-type version of human pol ι, using in vitro polymerase activity, enzyme kinetics, and binding assays. Two of the six POLI genetic variations studied here were found to significantly affect the in vitro enzymatic function of pol ι in translesion DNA synthesis and DNA substrate binding. These functional genetic variations of pol ι appear to be rare, similar to dysfunctional pol κ variants we reported recently,35 in that minor allele frequencies (MAFs) of the Δ1–25 and R96G pol ι variations are ∼0.1% and 0.6%, respectively, in public databases (Table 1). The biological significance of rare frequency genetic variations should not be overlooked in that recent reports suggest the possible relevance of rare genetic variations to complex human diseases. Rare genetic variations have been proposed as a potential source of missing disease heritability that has not been fully explained by common genetic variations,39 and the recent experimental reports support this view by providing evidence that rare genetic variations are abundantly present in human populations and are more likely to be misfunctional than common variations.40,41 Only 19 nonsynonymous germline variations were described in 2008 (including two reported cancer-related SNPs28,29), but now a total of 91 nonsynonymous germline variations have been listed in dbSNP, most of which seem to be rare in that MAFs of 89 variations are either <1% or unavailable yet. About 46 types of missense and nonsense somatic POLI gene mutations have also been described from various human cancer tissues such as endometrium, large intestine, and lung in the COSMIC database (www.sanger.ac.uk/genetics/CGP/cosmic/), but their functional effects have not been revealed yet. Although we focused on only six selected “putatively functional” coding variants, our biochemical investigation can be a useful initial step to find pol ι variations that have functional impacts and understanding their mechanistic implications. Eleven additional nonsynonymous coding POLI variations, located in polymerase core domains and also putatively deleterious, have been added in dbSNP since our study began, and those functional candidates are also under our investigation.
The six germline pol ι variants characterized in this study can be classified into three types according to the changes of relative polymerase efficiencies opposite G and the lesions in the presence of the added metal (Mg2+ or Mn2+) compared to wild-type (Tables 2–7). The first type is the defective variant (R96G), which is severely impaired in both Mg2+- and Mn2+-dependent polymerase efficiencies for both normal synthesis and lesion bypass. Surprisingly, this particular variant exhibited a substantial improvement in the Mn2+-dependent N2-EtG bypass, i.e., large increases (9-fold and 24-fold, respectively) in both efficiency and fidelity for Mn2+-dependent dCTP insertion opposite N2-EtG compared to wild-type enzyme (Table 5), from which we may speculate the likelihood of a beneficial effect of this variation to efficiently and faithfully bypass such a lesion at the expense of general diminution in polymerase function. The second type is the hyperactive variant (Δ1–25). This N-terminal truncation variant displayed considerable enhancement only in Mg2+-dependent polymerase efficiency but without any alteration in Mn2+-dependent efficiency. The last type is the “wild-type-like” variants (Δ17, I261M, E276K, and Y374N), which retain both normal and TLS polymerase efficiencies similar to those of wild-type in the presence of either Mg2+ or Mn2+. The fidelity of nucleotide insertion opposite G and the lesions does not appear to be altered in most variants except for the R96G variant when compared to wild-type (Tables 2–7). The R96G variant had a reduced fidelity in Mg2+-dependent 8-oxoG bypass due to a greater reduction of insertion efficiency for correct dCTP than for the other nucleotides, as well as an improved fidelity in Mn2+-dependent N2-EtG bypass (vide supra). Although all four studied missense variations (R96G, I261M, E276K, and Y374N) were predicted to be damaging by SIFT and/or PolyPhen, only the former variant was found to be deleterious, but the latter three variants were found to have nearly neutral effects on pol ι function in our study, indicating the substantial false-positive nature of in silico prediction. False-positive errors of 20 and 9% have been reported with SIFT and Polyphen, respectively.42 Taken together, our results suggest the necessity and importance of biochemical approaches to verify the functional alterations in genetic variants, although in silico predictions may still be useful for screening putatively damaging genetic variations for functional studies.
Three-dimensional structures of the catalytic core of pol ι have been determined in complex with various DNA substrates with/without incoming nucleotides, which would be useful for the mechanistic understanding of genetic variants (Figure 1).14,37,38,43−45 The catalytic core of pol ι contains the palm, fingers, thumb, and polymerase-associated domain (PAD) domains, forming the unique narrow active site that is not conducive to Watson–Crick base pairing.14,44 The mechanistic basis for our finding that the R96G variation caused a severe reduction in pol ι catalytic activity can be explained by a structural role of Arg96 in the active site as previously revealed.14,44 Thr90, Tyr93, and Arg96 from the fingers domain and Lys239 from the palm domain (the latter three of which are conserved in all Y-family DNA polymerases) function to stabilize the incoming nucleotide by making hydrogen bonds with the triphosphate moiety in the pol ι-DNA-dNTP ternary complex. In particular, Arg96 of pol ι undergoes a substantial conformational change (facing inward) upon nucleotide binding to form hydrogen bonds with the β- and γ-phosphates of the incoming nucleoside triphosphate. Our finding of defective function in the R96G variant is in good agreement with the previous report that Ala substitution of the homologous Arg67 residue in yeast pol η also considerably diminishes its polymerase activity, suggesting that this conserved Arg residue is commonly crucial for catalytic function in Y-family polymerases.46 However, it is not clear how the R96G variant unexpectedly displays an increased competence in an error-free Mn2+-dependent N2-EtG bypass. Pol ι normally accommodates the N2-EtG in the syn conformation paired with incoming dCTP in the active site.43 The R96G variation abolishes the long and positively charged side chain on Arg96 and thus might lead to an altered conformation that hinders nucleoside triphosphate binding and catalysis for most of the templates but is perhaps well suited only for Mn2+-assisted pairing between a template N2-EtG (syn) and an incoming dCTP (anti) in the active site pocket.
We established, for the first time, that the Δ1–25 variant (i.e., the former wild-type protein) has a considerably higher Mg2+-dependent polymerase activity (but with no alteration in Mn2+-dependent polymerase activity) and a much higher DNA binding affinity than wild-type pol ι. This unexpected biochemical trait is certainly due to the absence of the N-terminal extension of 25 amino acids (containing 12 acidic residues). As far as we know, all reported pol ι structures have been resolved only from the catalytic core fragment (amino acids 26–445) lacking the N-terminal 25 amino acids, and thus, the structural role of the N-terminal extension is not known yet. However, it is evident from our observations that this negatively charged N-terminal extension can interfere with both DNA substrate binding and Mg2+-dependent polymerase activity of pol ι, but these hindrances can be largely weakened by a low level of Mn2+. Only Mn2+ (at low concentration, 150 μM) but not Mg2+ seems to considerably overcome the inherently poor DNA binding trait of the wild-type pol ι, possibly by masking or neutralizing the negative charged N-terminal region of this enzyme and thus reducing the potential electrostatic repulsion from the negatively charged phosphosugar backbone of DNA substrates. Pol ι is also known to have a greater preference for Mn2+ over Mg2+ as a divalent metal for polymerase catalysis and shows maximal polymerase activity at a low concentration of Mn2+ (with the optimum around 50–250 μM).20 Taken together, we can postulate that the wild-type pol ι may require a low level of Mn2+ rather than Mg2+ as a metal for performing proper polymerase function for two reasons: (i) mediating efficient catalysis and (ii) facilitating tight DNA substrate binding, although the structural basis for such a scenario has not been revealed yet. These biochemical traits of wild-type pol ι are likely to be advantageous in that cells might be able to control the error-prone polymerase function of pol ι by varying the local concentration of metal near DNA damage sites in cell nucleus, which might permit the proficient TLS events by pol ι only in the presence of a low level of Mn2+. Although the intracellular level of Mn2+ is physiologically very low (0.1 to 40 μM),47−49 that of Mn2+ could be increased in some pathological cell conditions (e.g., Mn2+ overexposure or misregulated Mn2+ homeostasis),50,51 which might stimulate the activity of error-prone pol ι, as well as other Mn2+-dependent DNA-processing enzymes in the nucleus, and thus induce genomic instability. Mn2+ has also been known to alter both catalytic efficiency and fidelity of other DNA polymerases including pol β, pol λ, and pol μ, albeit at high concentrations.52−54 The effects might be related to their active site features with regard to the cofactor Mn2+, which has a slightly smaller ionic radius and a more relaxed coordination than Mg2+, and might allow different interactions with nucleoside triphosphate, DNA, and catalytic residues in the polymerase active site, which could alter polymerase function.55,56
It is very conceivable that the cellular pol ι-mediated TLS capacity could be substantially diminished or enhanced in individuals having these two identified dysfunctional POLI gene variations. If a cell possessed only the R96G pol ι variant (i.e., homozygote), then most of pol ι-mediated TLS events (except for the increased events of accurate N2-EtG bypass) would decrease in cells. Conversely, if the cell had only the Δ1–25 pol ι variant, then pol ι-mediated Mg2+-dependent TLS events would increase in cells. We might expect that high replication errors occur with hyperactive pol ι in normal DNA replication because pol ι is inherently error-prone in its general nature. However, predicting how these two POLI gene variations would lead to overall TLS-associated mutation outcomes in cells is not straightforward due to the complex TLS properties of pol ι and the cellular existence of other competitive TLS polymerases. Eukaryotic TLS process is carried out not by a single TLS polymerase but by a set of specialized TLS polymerases recruited at a site of DNA damage, although the extent of their individual participation varies depending on the lesion type. The mutation consequence from a particular DNA lesion in cells can be governed by the total set of enzymatic behavior of multiple TLS polymerases, mainly Y-family polymerases, utilized for the lesion substrate. Unlike Y-family pols η and κ, which are generally considered to be specialized for efficient and error-free bypasses at their cognate lesions, UV-induced pyrimidine dimers and bulky N2-G adducts, respectively, it is not clear what kind of DNA lesions could be the cognate substrate lesions for pol ι. There is much kinetic and structural evidence suggesting that some DNA lesions such as N2-EtG, O6-MeG, 8-oxoG, and abasic sites might be among the favored substrate lesions for pol ι, in that this enzyme can incorporate nucleotides opposite those lesions as efficiently as (or slightly less efficiently than) opposite an unmodified G by utilizing its unique active site and/or Hoogsteen base pairing but with different nucleotide selectivities.12,17,19,37,38,43,45 Pol ι seems to mediate relatively error-free bypass with some minor-groove N2-G lesions such as N2-EtG, while performing a relatively error-prone bypass opposite major-groove O6-alkylG adducts such as O6-MeG.12,17,37,43 In a support of this view, pol ι (as well as pol κ) has been implicated in the error-free bypass of N2-carboxyMeG and N2-carboxyEtG lesions in mouse cells.57 Pol ι has also been implicated to play a protective role from oxidative DNA damage, possibly by mediating both the error-free TLS and the base excision repair of 8-oxoG.38,58,59 However, the fidelity of 8-oxoG bypass by pol ι seems to be sequence context-dependent because the incorrect dGTP was incorporated more favorably than dCTP opposite template 8-oxoG in our different sequence context as observed here. Experimental evidence support the dual and conflicting “Janus” roles of pol ι in cancers: a protective role of pol ι in mouse lung and skin carcinogenesis22,23 and a hypermutagenic role of pol ι, upregulated in various human cancer tissues.24−27 Under these circumstances, it can be hypothesized that individual humans who possess the R96G or the Δ1–25 pol ι variation might have an altered but complex risk for mutation and cancer to various carcinogen exposures, depending on the DNA damage status in their target tissues. In this aspect, it is necessary to perform further studies to verify the in vivo impact of those functional pol ι variants in both cellular and organismal contexts. It is also worth examining other nonsynonymous coding variations located in protein interaction domains of pol ι to bind other proteins such as PCNA and ubiquitin, in that they might alter cellular TLS events such as polymerase switching and coordination. It is also plausible that noncoding functional variations in the gene regulatory regions of pol ι might be able to alter pol ι expression levels in cells and thus influence TLS events, although this aspect was out of scope for this study.
In conclusion, our results suggest that two germline genetic variations in human POLI gene may either hinder or promote the TLS capability of pol ι with various DNA lesions in vitro, possibly leading to different and distinctive mutation phenotypes in genetically affected cells and individuals, i.e., facilitating or protecting against mutagenesis/carcinogenesis after exposure to genotoxic carcinogens. The verification of two dysfunctional genetic variations for human pol ι in this study may provide insight into our understanding of individual differences in cellular TLS capacities to various carcinogen-derived DNA lesions. Such functional genetic alterations in TLS DNA polymerases might be expected to play some part in determining an individual’s genomic mutational susceptibility to specific carcinogens and the related cancer risk in human populations, although further in vivo or clinical investigations are needed to elucidate these associations.
Glossary
Abbreviations
- A
adenine
- BSA
bovine serum albumin
- C
cytosine
- Et
ethyl
- FAM
6-carboxyfluorescein
- G
guanine
- Me
methyl
- 8-oxoG
8-oxo-7,8-dihydroG
- PCR
polymerase chain reaction
- pol
DNA polymerase
- SDS
sodium dodecyl sulfate
- T
thymine
- TLS
translesion synthesis
Supporting Information Available
Analysis of human pol ι (1–445) wild-type and variant proteins by SDS-polyacrylamide gel electrophoresis (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant 2012R1A1A2042391) (to J.-Y.C.), Samsung Biomedical Research Institute grant, #SBRI SMX1132091 (to J.-Y.C.), and the National Institutes of Health Grants R01 CA183895 (to R.L.E.) and R01 ES010375 (to F.P.G.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2006) DNA Repair And Mutagenesis, 2nd ed., American Society for Microbiology Press, Washington, D.C. [Google Scholar]
- Giglia-Mari G.; Zotter A.; Vermeulen W. (2011) DNA damage response. Cold Spring Harb. Perspect. Biol. 3, a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J. H. (2009) DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- Jackson S. P.; Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D. O.; Agrawal M.; Shen J.; Terry M. B.; Zhang F. F.; Senie R. T.; Motykiewicz G.; Santella R. M. (2005) DNA repair capacity of lymphoblastoid cell lines from sisters discordant for breast cancer. J. Natl. Cancer Inst. 97, 127–132. [DOI] [PubMed] [Google Scholar]
- Gorlova O. Y.; Weng S. F.; Zhang Y.; Amos C. I.; Spitz M. R.; Wei Q. (2008) DNA repair capacity and lung cancer risk in never smokers. Cancer Epidemiol. Biomarkers Prev. 17, 1322–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. E.; Gorlova O. Y.; Ying J.; Qiao Y.; Weng S. F.; Lee A. T.; Gregersen P. K.; Spitz M. R.; Amos C. I.; Wei Q. (2013) Genome-wide association study reveals novel genetic determinants of DNA repair capacity in lung cancer. Cancer Res. 73, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberg A. J.; Jorgensen T. J.; Ruczinski I.; Wheless L.; Shugart Y. Y.; Berthier-Schaad Y.; Kessing B.; Hoffman-Bolton J.; Helzlsouer K. J.; Kao W. H.; Francis L.; Alani R. M.; Smith M. W.; Strickland P. T. (2013) DNA repair gene variants in relation to overall cancer risk: a population-based study. Carcinogenesis 34, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-Y., Eoff R. E., and Guengerich F. P. (2011) Bypass DNA polymerases, In Chemical Carcinogenesis (Penning T. M., Ed.) pp 345–373, Humana Press, New York. [Google Scholar]
- Choi J.-Y.; Angel K. C.; Guengerich F. P. (2006) Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase κ. J. Biol. Chem. 281, 21062–21072. [DOI] [PubMed] [Google Scholar]
- Choi J.-Y.; Guengerich F. P. (2005) Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J. Mol. Biol. 352, 72–90. [DOI] [PubMed] [Google Scholar]
- Choi J.-Y.; Guengerich F. P. (2006) Kinetic evidence for inefficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase ι. J. Biol. Chem. 281, 12315–12324. [DOI] [PubMed] [Google Scholar]
- Choi J.-Y.; Guengerich F. P. (2008) Kinetic analysis of translesion synthesis opposite bulky N2- and O6-alkylguanine DNA adducts by human DNA polymerase REV1. J. Biol. Chem. 283, 23645–23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D. T.; Johnson R. E.; Prakash S.; Prakash L.; Aggarwal A. K. (2004) Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature 430, 377–380. [DOI] [PubMed] [Google Scholar]
- Choi J.-Y.; Lim S.; Eoff R. L.; Guengerich F. P. (2009) Kinetic analysis of base-pairing preference for nucleotide incorporation opposite template pyrimidines by human DNA polymerase ι. J. Mol. Biol. 389, 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac K. N.; Ling H. (2009) Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase iota. EMBO J. 28, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-Y.; Chowdhury G.; Zang H.; Angel K. C.; Vu C. C.; Peterson L. A.; Guengerich F. P. (2006) Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 281, 38244–38256. [DOI] [PubMed] [Google Scholar]
- Vaisman A.; Frank E. G.; Iwai S.; Ohashi E.; Ohmori H.; Hanaoka F.; Woodgate R. (2003) Sequence context-dependent replication of DNA templates containing UV-induced lesions by human DNA polymerase iota. DNA Repair 2, 991–1006. [DOI] [PubMed] [Google Scholar]
- Choi J.-Y.; Lim S.; Kim E. J.; Jo A.; Guengerich F. P. (2010) Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J. Mol. Biol. 404, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E. G.; Woodgate R. (2007) Increased catalytic activity and altered fidelity of human DNA polymerase ι in the presence of manganese. J. Biol. Chem. 282, 24689–24696. [DOI] [PubMed] [Google Scholar]
- Dumstorf C. A.; Clark A. B.; Lin Q.; Kissling G. E.; Yuan T.; Kucherlapati R.; McGregor W. G.; Kunkel T. A. (2006) Participation of mouse DNA polymerase iota in strand-biased mutagenic bypass of UV photoproducts and suppression of skin cancer. Proc. Natl. Acad. Sci. U.S.A. 103, 18083–18088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkumo T.; Kondo Y.; Yokoi M.; Tsukamoto T.; Yamada A.; Sugimoto T.; Kanao R.; Higashi Y.; Kondoh H.; Tatematsu M.; Masutani C.; Hanaoka F. (2006) UV-B radiation induces epithelial tumors in mice lacking DNA polymerase eta and mesenchymal tumors in mice deficient for DNA polymerase iota. Mol. Cell. Biol. 26, 7696–7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi M.; Osanai M.; Hayashi Y.; Koentgen F.; Lee G. H. (2014) The error-prone DNA polymerase iota provides quantitative resistance to lung tumorigenesis and mutagenesis in mice. Oncogene 33, 3612–3617. [DOI] [PubMed] [Google Scholar]
- Yang J.; Chen Z.; Liu Y.; Hickey R. J.; Malkas L. H. (2004) Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 64, 5597–5607. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Zhang S.; Xie L.; Liu P.; Xie F.; Wu J.; Cao J.; Ding W. Q. (2012) Overexpression of DNA polymerase iota (Poliota) in esophageal squamous cell carcinoma. Cancer Sci. 103, 1574–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertella M. R.; Lau A.; O’Connor M. J. (2005) The overexpression of specialized DNA polymerases in cancer. DNA Repair 4, 583–593. [DOI] [PubMed] [Google Scholar]
- Yuan F.; Xu Z.; Yang M.; Wei Q.; Zhang Y.; Yu J.; Zhi Y.; Liu Y.; Chen Z.; Yang J. (2013) Overexpressed DNA polymerase iota regulated by JNK/c-Jun contributes to hypermutagenesis in bladder cancer. PLoS One 8, e69317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedeke M.; Linnert C. M.; Hofer M. D.; Surowy H. M.; Rinckleb A. E.; Hoegel J.; Kuefer R.; Rubin M. A.; Vogel W.; Maier C. (2009) Predisposition for TMPRSS2-ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol. Biomarkers Prev. 18, 3030–3035. [DOI] [PubMed] [Google Scholar]
- Sakiyama T.; Kohno T.; Mimaki S.; Ohta T.; Yanagitani N.; Sobue T.; Kunitoh H.; Saito R.; Shimizu K.; Hirama C.; Kimura J.; Maeno G.; Hirose H.; Eguchi T.; Saito D.; Ohki M.; Yokota J. (2005) Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int. J. Cancer 114, 730–737. [DOI] [PubMed] [Google Scholar]
- Ng P. C.; Henikoff S. (2001) Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V.; Bork P.; Sunyaev S. (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I. A.; Schmidt S.; Peshkin L.; Ramensky V. E.; Gerasimova A.; Bork P.; Kondrashov A. S.; Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-Y.; Guengerich F. P. (2004) Analysis of the effect of bulk at N2-alkylguanine DNA adducts on catalytic efficiency and fidelity of the processive DNA polymerases bacteriophage T7 exonuclease- and HIV-1 reverse transcriptase. J. Biol. Chem. 279, 19217–19229. [DOI] [PubMed] [Google Scholar]
- Boosalis M. S.; Petruska J.; Goodman M. F. (1987) DNA polymerase insertion fidelity: gel assay for site-specific kinetics. J. Biol. Chem. 262, 14689–14696. [PubMed] [Google Scholar]
- Song I.; Kim E. J.; Kim I. H.; Park E. M.; Lee K. E.; Shin J. H.; Guengerich F. P.; Choi J.-Y. (2014) Biochemical characterization of eight genetic variants of human DNA polymerase κ involved in error-free bypass across bulky N2-guanyl DNA adducts. Chem. Res. Toxicol. 27, 919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketkar A.; Zafar M. K.; Maddukuri L.; Yamanaka K.; Banerjee S.; Egli M.; Choi J.-Y.; Lloyd R. S.; Eoff R. L. (2013) Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity. Chem. Res. Toxicol. 26, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pence M. G.; Choi J.-Y.; Egli M.; Guengerich F. P. (2010) Structural basis for proficient incorporation of dTTP opposite O6-methylguanine by human DNA polymerase ι. J. Biol. Chem. 285, 40666–40672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac K. N.; Ling H. (2011) Unique active site promotes error-free replication opposite an 8-oxo-guanine lesion by human DNA polymerase iota. Proc. Natl. Acad. Sci. U.S.A. 108, 3210–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T. A.; Collins F. S.; Cox N. J.; Goldstein D. B.; Hindorff L. A.; Hunter D. J.; McCarthy M. I.; Ramos E. M.; Cardon L. R.; Chakravarti A.; Cho J. H.; Guttmacher A. E.; Kong A.; Kruglyak L.; Mardis E.; Rotimi C. N.; Slatkin M.; Valle D.; Whittemore A. S.; Boehnke M.; Clark A. G.; Eichler E. E.; Gibson G.; Haines J. L.; Mackay T. F.; McCarroll S. A.; Visscher P. M. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. R.; Wegmann D.; Ehm M. G.; Kessner D.; St. Jean P.; Verzilli C.; Shen J.; Tang Z.; Bacanu S. A.; Fraser D.; Warren L.; Aponte J.; Zawistowski M.; Liu X.; Zhang H.; Zhang Y.; Li J.; Li Y.; Li L.; Woollard P.; Topp S.; Hall M. D.; Nangle K.; Wang J.; Abecasis G.; Cardon L. R.; Zollner S.; Whittaker J. C.; Chissoe S. L.; Novembre J.; Mooser V. (2012) An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Ge D.; Maia J. M.; Zhu M.; Petrovski S.; Dickson S. P.; Heinzen E. L.; Shianna K. V.; Goldstein D. B. (2011) A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am. J. Hum. Genet. 88, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P. C.; Henikoff S. (2006) Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 7, 61–80. [DOI] [PubMed] [Google Scholar]
- Pence M. G.; Blans P.; Zink C. N.; Hollis T.; Fishbein J. C.; Perrino F. W. (2009) Lesion bypass of N2-ethylguanine by human DNA polymerase ι. J. Biol. Chem. 284, 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D. T.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. (2006) An incoming nucleotide imposes an anti to syn conformational change on the templating purine in the human DNA polymerase-iota active site. Structure 14, 749–755. [DOI] [PubMed] [Google Scholar]
- Nair D. T.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. (2009) DNA synthesis across an abasic lesion by human DNA polymerase iota. Structure 17, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. E.; Trincao J.; Aggarwal A. K.; Prakash S.; Prakash L. (2003) Deoxynucleotide triphosphate binding mode conserved in Y family DNA polymerases. Mol. Cell. Biol. 23, 3008–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash D. E.; Schramm V. L. (1982) Determination of free and bound manganese(II) in hepatocytes from fed and fasted rats. J. Biol. Chem. 257, 9261–9264. [PubMed] [Google Scholar]
- Markesbery W. R.; Ehmann W. D.; Alauddin M.; Hossain T. I. (1984) Brain trace element concentrations in aging. Neurobiol. Aging 5, 19–28. [DOI] [PubMed] [Google Scholar]
- Versieck J. (1985) Trace elements in human body fluids and tissues. Crit. Rev. Clin. Lab. Sci. 22, 97–184. [DOI] [PubMed] [Google Scholar]
- Crossgrove J.; Zheng W. (2004) Manganese toxicity upon overexposure. NMR Biomed. 17, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez N.; Diaz de la Loza Mdel C.; Andreson B.; Monje-Casas F.; Rothstein R.; Wellinger R. E. (2012) Impaired manganese metabolism causes mitotic misregulation. J. Biol. Chem. 287, 18717–18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. S.; Eichler D. C.; Korn D. (1977) Effect of Mn2+ on the in vitro activity of human deoxyribonucleic acid polymerase beta. Biochemistry 16, 4927–4934. [DOI] [PubMed] [Google Scholar]
- Blanca G.; Shevelev I.; Ramadan K.; Villani G.; Spadari S.; Hübscher U.; Maga G. (2003) Human DNA polymerase lambda diverged in evolution from DNA polymerase beta toward specific Mn(++) dependence: a kinetic and thermodynamic study. Biochemistry 42, 7467–7476. [DOI] [PubMed] [Google Scholar]
- Dominguez O.; Ruiz J. F.; Lain de Lera T.; Garcia-Diaz M.; Gonzalez M. A.; Kirchhoff T.; Martinez A. C.; Bernad A.; Blanco L. (2000) DNA polymerase mu (Pol mu), homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J. 19, 1731–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier H.; Sawaya M. R.; Wolfle W.; Wilson S. H.; Kraut J. (1996) Crystal structures of human DNA polymerase β complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry 35, 12742–12761. [DOI] [PubMed] [Google Scholar]
- Yang W.; Lee J. Y.; Nowotny M. (2006) Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell 22, 5–13. [DOI] [PubMed] [Google Scholar]
- Yuan B.; You C.; Andersen N.; Jiang Y.; Moriya M.; O’Connor T. R.; Wang Y. (2011) The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J. Biol. Chem. 286, 17503–17511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirouac K. N.; Ling H. (2011) Poli: Shining light on repair of oxidative DNA lesions and mutations. Cell Cycle 10, 1520–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta T. B.; Nakajima S.; Zlatanou A.; Despras E.; Couve-Privat S.; Ishchenko A.; Sarasin A.; Yasui A.; Kannouche P. (2008) Human DNA polymerase iota protects cells against oxidative stress. EMBO J. 27, 2883–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Wen L.; Gao X.; Jin C.; Xue Y.; Yao X. (2009) DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.