

Abstract
We have previously identified the pyrazolobenzothiazine scaffold as a promising chemotype against hepatitis C virus (HCV) NS5B polymerase, a validated and promising anti-HCV target. Herein we describe the design, synthesis, enzymatic, and cellular characterization of new pyrazolobenzothiazines as anti-HCV inhibitors. The binding site for a representative derivative was mapped to NS5B palm site I employing a mutant counterscreen assay, thus validating our previous in silico predictions. Derivative 2b proved to be the best selective anti-HCV derivative within the new series, exhibiting a IC50 of 7.9 μM against NS5B polymerase and antiviral effect (EC50 = 8.1 μM; EC90 = 23.3 μM) coupled with the absence of any antimetabolic effect (CC50 > 224 μM; SI > 28) in a cell based HCV replicon system assay. Significantly, microscopic analysis showed that, unlike the parent compounds, derivative 2b did not show any significant cell morphological alterations. Furthermore, since most of the pyrazolobenzothiazines tested altered cell morphology, this undesired aspect was further investigated by exploring possible perturbation of lipid metabolism during compound treatment.
Introduction
Hepatitis C virus (HCV) infection affects approximately 180 million people worldwide and is the main cause of hepatocellular carcinoma and liver transplantation in industrialized countries.1,2 Among several infectious diseases, chronic HCV infection is one of the hardest to treat. Currently there is no vaccine against HCV,3 and therapy consists of a NS3/4A protease inhibitor, i.e., boceprevir4 or telaprevir,5 administered in combination with interferon α (pegIFN-α) and ribavirin (RBV).6 This regimen improves the sustained virological response (SVR) to about 70% in the most prevalent HCV genotype in industrialized countries, namely, genotype 1, compared to the standard of care (pegIFN-α and RBV). However, the triple therapy still suffers from additional side effects, high cost, and increased pill burden. Thus, the development of a more adherent and efficacious therapy still remains an unmet medical need. A possibility is a combination of direct-acting antiviral agents (DAAs) targeting different HCV proteins that could eliminate the concomitant use of pegIFN-α and RBV.7,8 Almost all the nonstructural proteins (NSs) involved in HCV replication have been extensively studied and targeted to reach the goal, with several DAAs currently in different stages of clinical trials.9,10
The HCV NS5B RNA-dependent RNA polymerase (RdRp) is a validated and attractive target to identify new DAAs, given its key role in viral replication and significant differences from mammalian polymerases.11 In addition to the active site, NS5B harbors at least five allosteric sites: (i) thumb site I (TSI), (ii) thumb site II (TSII), (iii) palm site I (PSI), (iv) palm site II (PSII), and (v) palm site III (PSIII). NS5B inhibitors are classified as nucleoside inhibitors (NIs) or non-nucleoside inhibitors (NNIs) based on whether they bind to the active site or to one of the five allosteric sites, respectively.12−14 Tremendous efforts made by both pharmaceutical companies and academic groups culminated with the identification of DAAs targeting NS5B that are now under clinical evaluation either alone or in combined therapy.9,10 Unfortunately, the development of NS5B inhibitors suffers from high attrition rate and none of them has reached the market yet. Thus, the discovery of new chemotypes able to inhibit NS5B remains of great interest.
In this context, we have directed our efforts toward the identification of new anti-HCV chemotypes acting as NS5B NNIs.15−17 Employing a structure-based discovery approach, we recently reported on the identification of a novel series of pyrazolobenzothiazine-based anti-HCV compounds targeting the NS5B polymerase (Figure 1).17 A preliminary structure–activity relationship (SAR) profiling of this class of NNIs revealed that (i) the best substituent on the N-1-phenyl group of the pyrazolobenzothiazine nucleus was the fluorine atom with the meta position exhibiting the highest activity; (ii) the methanesulfonamide moiety played an important role, since its replacement with an amino group generated inactive molecules in both the anti-NS5B and anti-HCV replicon assays; (iii) an amide spacer was strongly preferred over an ester linkage. Accordingly, compound 1 (Figure 1) emerged as the most active hit within this first series of pyrazolobenzothiazines.17
Figure 1.

(Left) General formula of our first series of anti-NS5B pyrazolobenzothiazines. (Right) Hit compound 1.
Specifically, docking of compound 1 within PSI revealed that (i) the benzene ring and the N-1-phenyl moiety of the pyrazolobenzothiazine nucleus filled two hydrophobic pockets in the PSI; (ii) the sulfonyl function engaged in a direct hydrogen bond with Tyr448 and in a water-mediated interaction with Gly449; (iii) the p-methanesulfonamide phenyl moiety at the C-3 amide group established several polar interactions with the surrounding NS5B residues.17
On the basis of this analysis, we describe herein the design and synthesis of novel pyrazolobenzothiazine derivatives in an attempt to better outline their SAR profile and optimize their biological activities. Furthermore, we report on the effect of these compounds on HCV NS5B polymerase inhibition and on HCV RNA replication. Employing mutant counterscreen assays, we have investigated the interaction of one of the most potent compounds with the NS5B binding pockets to validate its putative binding site, i.e., PSI. Finally, we describe molecular modeling, in vitro ADME, and drug-induced phospholipidosis investigations, aimed at gaining more detailed insight into NS5B pyrazolobenzothiazine inhibitors.
Results and Discussions
Design of the New Pyrazolobenzothiazines
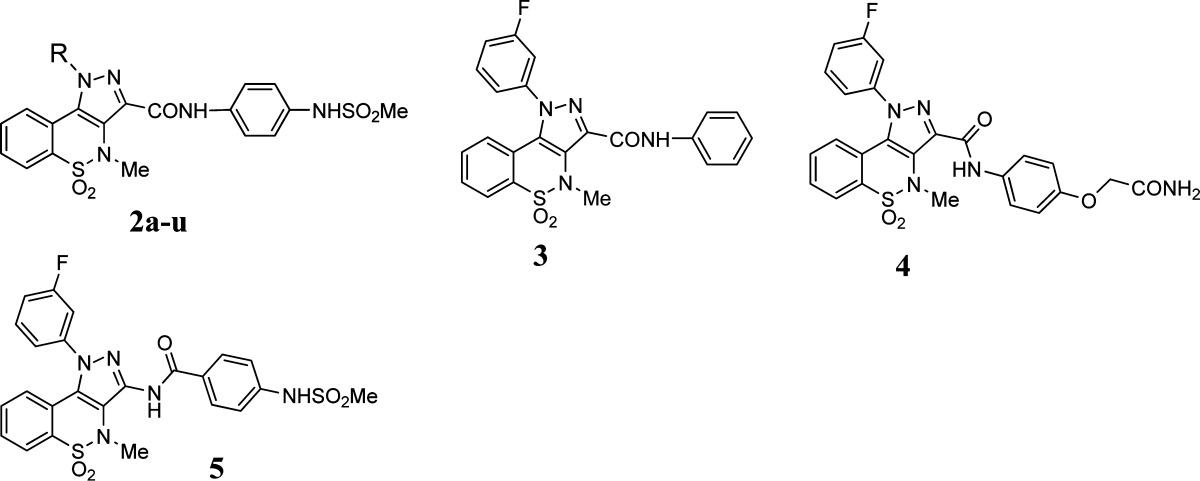
Employing pyrazolobenzothiazine derivative 1 as our reference, we designed and synthesized derivatives 2a–u (Scheme 1 and Table 1) to explore the influence of the interaction of the lipophilic tail at the N-1 position with the hydrophobic pocket in the PSI. In particular, we replaced the m-fluorine atom of hit 1 with the more lipophilic chlorine (2a) and bromine (2d) substituents and also shifted these two new halogens from the meta to the para (2b, 2e) and the ortho (2c, 2f) positions, respectively. The fluorine atom of 1 was also replaced by other groups with varying degrees of lipophilicity such as methyl (2g), methoxy (2j), or a trifluoromethyl (2l). These groups were also placed in para (2h, 2k, and 2m) and ortho (2i, 2n) positions, with the exception of the o-methoxy group because in this case the requisite starting material was not available by our common suppliers. Further modifications carried out on the N-1 tail of the pyrazolobenzothiazine scaffold entailed the introduction of an additional fluorine atom to obtain the meta/para-disubstituted fluorine derivative (2o). Other disubstitution patterns, selected on the basis of commercially available phenylhydrazines as starting material, were also investigated by designing meta/para-disubstituted derivatives such as dichloro (2p), dimethyl (2q), chloro/methyl (2r), and trifluomethyl/chloro (2s) compounds. Furthermore, the phenyl ring at the N-1 position was either replaced by a cyclohexyl fragment to obtain compound 2t or completely removed in order to realize the N-1 hydrogen derivative 2u. In order to definitively confirm the importance of the methanesulfonamide moiety as an essential pharmacophoric element,17 it was removed (derivative 3) or replaced by an oxyacetamide fragment (derivative 4) (Scheme 2 and Table 1); the latter substituent has been reported as a suitable group for anti-HCV activity and in vivo properties in the well-known anti-NS5B benzothiadiazine series described in the literature.18 Finally, with the aim of exploring the influence of the amide on the inhibitory activity of our series of pyrazolobenzothiazine derivatives, it was inverted to give derivative 5 (Scheme 2 and Table 1).
Scheme 1.

Table 1. Biological Activities of the Target Pyrazolobenzothiazines 2a–u, 3, 4, and 5.
| cell-based replicon assay |
||||||
|---|---|---|---|---|---|---|
| compd | R | anti-NS5B assay, IC50 a (μM) | EC50 b (μM) | EC90 c (μM) | CC50 d (μM) | SI e |
| 2a | (3-Cl)Ph | 4.3 ± 0.2 | 9.8 ± 3.2 | 41 ± 11 | >179 | >18 |
| 2b | (4-Cl)Ph | 7.9 ± 0.3 | 8.1 ± 4.3 | 23.3 ± 11.4 | >224 | >28 |
| 2c | (2-Cl)Ph | 17.8 ± 0.5 | 7.4 ± 2.1 | 20.8 ± 3.9 | >224 | >30 |
| 2d | (3-Br)Ph | 7.7 ± 0.2 | 8.3 ± 0.5 | 26 ± 5 | >166 | >20 |
| 2e | (4-Br)Ph | 12.1 ± 0.5 | 7.4 ± 0.8 | 21 ± 5 | >166 | >22 |
| 2f | (2-Br)Ph | 26.5 ± 3.3 | 12.0 ± 0.4 | NDg | 107 ± 34 | 8.9 |
| 2g | (3-Me)Ph | 21.2 ± 4.5 | 5.1 ± 0.6 | 11.8 ± 1.2 | >233 | >46 |
| 2h | (4-Me)Ph | 14.7 ± 0.2 | 4.8 ± 1.7 | 19.6 ± 11.8 | >186 | 39 |
| 2i | (2-Me)Ph | 30.8 ± 3.8 | 14.3 ± 1.1 | NDg | >233 | >16 |
| 2j | (3-OMe)Ph | NAf | 11 ± 2 | NDg | >181 | >16 |
| 2k | (4-OMe)Ph | NAf | 8.1 ± 1.5 | 25.0 ± 7.3 | >226 | >28 |
| 2l | (3-CF3)Ph | 23.7 ± 0.9 | 7.7 ± 1.9 | 33.2 ± 24.9 | >211 | >28 |
| 2m | (4-CF3)Ph | 36.3 ± 4.2 | 7.3 ± 0.8 | 31.2 | >169 | >23 |
| 2n | (2-CF3)Ph | NAf | 25.0 ± 3 | NDg | >169 | >7 |
| 2o | (3,4-diF)Ph | 25.4 ± 3.8 | 13.9 ± 0.4 | NDg | 138 ± 24 | 9.9 |
| 2p | (3,4-diCl)Ph | 2.7 ± 0.3 | 9.7 ± 1.8 | NDg | >168 | >17 |
| 2q | (3,4-diMe)Ph | NAf | 6.2 ± 0.3 | 20 ± 2 | >181 | >29 |
| 2r | (3-Cl,4-Me)Ph | 8.0 ± 1.0 | 7.0 ± 2.0 | 37 ± 4 | >175 | >25 |
| 2s | (3-CF3,4-Cl)Ph | NAf | 6.4 ± 1.7 | NDg | >160 | >25 |
| 2t | Cy | 43.0 ± 0.7 | 4.5 ± 0.6 | 11 ± 2 | 44 ± 38 | 9.7 |
| 2u | H | NAf | 88.2 ± 42.8 | 65.2 | 155 ± 50.5 | 2.5 |
| 3 | NAf | 4.5 ± 0.6 | NDg | 56 | 12 | |
| 4 | NAf | 15 ± 2 | 34 ± 7 | >192 | >13 | |
| 5 | NAf | 80.8 ± 9.4 | NDg | 160 ± 18.2 | 2.3 | |
| 1h | (3-F)Ph | 21.0 ± 2.8 | 7.5 ± 2.5 | 42.0 ± 2.0 | >370 | >49 |
IC50 = concentration of compound that inhibits 50% enzyme activity in vitro. The reported values represent the mean ± SD of data derived from two independent experiments performed in duplicate.
EC50 = the effective concentration that is required to inhibit virus replication by 50% as quantified by luciferase reporter signal. The reported values represent the mean ± SD of data derived from at least three dose–response curves.
EC90 = the effective concentration that is required to inhibit virus replication by 90% as quantified by luciferase reporter signal. The reported values represent the mean ± SD of data derived from at least three independent dose–response curves.
CC50 = concentration that is required to reduce the bioreduction of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) into formazan by 50% and is representative of the antimetabolic effect of the compound on the host cell. The reported value represents the mean ± SD of data derived from at least three dose–response curves.
SI = selectivity index (ratio of CC50 to EC50).
NA = not active. These compounds were not able to reach ≥50% inhibition of NS5B RdRp activity at 50 μM.
ND = not determined. These compounds were not able to reach 90% inhibition of HCV replication at any concentration employed.
Data from ref (17).
Scheme 2.

Chemistry
The synthetic pathways utilized in the preparation of the desired pyrazolobenzothiazines 2a–u, and 3–5 are outlined in Schemes 1 and 2, respectively. Through a regioselective condensation17 of key synthon 6(19) with the appropriate hydrazine hydrochloride, the N-1 phenylpyrazolobenzothiazine esters 7a–c,197d–g, 7h,197i–k, 7l,197m–s, the N-1 cyclohexyl pyrazolobenzothiazine 7t, and the N-1 unsubstituted pyrazolobenzothiazine 7u were prepared and subsequently hydrolyzed under basic conditions. The 3-carboxylic acids 8a–k, 8l,19 and 8m–u thus obtained were then converted to the corresponding acyl chloride, under reflux of SOCl2, and immediately reacted with N-(4-aminophenyl)methanesulfonamide,20 using Et3N as scavenger, to obtain the target amide derivatives 2a–u (Scheme 1). Next, by use of the latter procedure, the acid 9(17) (Scheme 2) was reacted with aniline or 2-(4-aminophenoxy)acetamide21 to give the target 3 or 4, respectively.
The synthesis of the inverse amide 5 (Scheme 2) entailed the conversion of the 3-carboxylic acid 9(17) into the corresponding unstable azide intermediate which was immediately decomposed to the 3-aminopyrazolobenzothiazine 10 through a Curtius reaction. Derivative 10 was then reacted with p-nitrobenzoyl chloride in dry pyridine to afford the nitro intermediate 11, which was reduced under catalytic hydrogenation to the corresponding amino derivative 12. Finally, mesylation of the latter compound gave the desired target derivative 5.
Anti-NS5B Activity
The anti-NS5B activity of the target compounds 2a–u and 3–5 (Table 1) was determined using recombinant NS5BCΔ21 1b in a primer dependent elongation assay as described previously (see also Supporting Information).16,17,22−24
All compounds were first screened at 50 μM to identify NS5B inhibitor candidates exhibiting ≥50% inhibition of NS5B RdRp activity at this concentration. This investigation led to the identification of 15 compounds (2a–i, 2l, 2m, 2o, 2p, 2r, 2t) satisfying this criterion, while nine compounds (2j, 2k, 2n, 2q, 2s, 2u, 3–5) exhibited at the same concentration lower inhibition of NS5B RdRp activity; the latter compounds were thus considered as inactive.
The fifteen selected compounds were further screened for their NS5B inhibition potency and yielded IC50 values ranging from 2.7 to 43 μM.
Compounds 2a (IC50 = 4.3 μM), characterized by the presence of a N-1 m-chlorophenyl residue, exhibited the highest NS5B inhibitory activity within the monosubstituted derivatives, displaying about 5-fold higher activity than the parent compound 1 (IC50 = 21.0 μM). Further analysis of the monosubstituted miniseries (compounds 2a–n) clearly showed that the meta and para substitutions at the N-1 phenyl ring of the pyrazolobenzothiazine were preferred over the ortho substitution irrespective of the nature of the substituent. In particular, the m-chlorine (2a) and p-chlorine (2b) derivatives were about 4-fold and 2-fold more active than the corresponding o-chlorine derivative 2c, respectively. From a head to head comparison between the m-chloro derivative 2a and hit compound 1, it was clear that chlorine was a good replacement for fluorine. This pattern was consistent in the N-1 bromophenyl substituted derivatives 2d–f. The presence of a methyl group (derivatives 2g–i) or a trifluoromethyl group (derivatives 2l and 2m) yielded less active compounds, although the impact of their positions on the activity was less evident. The presence of a more polar methoxyl group (2j and 2k) resulted in inactive compounds, highlighting the importance of a lipophilic substituent at the N-1 phenyl ring. The importance of the lipophilic and bulky nature of the N-1 substituent was also confirmed by the very low activity shown by N-1 cyclohexyl derivative 2t and by the totally inactive N-1 unsubstituted derivative 2u.
The meta/para-disubstituted derivatives designed with the objective to better fill the hydrophobic cavity on NS5B polymerase showed variable activities. In particular, the dichloro and the chloro/methyl N-1 phenyl substituted derivatives 2p and 2r exhibited enhanced activity (IC50 of 2.7 and 8.0 μM, respectively), with derivative 2p representing the best hit in anti-NS5B functional assay. By contrast, the difluoro N-1 phenyl substituted derivative 2o was less active than compound 1. These data further demonstrated that chlorine is a good replacement for fluorine at the meta and/or para position.
We previously reported that replacement of the amide linkage with an ester function was detrimental for the anti-NS5B activity.17 Consistent with this trend, the reverse amide of starting hit 1 yielded the inactive derivative 5, thus emphasizing the crucial role of this linker for this series of NNIs. In a SAR study on some benzothiazine and benzothiadiazine PSI-NNIs, it was observed that replacement of the methanesulfonyl moiety with the oxyacetamide chain conferred better pharmacological properties while maintaining potent anti-NS5B inhibition.18 Paradoxically though, derivative 4 bearing an oxyacetamide moiety on the phenyl ring was inactive. Furthermore, as expected because of our previous observations,17 deletion of the sole methanesulfonamide fragment on the hit compound 1 also yielded the inactive compound 3.
Mapping the Inhibitor Binding Site on NS5B
In our previous studies, we reported that pyrazolobenzothiazine-based inhibitors should bind NS5B PSI based on molecular docking studies.17
With the objective of confirming this in silico prediction, we carried out the mutant counterscreen assay25−27 with 2a as a representative member of this class of NS5B inhibitors. In this assay, recombinant NS5BCΔ21 mutant proteins P495L, M423T, and M414T are employed as screens for TSI, TSII, and PSI binders, respectively.25−27
Each of these proteins carries a mutation at an amino acid that is critical for the binding of inhibitors to each of the aforementioned allosteric sites, thus impairing potency of the inhibitor if these residues are involved. Consistent with our hypothesis, the inhibitory potency of compound 2a was impaired by mutation at M414T residue of NS5B but not with P495L or M423T mutants (Table 2). This was evident from the ∼30-fold higher IC50 value against M414T NS5B relative to wild-type NS5B. By contrast, the IC50 value of 2a either remained unchanged or exhibited an approximate 2-fold increase with NS5B mutant P495L or M423T, respectively. These data thus validated our computational predictions and provided evidence for 2a binding at PSI of NS5B.
Table 2. NS5B Mutant Inhibition Parameters for Derivative 2aa.
| binding site | NS5B mutant | IC50 (μM) | fold change |
|---|---|---|---|
| PSI | M414T | 127.7 ± 5.5 | 29.7 |
| TSI | P495L | 5.1 ± 0.3 | 1.2 |
| TSII | M423T | 9.3 ± 1.5 | 2.2 |
The IC50 values were evaluated against the indicated NS5B mutants as described for the wild-type NS5B and represents an average ± SD of two independent experiments in duplicate. Fold change is indicated relative to NS5B WT (IC50 = 4.3 μM).
In silico simulations were then performed to confirm the binding mode previously proposed for this class of PSI-NNIs.17 Toward this end, derivative 2a was submitted to molecular modeling studies. In a recent paper we rationalized the influence of water molecules and protein flexibility on docking studies, putting particular emphasis on the role of specific water molecules found to mediate hydrogen bond interactions of NNIs with residues Gln446 and/or Gly449 in several experimental structures.28
Pyrazolobenzothiazine derivative 2a was docked into NS5B polymerase (PDB code 3SKE)29 by means of the Glide software,30 yielding a bound pose that was very similar (heavy-atom rmsd of 1.9 Å) to the one we obtained in our previous paper for derivative 1 (Figure 1),17 a strict analogue of 2a. The 3SKE crystal structure was chosen as target because in its waterless form it was found to be one of the best performing in docking-based virtual screening simulations.28 We considered the absence of explicit waters a good premise to avoid biasing in the study of water–ligand–protein interactions. The obtained 3SKE/2a complex was then submitted to a 10 ns long molecular dynamics (MD) study31 with the aim of investigating the binding mode dynamically. From the heavy-atom rmsd analysis, the system was equilibrated after the first 4 ns of simulation, and thus, this equilibration time was not considered in the binding mode analysis.
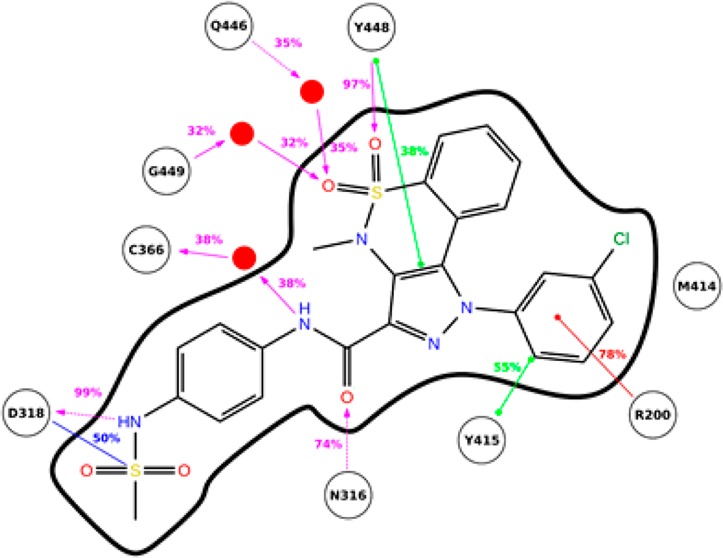
The binding of 2a to NS5B is characterized by several interactions (Figure 2). The sulfone group of the exocyclic sulfonamide is engaged in an ion–dipole interaction with the side chain of Asp318, which in turn accepts an H-bond from the sulfonamidic nitrogen. One of the two endocyclic sulfonamide oxygens establishes hydrogen-bonding with the backbone nitrogen of Tyr448, while the other one is engaged in water-mediated interactions with Gln446 and/or Gly449. The amide group of 2a is involved in two H-bond interactions: a water-mediated one between the backbone oxygen of Cys366 and the amidic nitrogen and a direct one between the amide oxygen and the side chain of Asn316. Furthermore, compound 2a is involved in three π interactions: the tricyclic system stacks with the aromatic ring of Tyr448, while the m-chlorophenyl moiety interacts with the aromatic ring of Tyr415 and the cationic side chain of Arg200.
Figure 2.

Binding mode analysis of pyrazolobenzothiazine derivative 2a, depicted together with interacting NS5B residues and water molecules. Main interactions are represented schematically with their occupancies calculated in the time window 4–10 ns.
Replicon System Assays
The target compounds 2a–u and 3–5 were evaluated for their anti-HCV activity (EC50, EC90) in Huh-5-2 cells carrying a genotype 1b HCV subgenomic replicon (Table 1). The antimetabolic effect (CC50) of the compounds was investigated in parallel employing the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Only compounds that showed ≥70% inhibition of virus RNA replication without exhibiting any adverse antimetabolic effect at the concentration tested were considered to be endowed with selective anti-HCV activity. In fact, any compound that can induce a cytostatic effect on cell growth could result indirectly in inhibition of HCV subgenomic replicon replication.32,33 The selectivity index (SI) was calculated as well to estimate the therapeutic potential of the compounds in this system. Experimental details for the cellular assays are according to procedures described previously (see also Supporting Information).15−17,34
Furthermore, to identify possible pleiotropic effects exerted by the compounds, the wells were subjected to microscopic examination to discern subtle changes in cellular phenotypes that may have resulted from compound treatment and that could be considered as a hallmark for off-target effects of the compounds.
The cell-based assays (Table 1) revealed that with the exception of derivatives 2n, 2u, and 5, all remaining compounds were active against HCV, displaying EC50 values of ≤15 μM. However, because of either a significant toxic effect as apparent from the MTS assay or low SI values (i.e., ≤10), compounds 2f, 2o, 2t, and 3 were considered as false positives (EC50 not reliable). After dose–response curve analysis, derivatives 2c, 2i, 2m, 2p, 2r, 2s, and 4 were also deemed as unsuitable, as they displayed antimetabolic effect (data not shown). Hence, only derivatives 2a, 2b, 2d, 2e, 2g, 2h, 2k, 2l produced selective anti-HCV activity without any antimetabolic effect. After microscopic analysis aimed at visualizing minor changes in the cellular morphology, only pyrazolobenzothiazines 2b, 2k, and to less extent 2h proved to be true anti-HCV agents, as they did not display any significant effects on cell morphology after 72 h of compound treatment. Interestingly, all three of these derivatives were characterized by the presence of a para-substituent at the N-1 phenyl ring. Target compound 2b, bearing a p-chlorine atom on the N-1 phenyl ring, was endowed with comparable inhibitory activity both against NS5B (IC50 = 7.9 μM) and in cell based HCV replication assay (EC50 = 8.1 μM and EC90 = 23.3 μM). In the case of derivative 2h, the anti-HCV activity was better than the anti-NS5B activity, but this result can be easily explained by the presence of a slight pleiotropic effect that alters the cellular environment and negatively affects HCV replication. Strangely, compound 2k, which did not display anti-NS5B activity (≤50% inhibition at 50 μM), showed good anti-HCV activity, as is evident from its EC50 value of 8.1 μM in a cell based assay.
In a head to head comparison, the best selective anti-HCV derivative within this new series (compound 2b) exhibited equivalent activity as reference compound 1. Significantly, it is important to note that, unlike the parent compound, derivative 2b did not exhibit any cell morphological alterations, thus emerging as a more promising antiviral agent.
Selective HCV Inhibition Assessment and Physicochemical and ADME Properties Characterization
Any compound tested on a HCV replicon system can inadvertently inhibit heterologous elements such as the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) or the neomycin phosphotransferase (neo) that is required for the proper translation of the HCV polyprotein or to keep the HCV subgenomic replicon persistently in cells, respectively.35 To explore this unspecific effect in our pyrazolobenzothiazine series, compound 2a was selected as a representative member because it was one of the best derivatives in both enzymatic and cellular assays and also available in large quantities. Thus, compound 2a was tested in different replicon systems. On the one hand, we used Huh-9-13 HCV replicons that have the same genetic makeup as Huh-5-2 replicons but differ in (i) their composition of cell culture adaptive mutations, (ii) the absence of a luciferase reporter gene, and (iii) the higher HCV RNA content compared to Huh-5-2 cells. In addition, we employed a subgenomic replicon contained in HuH6 cells, where the replicon has a similar genetic makeup as in Huh-5-2 cells, but (i) it replicates in a different cell line (HuH6 cells instead of Huh-7 cells) and (ii) it carries different adaptive mutations. Moreover, in HCV subgenomic replicon containing HuH6 cells, the replicon replication is not inhibited by IFN-γ and viral RNA replication is independent from cell proliferation.32 Compound 2a proved to be active in both Huh-9-13 and HuH6 HCV subgenomic replicon bearing cells, exhibiting EC50 values of 7.6 and 14 μM, respectively, as determined by RT-qPCR. In both replicon containing cells, 2a did not show any effect on the proliferation and metabolism of the host cell as determined by the MTS assay (CC50 > 90 μM). The values obtained are in the same range as for the Huh-5-2 HCV subgenomic replicon containing cells, thus excluding any inhibitory effect on the luciferase expressed by the HCV subgenomic replicons in Huh-5-2 cells.
Derivative 2a was also selected for additional profiling of our pyrazolobenzothiazine PSI-NNIs. Toward this end, log P, thermodynamic and kinetic water solubility (log S), parallel artificial membrane permeability (PAMPA), membrane retention (MR), and human liver microsome (HLM) stability were experimentally determined (Table 3).
Table 3. In Vitro Physicochemical and ADME Properties of Derivative 2a.
| water solubility, log S (S in mol L–1) |
PAMPA-GI,bPapp × 10–6 (cm/s) (% MR)c |
|||||
|---|---|---|---|---|---|---|
| compd | log P a | thermodynamic | kinetic | 5 h | 22 h | HLMd (%) |
| 2a | 2.11 | –8.40 | –6.56 | 2.30 (7.3) | 4.20 (3.2) | 99.9 |
Lipophilicity was measured through the shake flask method.
Permeability assay at different incubation times in gastrointestinal model.
% MR: percent membrane retention.
Metabolic stability after 60 min in presence of human liver microsomes expressed as percentage of unmodified parent drug.
The log P value turned out to be in the optimal range for druglike compounds. The thermodynamic solubility value varies with the crystal form of the solid (amorphous, crystalline, different polymorphs, hydrates, and solvates). Kinetic solubility has two distinguishing characteristics: the first one is that the compound initially is fully dissolved in an organic solvent (DMSO) and then added to the water. Desired log S for a drug candidate should be between −4 and −6, whereas log values of <−6 indicate low solubility. Both low thermodynamic and kinetic solubility values were determined for this derivative, and this behavior may be partially due to a high molecular packing effect. To evaluate membrane permeability, we have used the PAMPA assay proposed by Kansy in 1998.36 Accordingly, several studies indicate that PAMPA permeability is correlated with both Caco-2 cell permeability and human intestinal absorption.37,38 The PAMPA experiments on 2a revealed a low permeability, which cannot be correlated to a high membrane retention as shown by the low MR value. Notably, 2a turned out to be a metabolically stable NS5B inhibitor, given the high HLM stability value, indicating that no metabolites were detected after 60 min of exposure to cytochrome metabolism.
Assessment of Potential Off-Target Effects of the Compounds Studied
As reported in the previous paragraph, microscopic observations had highlighted that, with the exceptions of hit compounds 2b, 2h, and 2k, most of the pyrazolobenzothiazines tested altered cell morphology. These morphological alterations may be a hallmark of potential off-target or pleiotropic effects of the compounds studied. Among the derivatives showing this undesired aspect, compound 2a was selected as a representative analogue for further investigations.
A potential off-target effect observed during compound treatment may due to perturbation of cellular lipid metabolism,39 e.g., inhibition of fatty acid oxidation, which may affect HCV replication.40 Candidate drugs can perturb the lipid metabolism of a cell in different ways, for example, by inducing phospholipidosis and steatosis.39 To assess if our pyrazolobenzothiazines could alter the cellular lipid metabolism, we used differential vital stainings that could indicate the ability of 2a to induce phospholipidosis (Nile Red stain and monodansylcadaverine) or drug-induced steatosis (Nile Red stain and LipidTOX red stain). The aim of monodansylcadaverine staining was to screen for the presence of multilamellar bodies, a hallmark of phospholipidosis. Cell treatment with 90 μM derivative 2a showed that the compound did not induce multilamellar bodies (Figure 3).
Figure 3.

Huh-9-13 cells were stained with monodansylcadaverine (MDC) (a–c), Nile Red (d–f), or LipidTOX red (g–i) after mock treatment (DMSO) or treatment with either the HCV NS3 protease inhibitor VX-950 (at 15 μM) or compound 2a (at 90 μM). Monodansylcadaverine stains multilamellar bodies and autophagic vacuoles (blue). Nile Red stains both neutral lipids (yellow/gold color) and phospholipids (orange/red color). LipidTOX red stains neutral lipids (red).
Finally LipidTOX red stain and Nile Red stain were performed with the aim to evaluate if compound 2a was able to induce intracellular accumulation of neutral lipids and/or phospholipids. Overall these two assays demonstrated that derivative 2a (at 90 μM) significantly altered the neutral lipids content of the treated cells (Figure 3) as apparent from the increase of neutral lipid stain (yellow/gold color) in Nile Red stained cells and the size of the lipid droplets in LipidTOX red stain. These observations suggest that the alteration of cell morphology induced by 2a, and probably by most of the other pyrazolobenzothiazine derivatives, might be partially due to drug-induced steatosis. A plausible explanation might be the inhibition of mitochondrial β-oxidation by pyrazolobenzothiazines, although other mechanisms of drug-induced steatosis could be equally feasible.41
In depth analysis of the data obtained by different stainings performed on compound 2b highlighted that this derivative also induced pospholipidosis/steatosis (i.e., pleiotropic effect) but with an altered effect compared to 2a (see Supporting Information). Notably, compound 2b did not show any significant alteration in cell morphology as a consequence of pleiotropic effect at all time points tested.
Conclusions
We have designed and synthesized novel pyrazolobenzothiazine derivatives with the aim of both tracing a clearer SAR for this class of NS5B NNIs and identifying compounds with improved anti-HCV profile. Biochemical studies and mutant counterscreen assay confirmed that our pyrazolobenzothiazines act as NS5B PSI inhibitors. Most derivatives exhibited EC50 values of ≤15 μM against HCV in the cell-based replicon assay. Notably, pyrazolobenzothiazine NS5B inibitors 2b and 2h proved to be true anti-HCV agents, as they did not display any antimetabolic and cell morphology alteration effects. Thus, these new derivatives represent an important improvement with respect to the previously reported parent compounds.
A combination of HCV replicons with the same genetic makeup but different cell culture adaptive mutations in several cell types was also employed to understand the effect of compound 2a, as a representative analogue, on HCV RNA replication, host cell proliferation, and metabolism.
Finally, compound 2a was also selected among the derivatives showing the undesired pleiotropic effect to assess possible perturbation of cellular lipid metabolism during pyrazolobenzothiazine treatment. Differential staining with dyes such as LipidTOX red and Nile Red revealed that the alteration of cell morphology induced by the pyrazolobenzothiazine 2a might be partially due to drug-induced steatosis. Additional experimental studies highlighted physicochemical and ADME issues for the representative derivative 2a.
These findings argue in favor of including phospholipidosis/drug-induced steatosis and pharmacokinetic profile as factors during further chemical optimization of the pyrazolobenzothiazine scaffold to obtain more selective and potent anti-HCV inhibitors and to prevent any side effect of the compounds on hepatic tissue.
Experimental Section
Chemistry
Reagents and solvents were purchased from common commercial suppliers and were used as such unless otherwise indicated. Organic solutions were dried over anhydrous Na2SO4 and concentrated with a rotary evaporator at low pressure. All reactions were routinely checked by thin-layer chromatography (TLC) on silica gel 60 F254 (Merck) and visualized by using UV or iodine. Column chromatography separations were carried out with Merck silica gel 60 (mesh 70–230). Flash chromatography separations were carried out with Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Büchi Electrotermal model 9100) and are uncorrected. Yields were of purified products and were not optimized. 1H NMR spectra were recorded at 200 or 400 MHz (Bruker Avance DRX-200 or -400, respectively), while 13C NMR spectra were recorded at 100 MHz (Bruker Avance DRX-400). Chemical shifts (δ) are given in ppm relative to TMS and calibrated using residual undeuterated solvent as internal reference. Spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR, and the spectral data are consistent with the assigned structures. The purity of the tested compounds was evaluated by combustion analysis using a Fisons elemental analyzer, model EA1108CHN, and data for C, H, and N are within 0.4% of the theoretical values (≥95% sample purity).
General Procedure for Condensation of Key Intermediate 6 with Hydrazine Hydrochlorides. Method A
A hot solution of the appropriate hydrazine hydrochloride (1.2 mmol) dissolved in hot MeOH (2 mL) was added at once to a refluxing solution of key intermediate 6(19) (1 mmol) in MeOH (20 mL). The mixture was maintained at reflux for 2–20 h, then concentrated under reduced pressure to half volume and cooled to room temperature. The crystallized solid was filtered under vacuum to give the pyrazolobenzothiazine 3-carboxymethyl esters 7d–g, 7i–k, and 7m–u used as is in the next reaction step.
Methyl 1-(3-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7d)
Following the general procedure method A and using the 3-(bromophenyl)hydrazine hydrochloride, compound 7d was obtained in 87% yield (reaction time, 15 h) as a whitish amorphous solid: mp 246–248 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 7.00 (dd, J = 1.6 and 7.6 Hz, 1H, H-9), 7.55–7.60 (m, 2H, H-5′ and H-6′), 7.70–7.80 (m, 2H, H-7 and H-8), 7.85–7.90 (m, 2H, H-2′ and H-4′), 8.00 (dd, J = 1.4 and 7.0 Hz, 1H, H-6).
Methyl 1-(4-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7e)
Following the general procedure method A and using the 4-(bromophenyl)hydrazine hydrochloride, compound 7e was obtained in 50% yield (reaction time, 3 h) as an orange amorphous solid: mp 196–199 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 7.05 (dd, J = 1.3 and 7.3 Hz, 1H, H-9), 7.55–7.60 (m, 2H, H-3′ and H-5′), 7.65–7.75 (m, 2H, H-7 and H-8), 7.80–7.90 (m, 2H, H-2′ and H-6′), 8.00 (dd, J = 1.5 and 7.6 Hz, 1H, H-6).
Methyl 1-(2-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7f)
Following the general procedure method A and using the 2-(bromophenyl)hydrazine hydrochloride, compound 7f was obtained in 90% yield (reaction time, 4 h) as an orange amorphous solid: mp 215–218 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 6.75 (d, J = 7.9 Hz, 1H, H-9), 7.60–7.75 (m, 4H, H-7, H-8, H-4′, and H-5′), 7.85 (d, J = 7.7 Hz, 1H, H-6′), 7.95 (d, J = 7.8 Hz, 1H, H-3′), 8.00 (d, J = 7.7 Hz, 1H, H-6).
Methyl 4-Methyl-1-(3-methylphenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7g)
Following the general procedure method A and using the 3-(methylphenyl)hydrazine hydrochloride, compound 7g was obtained in 80% yield (reaction time, 14 h) as a whitish amorphous solid: mp 199–200 °C. 1H NMR (200 MHz, acetone-d6): δ 2.50 (s, 3H, CH3), 3.30 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.10–7.15 (m, 1H, H-9), 7.35–7.60 (m, 4H, H-2′, H-4′, H-5′, and H-6′), 7.65 (dt, J = 1.6 and 7.9 Hz, 1H, H-8), 7.70 (dt, J = 1.3 and 7.9 Hz, 1H, H-7), 8.00–8.10 (m, 1H, H-6).
Methyl 4-Methyl-1-(2-methylphenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7i)
Following the general procedure method A and using the 2-(methylphenyl)hydrazine hydrochloride, compound 7i was obtained in 90% yield (reaction time, 10 h) as a whitish amorphous solid: mp 180–182 °C. 1H NMR (400 MHz, acetone-d6): δ 2.00 (s, 3H, CH3), 3.30 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 6.85 (d, J = 8.0 Hz, 1H, H-9), 7.50–7.65 (m, 5H, H-8, H-3′, H-4, H-5′, and H-6′), 7.70 (dt, J = 1.1 and 7.8 Hz, 1H, H-7), 8.00 (dd, J = 0.9 and 7.8 Hz, 1H, H-6).
Methyl 1-(3-Methoxyphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7j)
Following the general procedure method A and using the 3-(methoxyphenyl)hydrazine hydrochloride, compound 7j was obtained in 60% yield (reaction time, 10 h) as yellow amorphous solid: mp 197–199 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.13 (s, 3H, NCH3), 3.75 (s, 3H, OCH3), 3.90 (s, 3H, CO2CH3), 6.95 (dd, J = 1.3 and 8.0 Hz, 1H, H-9), 6.95–7.05 (m, 1H, H-4′), 7.13 (t, J = 2.1 Hz, 1H, H-2′), 7.15–7.20 (m, 1H, H-6′), 7.50 (t, J = 8.1 Hz, 1H, H-5′), 7.65 (dt, J = 1.4 and 8.0 Hz, 1H, H-8), 7.70 (dt, J = 1.3 and 7.2 Hz, 1H, H-7), 7.90 (dd, J = 1.4 and 7.2 Hz, 1H, H-6).
Methyl 1-(4-Methoxyphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7k)
Following the general procedure method A and using the 4-(methoxyphenyl)hydrazine hydrochloride, compound 7k was obtained in 90% yield (reaction time, 15 h) as a white amorphous solid: mp 206–208 °C. 1H NMR (400 MHz, acetone-d6): δ 3.25 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 4.00 (s, 3H, CO2CH3), 7.15 (dd, J = 1.4 and 7.5 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3′ and H-5′), 7.50–7.55 (m, 2H, H-2′ and H-6′), 7.60–7.80 (m, 2H, H-7 and H-8), 8.05 (m, 1H, H-6).
Methyl 4-Methyl-1-[4-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7m)
Following the general procedure method A and using the 4-(trifluoromethylphenyl)hydrazine hydrochloride, compound 7m was obtained in 60% yield (reaction time, 3.5 h) as a white amorphous solid: mp 189–191 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.10 (dd, J = 2.1 and 7.0 Hz, 1H, H-9), 7.65–7.75 (m, 2H, H-7 and H-8), 7.80–7.85 (m, 2H, H-2′ and H-6′), 7.95–8.10 (m, 3H, H-6, H-3′, and H-5′).
Methyl 4-Methyl-1-[2-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7n)
Following the general procedure method A and using the 2-(trifluoromethylphenyl)hydrazine hydrochloride, compound 7n was obtained in 70% yield (reaction time, 4 h) as an orange amorphous solid: mp 222–225 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 6.60 (d, J = 7.6 Hz, 1H, H-9), 7.60 (dt, J = 1.3 and 7.6 Hz, 1H, H-8), 7.70 (dt, J = 1.0 and 7.6 Hz, 1H, H-7), 7.80 (d, J = 7.2 Hz, 1H, H-3′), 7.95–8.05 (m, 3H, H-4′, H-5′, and H-6′), 8.10 (dd, J = 1.3 and 7.6 Hz, 1H, H-6).
Methyl 1-(3,4-Difluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7o)
Following the general procedure method A and using the 3,4-(difluorophenyl)hydrazine hydrochloride, compound 7o was obtained in 80% yield (reaction time, 4 h) as a white amorphous solid: mp 241–243 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.10 (dd, J = 1.0 and 8.2 Hz, 1H, H-9), 7.40–7.50 (m, 1H, H-6′), 7.65–7.75 (m, 3H, H-7, H-8, and H-5′), 7.85–7.95 (m, 1H, H-2′), 8.05 (dd, J = 1.3 and 7.7 Hz, 1H, H-6).
Methyl 1-(3,4-Dichlorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7p)
Following the general procedure method A and using the 3,4-(dichlorophenyl)hydrazine hydrochloride, compound 7p was obtained in 80% yield (reaction time, 3 h) as a whitish amorphous solid: mp 300–302 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 7.15 (dd, J = 1.0 and 7.4 Hz, 1H, H-9), 7.60 (dd, J = 2.6 and 8.6 Hz, 1H, H-6′), 7.45–7.55 (m, 2H, H-7 and H-8), 7.85 (d, J = 8.6 Hz, 1H, H-5′), 8.00–8.05 (m, 2H, H-6 and H-2′).
Methyl 1-(3,4-Dimethylphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7q)
Following the general procedure method A and using the 3,4-(dimethylphenyl)hydrazine hydrochloride, compound 7q was obtained in 50% yield (reaction time, 4 h) as a whitish amorphous solid: mp 245–247 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.20 and 2.30 (s, each 3H, CH3), 2.75 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 6.95 (dd, J = 1.0 and 7.7 Hz, 1H, H-9), 7.20 (dd, J = 2.3 and 7.9 Hz, 1H, H-6′), 7.30–7.40 (m, 2H, H-2′ and H-5′), 7.55–7.65 (m, 2H, H-7 and H-8), 7.90 (dd, J = 1.3 and 6.9 Hz, 1H, H-6).
Methyl 1-(3-Chloro-4-methylphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7r)
Following the general procedure method A and using the 1-(3-chloro-4-methylphenyl)hydrazine hydrochloride, compound 7r was obtained in 80% yield (reaction time, 4 h) as a whitish amorphous solid: mp 285–287 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.45 (s, 3H, CH3), 3.10 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.00 (dd, J = 1.6 and 7.1 Hz, 1H, H-9), 7.40 (dd, J = 2.2 and 8.1 Hz, 1H, H-6′), 7.55 (d, J = 8.1 Hz, 1H, H-5′), 7.60–7.65 (m, 3H, H-7, H-8, and H-2′), 7.90 (dd, J = 1.6 and 6.8 Hz, 1H, H-6).
Methyl 1-[4-Chloro-3-(trifluoromethyl)phenyl]-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7s)
Following the general procedure method A and using the 1-(4-chloro-3-trifluoromethylphenyl)hydrazine hydrochloride, compound 7s was obtained in 50% yield (reaction time, 2 h) as a whitish amorphous solid: mp 291–293 °C. 1H NMR (400 MHz, DMSO-d6) δ 3.10 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.10 (d, J = 7.7 Hz, 1H, H-9), 7.60–7.75 (m, 2H, H-7 and H-8), 7.85 (dd, J = 2.4 and 8.6 Hz, 1H, H-6′), 7.90 (d, J = 8.6 Hz, 1H, H-5′), 8.00 (dd, J = 1.0 and 7.3 Hz, 1H, H-6), 8.10 (d, J = 2.4 Hz, 1H, H-2′).
Methyl 1-Cyclohexyl-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7t)
Following the general procedure method A and using the cyclohexylhydrazine hydrochloride, compound 7t was obtained in 50% yield (reaction time, 20 h) as a white solid: mp 198–200 °C. 1H NMR (200 MHz, acetone-d6): δ 1.25–1.85 (m, 4H, Cy-CH2), 1.90–2.35 (m, 6H, Cy-CH2), 3.20 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 4.70–5.00 (m, 1H, Cy-CH), 7.75–7.85 (m, 1H, H-8), 7.90–8.05 (m, 3H, H-6, H-7, and H-9).
Methyl 4-Methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-Dioxide (7u)
Following the general procedure method A and using the hydrazine hydrochloride, compound 7u was obtained as a yellow solid in 70% yield (reaction time, 5 h) after purification by flash column chromatography, eluting with CH2Cl2/MeOH (95:5): mp 218–220 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.25 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.70 (t, J = 7.4 Hz, 1H, H-7), 7.80–8.00 (m, 2H, H-8 and H-9), 8.05 (d, J = 7.4 Hz, 1H, H-6).
General Procedure of Basic Hydrolysis. Method B
A stirred mixture of appropriate pyrazolobenzothiazine methyl esters 7a–c,197d–g, 7h,197i–k, and 7m–u (1 mmol) in aqueous 10% NaOH (6 mL) and MeOH (6 mL) was refluxed for 1 h and then concentrated to one-third of the volume under reduced pressure. The mixture was poured into ice/water and acidified with 2 N HCl (pH 3). The precipitate formed was then filtered under vacuum, washed with Et2O, and dried to give the pyrazolobenzothiazine-3-carboxylic acids 8a–k, and 8m–u used as is in the next reaction step.
1-(3-Chlorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8a)
Following the general procedure method B, compound 8a was obtained from 7a(19) in 85% yield as a white solid: mp 247–248 °C. 1H NMR (200 MHz, acetone-d6): δ 3.40 (s, 3H, NCH3), 7.25 (dd, J = 0.8 and 6.8 Hz, 1H, H-9), 7.60–7.85 (m, 6H, H-7, H-8, H-2′, H-4′, H-5′, and H-6′), 8.05 (dd, J = 1.2 and 7.5 Hz, 1H, H-6).
1-(4-Chlorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8b)
Following the general procedure method B, compound 8b was obtained from 7b(19) in 80% yield as a white solid: mp 259–260 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 7.20–7.25 (m, 1H, H-9), 7.65–7.85 (m, 6H, H-7, H-8, H-2′, H-3′, H-5′, and H-6′), 8.05–8.10 (m, 1H, H-6).
1-(2-Chlorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8c)
Following the general procedure method B, compound 8c was obtained from 7c(19) in 75% yield as a white solid: mp 224–226 °C. 1H NMR (200 MHz, acetone-d6): δ 3.40 (s, 3H, NCH3), 6.95 (dd, J = 1.1 and 6.9 Hz, 1H, H-9), 7.65 (dt, J = 1.5 and 6.9 Hz, 1H, H-8), 7.70–7.85 (m, 4H, H-7, H-3′, H-4′, and H-5′), 7.90–7.95 (m, 1H, H-6′), 8.05 (dd, J = 1.5 and 8.1 Hz, 1H, H-6).
1-(3-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8d)
Following the general procedure method B, compound 8d was obtained from 7d in 90% yield as a white solid: mp 228–230 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.25 (s, 3H, NCH3), 7.05 (dd, J = 1.3 and 7.4 Hz, 1H, H-9), 7.50–7.60 (m, 2H, H-5′ and H-6′), 7.65–7.75 (m, 2H, H-7 and H-8), 7.80–7.90 (m, 2H, H-2′ and H-4′), 8.00 (dd, J = 1.7 and 7.0 Hz, 1H, H-6).
1-(4-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8e)
Following the general procedure method B, compound 8e was obtained from 7e in 80% yield as a white solid: mp 255–257 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 3H, NCH3), 7.05 (d, J = 7.5 Hz, 1H, H-9), 7.50–7.60 (m, 2H, H-3′ and H-5′), 7.65–7.80 (m, 2H, H-7 and H-8), 7.80–7.85 (m, 2H, H-2′ and H-6′), 8.05 (d, J = 7.0 Hz, 1H, H-6).
1-(2-Bromophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8f)
Following the general procedure method B, compound 8f was obtained from 7f in 85% yield as a white solid: mp 274–276 °C. 1H NMR (400 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 6.90 (d, J = 7.6 Hz, 1H, H-9), 7.60 (dt, J = 1.2 and 8.0 Hz, 1H, H-4′), 7.70–7.80 (m, 3H, H-7, H-8, and H-5′), 7.80 (dd, J = 1.2 and 6.0 Hz, 1H, H-6′), 7.95 (dd, J = 1.3 and 8.0 Hz, 1H, H-3′), 8.00 (dd, J = 0.7 and 7.8 Hz, 1H, H-6).
4-Methyl-1-(3-methylphenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8g)
Following the general procedure method B, compound 8g was obtained from 7g in 80% yield as a white solid: mp 238–240 °C. 1H NMR (400 MHz, acetone-d6): δ 2.50 (s, 3H, CH3), 3.25 (s, 3H, NCH3), 7.10 (d, J = 7.9 Hz, 1H, H-9), 7.40–7.60 (m, 4H, H-2′, H-4′, H-5′, and H-6′), 7.65 (dt, J = 1.0 and 7.6 Hz, 1H, H-8), 7.75 (dt, J = 1.1 and 7.6 Hz, 1H, H-7), 8.00 (dd, J = 1.0 and 7.6 Hz, 1H, H-6).
4-Methyl-1-(4-methylphenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8h)
Following the general procedure method B, compound 8h was obtained from 7h(19) in 80% yield as a white solid: mp 260–261 °C. 1H NMR (200 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 3.15 (s, 3H, NCH3), 7.10 (dd, J = 1.2 and 7.0 Hz, 1H, H-9), 7.40–7–80 (m, 4H, H-2′, H-3′, H-5′, and H-6′), 7.80–7.90 (m, 2H, H-7 and H-8), 8.00 (d, J = 1.2 and 7.1 Hz, 1H, H-6).
4-Methyl-1-(2-methylphenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8i)
Following the general procedure method B, compound 8i was obtained from 7i in 70% yield as a white solid: mp 214–215 °C. 1H NMR (400 MHz, acetone-d6): δ 2.00 (s, 3H, CH3), 3.25 (s, 3H, NCH3), 6.80 (dd, J = 1.0 and 7.0 Hz, 1H, H-9), 7.50–7.60 (m, 5H, H-8, H-3′, H-4′, H-5′, and H-6′), 7.70 (dt, J = 1.0 and 7.7 Hz, 1H, H-7), 8.00 (dd, J = 0.8 and 7.7 Hz, 1H, H-6).
1-(3-Methoxyphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8j)
Following the general procedure method B, compound 8j was obtained from 7j in 90% yield as a whitish solid: mp 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 3H, NCH3), 3.80 (s, 3H, OCH3), 6.90 (d, J = 7.6 Hz, 1H, H-9), 7.05 (dd, J = 1.7 and 7.4 Hz, 1H, H-4′), 7.15 (t, J = 2.2 Hz, 1H, H-2′), 7.20 (dd, J = 2.2 and 8.2 Hz, 1H, H-6′), 7.50 (t, J = 8.2 Hz, 1H, H-5′), 7.55–7.65 (m, 2H, H-7 and H-8), 7.90 (dd, J = 1.0 and 7.8 Hz, 1H, H-6).
1-(4-Methoxyphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8k)
Following the general procedure method B, compound 8k was obtained from 7k in 90% yield as a whitish solid: mp 244–245 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 3.95 (s, 3H, OCH3), 7.10 (dd, J = 1.3 and 7.7 Hz, 1H, H-9), 7.20–7.30 (m, 2H, H-3′ and H-5′), 7.55–7.60 (m, 2H, H-2′ and H-6′), 7.65 (dt, J = 1.4 and 7.7 Hz, 1H, H-8), 7.75 (dt, J = 1.3 and 7.6 Hz, 1H, H-7), 8.05 (dd, J = 1.4 and 7.6 Hz, H-6).
4-Methyl-1-[4-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8m)
Following the general procedure method B, compound 8m was obtained from 7m in 90% yield as a whitish solid: mp 187–189 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 7.10 (d, J = 7.4 Hz, 1H, H-9), 7.65–7.75 (m, 2H, H-7 and H-8), 7.80–7.85 (m, 2H, H-2′ and H-6′), 8.00–8.05 (m, 3H, H-6, H-3′, and H-5′).
4-Methyl-1-[2-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8n)
Following the general procedure method B, compound 8n was obtained from 7n in 70% yield as a whitish solid: mp 228–230 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 6.60 (d, J = 7.9 Hz, 1H, H-9), 7.60 (dt, J = 1.3 and 7.9 Hz, 1H, H-8), 7.65 (dt, J = 1.0 and 7.7 Hz, 1H, H-7), 7.85 (d, J = 7.2 Hz, 1H, H-3′), 7.90–8.00 (m, 3H, H-4′, H-5′, and H-6′), 8.05 (dd, J = 1.3 and 7.6 Hz, 1H, H-6).
1-(3,4-Difluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8o)
Following the general procedure method B, compound 8o was obtained from 7o in 80% yield as a whitish solid: mp 244–246 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 3H, NCH3), 6.95–7.05 (m, 1H, H-9), 7.35–7.45 (m, 1H, H-6′), 7.60–7.75 (m, 3H, H-7, H-8, and H-2′), 7.85–7.90 (m, 1H, H-5′), 7.95–8.00 (m, 1H, H-6), 13.50 (bs, 1H, COOH).
1-(3,4-Dichlorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8p)
Following the general procedure method B, compound 8p was obtained from 7p in 90% yield as a whitish solid: mp 245–247 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 7.15 (dd, J = 1.5 and 7.5 Hz, 1H, H-9), 7.55 (dd, J = 2.6 and 8.7 Hz, 1H, H-6′), 7.65–7.80 (m, 2H, H-7 and H-8), 7.85 (d, J = 8.7 Hz, 1H, H-5′), 7.90–8.05 (m, 2H, H-6 and H-2′).
1-(3,4-Dimethylphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8q)
Following the general procedure method B, compound 8q was obtained from 7q in 80% yield as a whitish solid: mp 258–260 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.25 and 2.30 (s, each 3H, CH3), 3.15 (s, 3H, NCH3), 6.90 (d, J = 7.6 Hz, 1H, H-9), 7.23 (dd, J = 2.0 and 7.9 Hz, 1H, H-6′), 7.30–7.35 (m, 2H, H-2′ and H-5′), 7.60–7.70 (m, 2H, H-7 and H-8), 7.90 (d, J = 7.6 Hz, 1H, H-6).
1-(3-Chloro-4-methylphenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8r)
Following the general procedure method B, compound 8r was obtained from 7r, in 80% yield as a whitish solid: mp 244–246 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 3.20 (s, 3H, NCH3), 7.00 (d, J = 7.5 Hz, 1H, H-9), 7.35–7.40 (m, 1H, H-6′), 7.55 (d, J = 7.5 Hz, 1H, H-5′), 7.60–7.70 (m, 3H, H-7, H-8, and H-2′), 7.90–8.00 (m, 1H, H-6).
1-[4-Chloro-3-(trifluoromethyl)phenyl]-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8s)
Following the general procedure method B, compound 8s was obtained from 7s, in 75% yield as a whitish solid: mp 242–244 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 7.10 (d, J = 7.7 Hz, 1H, H-9), 7.60–7.75 (m, 2H, H-7 and H-8), 7.84 (dd, J = 2.4 and 8.6 Hz, 1H, H-6′), 7.90 (d, J = 8.6 Hz, 1H, H-5′), 8.00 (dd, J = 1.0 and 7.3 Hz, 1H, H-6), 8.10 (d, J = 2.4 Hz, 1H, H-2′), 13.75 (bs, 1H, COOH).
1-Cyclohexyl-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8t)
Following the general procedure method B, compound 8t was obtained from 7t, in 70% yield as a white solid: mp 229–231 °C. 1H NMR (200 MHz, CDCl3): δ 1.30–1.70 (m, 4H, Cy-CH2), 2.00–2.45 (m, 6H, Cy-CH2), 3.30 (s, 3H, NCH3), 4.50–4.75 (m, 1H, Cy-CH), 7.60–7.90 (m, 3H, H-7, H-8, and H-9), 8.10 (d, J = 7.7 Hz, 1H, H-6).
4-Methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic Acid 5,5-Dioxide (8u)
Following the general procedure method B, compound 8u was obtained from 7u, in 84% yield as yellow solid: mp 169–172 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 3H, NCH3), 7.60–7.65 (m, 1H, H-8), 7.80–7.95 (m, 2H, H-7 and H-9), 8.05 (d, J = 7.7 Hz, 1H, H-6), 14.50 (s, 1H, pyrazolo-NH).
General Procedure for Amidation Reaction with N-(4-aminophenyl)methanesulfonamide. Method C
A mixture of the acid intermediates 8a–u (1 mmol) in an excess of SOCl2 was refluxed under stirring for 1 h. The solvent was distilled under reduced pressure, and the residues were washed three times with dry benzene. The corresponding pyrazolobenzothiazine-3-carbonyl chloride was immediately dissolved in dry DMF (2 mL) and added dropwise, under nitrogen flux, to a solution of N-(4-aminophenyl)methanesulfonamide20 (2 mmol) and Et3N (2 mmol) in dry DMF (7 mL). The mixture was heated at 40 °C for 24 h. The solvent was then concentrated to half volume, and the mixture was poured into ice–water and acidified with 2 N HCl (pH 3). The precipitate formed was filtered under vacuum. The solid obtained was crystallized by EtOH/DMF (2:1) to give the target pyrazolobenzothiazine-3-carboxyamides 2a–s or purified by flash column chromatography in the cases of compounds 2t and 2u.
1-(3-Chlorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2a)
Following the general procedure method C, compound 2a was obtained, from 8a, in 40% yield as a white solid: mp 297–299 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.90 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.05 (dd, J = 1.3 and 7.9 Hz, 1H, H-9), 7.10–7.20 (m, 2H, H-3″ and H-5″), 7.50–7.55 (m, 1H, H-4′), 7.60 (t, J = 7.8 Hz, 1H, H-5′), 7.65–7.75 (m, 5H, H-7, H-8, H-6′, H-2″, and H-6″), 7.85 (t, J = 2.0 Hz, 1H, H-2′), 8.00 (dd, J = 1.5 and 7.0 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.12, 39.16, 121.26, 122.15, 123.33, 124.87, 124.95, 125.16, 126.18, 126.82, 130.48, 130.57, 130.76, 130.85, 132.04, 133.49, 134.65, 134.70, 135.02, 139.73, 140.37, 158.69. Anal. Calcd for C24H20ClN5O5S2: C, 51.66; H, 3.61; N, 12.55. Found: C, 51.42; H, 3.80; N, 12.45.
1-(4-Chlorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2b)
Following the general procedure method C, compound 2b was obtained, from 8b, in 30% yield as a white solid: mp 284–286 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.90 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.00 (d, J = 7.7 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3″ and H-5″), 7.65–7.75 (m, 6H, H-7, H-8, H-2′, H-3′, H-5′, and H-6′), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.05 (dd, J = 1.5 and 7.8 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.15, 39.56, 121.30, 122.12, 123.39, 124.95, 125.16, 126.77, 127.97, 130.55, 130.59, 130.69, 130.87, 133.52, 134.68, 134.92, 135.06, 138.09, 139.79, 158.76. Anal. Calcd for C24H20ClN5O5S2: C, 51.66; H, 3.61; N, 12.55. Found: C, 51.39; H, 3.86; N, 13.05.
1-(2-Chlorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2c)
Following the general procedure method C, compound 2c was obtained, from 8c, in 30% yield as a white solid: mp 250–252 °C. 1H NMR (400 MHz, CDCl3): δ 3.00 (s, 3H, SO2CH3), 3.50 (s, 3H, NCH3), 6.55 (s, 1H, SO2NH), 6.80 (dd, J = 7.9 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3″ and H-5″), 7.45 (t, J = 7.9 Hz, 1H, H-8), 7.55–7.75 (m, 7H, H-7, H-3′, H-4′, H-5′, H-6′, H-2″, and H-6″), 8.05 (d, J = 7.8 Hz, 1H, H-6) 8.75 (s, 1H, CONH). 13C NMR (CDCl3): δ 39.12, 39.16, 121.18, 122.47, 122.91, 123.52, 125.05, 126.09, 128.45, 129.26, 129.76, 131.15, 131.48, 131.91, 132.04, 132.30, 132.40, 132.74, 135.23, 137.00, 139.10, 157.79. Anal. Calcd for C24H20ClN5O5S2: C, 51.66; H, 3.61; N, 12.55. Found: C, 51.93; H, 3.89; N, 12.30.
1-(3-Bromophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2d)
Following the general procedure method C, compound 2d was obtained, from 8d, in 60% yield as a white solid: mp 297–300 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.05 (dd, J = 1.3 and 7.4 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.55–7.65 (m, 2H, H-4′ and H-5′), 7.70–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.85–7.90 (m, 1H, H-6′), 8.00–8.05 (m, 2H, H-6 and H-2′), 9.65 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.13, 39.32, 121.25, 122.16, 122.85, 123.33, 124.95, 125.16, 125.21, 126.81, 128.93, 130.55, 130.76, 130.83, 132.23, 133.36, 133.48, 134.71, 135.01, 139.73, 140.42, 158.68. Anal. Calcd for C24H20BrN5O5S2: C, 47.85; H, 3.35; N, 11.62. Found: C, 48.08; H, 3.62; N, 11.30.
1-(4-Bromophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2e)
Following the general procedure method C, compound 2e was obtained, from 8e, in 30% yield as a white solid: mp 243–245 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.05 (dd, J = 1.6 and 7.6 Hz, 1H, H-9), 7.15–7.25 (m, 2H, H-3″ and H-5″), 7.60–7.65 (m, 2H, H-3′ and H-5′), 7.70–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.85–7.90 (m, 2H, H-2′ and H-6′), 8.00–8.05 (m, 1H, H-6), 9.65 (s, 1H, SO2NH), 10.45 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.15, 39.36, 121.28, 122.11, 123.39, 123.41, 124.98, 125.16, 126.81, 128.15, 130.53, 130.70, 130.85, 133.48, 133.54, 134.70, 135.05, 138.51, 139.83, 158.75. Anal. Calcd for C24H20BrN5O5S2: C, 47.85; H, 3.35; N, 11.62. Found: C, 47.73; H, 3.26; N, 11.80.
1-(2-Bromophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2f)
Following the general procedure method C, compound 2f was obtained, from 8f, in 50% yield as a white solid: mp 240–242 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.95 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 6.80 (d, J = 7.6 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.65–7.80 (m, 6H, H-7, H-8, H-4′, H-5′, H-2″, and H-6″), 7.95 (dd, J = 1.0 and 7.3 Hz, 1H, H-3′), 8.00 (dd, J = 1.1 and 7.2 Hz, 1H, H-6′), 8.05 (dd, J = 1.0 and 7.8 Hz, 1H, H-6), 9.65 (s, 1H, SO2NH), 10.45 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.37, 39.39, 121.24, 121.34, 122.30, 123.22, 123.50, 125.22, 125.96, 130.15, 130.63, 130.87, 130.90, 131.88, 133.28, 133.68, 134.38, 134.67, 135.03, 138.52, 139.71, 158.67. Anal. Calcd for C24H20BrN5O5S2: C, 47.85; H, 3.35; N, 11.62. Found: C, 47.90; H, 3.40; N, 11.50.
4-Methyl-1-(3-methylphenyl)-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2g)
Following the general procedure method C, compound 2g was obtained, from 8g, in 60% yield as yellowish solid: mp 283–285 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.40 (s, 3H, CH3), 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.05 (d, J = 7.6 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3″ and H-5″), 7.45 (d, J = 7.7 Hz, 1H, H-4′), 7.50–7.60 (m, 3H, H-2, H-5′, and H-6′), 7.70 (dt, J = 1.4 and 7.8 Hz, 1H, H-8), 7.75 (dt, J = 1.2 and 7.8 Hz, 1H, H-7), 7.80–7.85 (m, 2H, H-2″ and H-6″), 8.05 (dd, J = 1.4 and 7.8 Hz, 1H, H-6), 9.75 (s, 1H, SO2NH), 10.60 (s, 1H, NHCO). 13C NMR (DMSO-d6): δ 21.21, 39.22, 39.33, 121.28, 122.12, 123.34, 123.61, 124.71, 125.14, 126.52, 126.62, 130.25, 130.46, 130.57, 130.81, 131.16, 133.35, 134.62, 135.11, 139.27, 139.34, 140.46, 158.86. Anal. Calcd for C25H23N5O5S2: C, 55.85; H, 4.31; N, 13.03. Found: C, 56.01; H, 4.50; N, 12.85.
4-Methyl-1-(4-methylphenyl)-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2h)
Following the general procedure method C, compound 2h was obtained, from 8h, in 40% yield as a white solid: mp 279–281 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.50 (s, 3H, CH3), 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 6.95 (dd, J = 1.0 and 7.7 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3″ and H-5″), 7.40–7.45 (m, 2H, H-3′ and H-5′), 7.50–7.55 (m, 2H, H-2′ and H-6′), 7.65 (dt, J = 1.4 and 7.7 Hz, 1H, H-8), 7.70 (dt, J = 1.0 and 7.7 Hz, 1H, H-7), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.05 (dd, J = 1.4 and 7.7 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.45 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 21.28, 39.33, 39.51, 121.29, 122.11, 123.65, 124.68, 125.14, 126.09, 126.46, 130.47, 130.53, 130.83, 130.90, 133.36, 134.62, 135.12, 136.92, 139.32, 140.33, 158.88. Anal. Calcd for C25H23N5O5S2: C, 55.85; H, 4.31; N, 13.03. Found: C, 55.71; H 4.25; N 13.18.
4-Methyl-1-(2-methylphenyl)-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2i)
Following the general procedure method C, compound 2i was obtained, from 8i, in 60% yield as a white solid: mp 251–253 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.00 (s, 3H, CH3), 3.00 (s, 3H, SO2CH3), 3.40 (s, 3H, NCH3), 6.75 (d, J = 7.9 Hz, 1H, H-9), 7.25–7.30 (m, 2H, H-3″ and H-5″), 7.45–7.75 (m, 6H, H-7, H-8, H-3′, H-4′, H-5′,and H-6′), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.05 (d, J = 7.7 Hz, 1H, H-6), 9.65 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 17.08, 39.27, 39.32, 121.25, 122.19, 123.44, 123.57, 125.14, 125.81, 128.12, 128.20, 130.74, 130.94, 131.31, 131.48, 132.07, 133.63, 134.60, 135.10, 135.42, 138.58, 139.34, 158.84. Anal. Calcd for C25H23N5O5S2: C, 55.85; H, 4.31; N, 13.03. Found: C, 56.14; H, 4.60; N, 12.75.
1-(3-Methoxyphenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2j)
Following the general procedure method C, compound 2j was obtained, from 8j, in 30% yield as a white solid: mp 286–288 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 3.80 (s, 3H, OCH3), 7.05 (dd, J = 1.1 and 7.0 Hz, 1H, H-9), 7.15 (dd, J = 2.0 and 8.0 Hz, 1H, H-4′), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.25 (dd, J = 2.0 and 8.2 Hz, 1H, H-6′), 7.30 (t, J = 2.0 Hz, 1H, H-2′), 7.55 (t, J = 8.2 Hz, 1H, H-5′), 7.65–7.75 (m, 2H, H-7 and H-8), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.05 (dd, J = 1.4 and 7.5 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.22, 39.35, 56.21, 111.87, 116.50, 118.35, 121.28, 122.14, 123.53, 124.81, 125.12, 126.52, 130.52, 130.61, 130.81, 131.32, 133.38, 134.65, 135.09, 139.35, 140.33, 158.84, 160.70. Anal. Calcd for C25H23N5O6S2: C, 54.24; H, 4.19; N, 12.65. Found: C, 54.48; H, 4.32; N, 12.38.
1-(4-Methoxyphenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2k)
Following the general procedure method C, compound 2k was obtained, from 8k, in 40% yield as a white solid: mp 269–271 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 6.95 (dd, J = 1.2 and 7.5 Hz, 1H, H-9), 7.15–7.25 (m, 4H, H-3′, H-5′, H-3″, and H-5″), 7.55–7.60 (m, 2H, H-2′ and H-6′), 7.65–7.75 (m, 2H, H-7 and H-8), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.00 (dd, J = 1.4 and 7.6 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.23, 39.34, 56.13, 115.53, 121.29, 122.12, 123.69, 124.58, 125.15, 126.24, 127.90, 130.49, 130.64, 130.88, 132.21, 133.40, 134.60, 135.15, 139.14, 158.91, 160.65. Anal. Calcd for C25H23N5O6S2: C, 54.24; H, 4.19; N, 12.65. Found: C, 53.04; H, 4.06; N, 12.78.
4-Methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1-[3-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2l)
Following the general procedure method C, compound 2l was obtained, from 8l,19 in 60% yield as a white solid: mp 200–202 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.00 (d, J = 7.6 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.65–7.75 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.80–7.90 (m, 2H, H-5′ and H-6′), 7.95–8.00 (m, 2H, H-6 and H-4′), 8.15–8.20 (bs, 1H, H-2′), 9.70 (s, 1H, SO2NH), 10.60 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.15, 39.33, 121.27, 122.19, 123.09–123.13 (m, C-2′), 123.28, 123.90 (q, JC–F = 270 Hz, CF3), 124.92, 125.22, 126.96, 127.07–127.11 (m, C-4′), 130.20, 130.68, 130.82, 131.14 (q, JC–F = 32 Hz, C-3′), 131.82, 133.45, 134.73, 134.99, 139.81, 140.00, 158.69. Anal. Calcd for C25H20F3N5O5S2: C, 50.76; H, 3.41; N, 11.84. Found: C, 50.80; H, 3.46; N, 11.74.
4-Methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1-[4-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2m)
Following the general procedure method C, compound 2m was obtained, from 8m, in 60% yield as a white solid: mp 285–286 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.95 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 7.10 (d, J = 7.4 Hz, 1H, H-9), 7.15–7.25 (m, 2H, H-3″ and H-5″), 7.70–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.90–7.95 (m, 2H, H-2′ and H-6′), 8.00–8.10 (m, 3H, H-6, H-3′, and H-5′), 9.60 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.00, 39.11, 121.27, 122.09, 123.29, 125.17–125.22 (m, C-2′ and C-6′), 126.10 (q, JC–F = 300 Hz, CF3), 126.69, 127.16, 127.70–127.75 (m, C-3′ and C-5′), 130.49 (q, J = 32 Hz, C-4′), 130.57, 130.80, 130.83, 133.59, 134.75, 134.98, 140.24, 142.36, 158.67. Anal. Calcd for C25H20F3N5O5S2: C, 50.76; H, 3.41; N, 11.84. Found: C, 50.96; H, 3.58; N, 11.66.
4-Methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1-[2-(trifluoromethyl)phenyl]-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2n)
Following the general procedure method C, compound 2n was obtained, from 8n, in 30% yield as a white solid: mp 255–257 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 6.70 (d, J = 8.0 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.65 (dt, J = 1.2 and 7.9 Hz, 1H, H-8), 7.70 (dt, J = 1.2 and 7.9 Hz, 1H, H-7), 7.75–7.80 (m, 2H, H-2″, and H-6″), 7.95–8.05 (m, 4H, H-3′, H-4′, H-5′, and H-6′), 8.10–8.15 (m, 1H, H-6), 9.70 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.33, 39.37, 121.21, 122.31, 123.05 (q, JC–F = 270 Hz, CF3), 123.28, 123.68, 125.26, 126.03, 126.68 (q, JC–F = 30 Hz, C-2′), 128.63–128.68 (m, C-6′), 130.84, 130.98, 131.28, 132.54, 132.57, 133.62, 134.69, 134.96, 135.24, 136.62, 139.63, 158.61. Anal. Calcd for C25H20F3N5O5S2: C, 50.76; H, 3.41; N, 11.84. Found: C, 50.67; H, 3.36; N, 12.00.
1-(3,4-Difluorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2o)
Following the general procedure method C, compound 2o was obtained, from 8o, in 70% yield as a white solid: mp 290–292 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.95 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.10 (d, J = 7.5 Hz, 1H, H-9), 7.15–7.25 (m, 2H, H-3″ and H-5″), 7.50–7.55 (m, 1H, H-6′), 7.65–7.85 (m, 5H, H-7, H-8, H-2′, H-2″, and H-6″), 7.95–8.05 (m, 2H, H-6 and H-5′), 9.65 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.20, 39.30, 116.62 (d, JC–F = 20 Hz, C-2′), 119.27 (d, JC–F = 18.6 Hz, C-5′), 121.26, 122.13, 123.23, 123.65–123.80 (m, C-6′), 125.00, 125.18, 126.62, 130.77, 130.83, 130.85, 133.62, 134.69, 135.02, 135.75 (dd, JC–F = 3.4 and 8.3 Hz, C-1′), 139.69, 150.10 (dd, JC–F = 13.1 and 247.5 Hz, C-4′), 150.80 (dd, JC–F = 12.2 and 248 Hz, C-3′), 158.67. Anal. Calcd for C24H19F2N5O5S2: C, 51.51; H, 3.42; N, 12.52. Found: C, 51.76; H, 3.67; N, 12.22.
1-(3,4-Dichlorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2p)
Following the general procedure method C, compound 2p was obtained, from 8p, in 40% yield as a white solid: mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 7.20–7.25 (m, 3H, H-9, H-3″, and H-5″), 7.60 (dd, J = 2.3 and 8.6 Hz, 1H, H-6′), 7.70–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.85 (d, J = 8.6 Hz, 1H, H-5′), 8.00 (dd, J = 1.1 and 7.2 Hz, 1H, H-6), 8.10 (d, J = 2.3 Hz, 1H, H-2′), 9.60 (s, 1H, SO2NH), 10.45 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.09, 39.33, 121.25, 122.15, 123.20, 125.15, 125.21, 126.14, 126.95, 128.07, 130.61, 130.82, 130.83, 132.21, 132.93, 133.09, 133.63, 134.74, 134.97, 138.80, 139.90, 158.61. Anal. Calcd for C24H19Cl2N5O5S2: C, 48.65; H, 3.23; N, 11.82. Found: C, 48.33; H, 2.29; N, 12.06.
1-(3,4-Dimethylphenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2q)
Following the general procedure method C, compound 2q was obtained, from 8q, in 40% yield as a white solid: mp 301–303 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.25 and 2.30 (s, each 3H, CH3), 2.95 (s, 3H, SO2CH3), 3.20 (s, 3H, NCH3), 7.00 (d, J = 8.0 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.30 (dd, J = 2.0 and 8.0 Hz, 1H, H-6′), 7.40 (d, J = 8.0 Hz, 1H, H-5′), 7.45 (d, J = 2.0 Hz, 1H, H-2′), 7.65–7.75 (m, 2H, H-7 and H-8), 7.75–7.80 (m, 2H, H-2″ and H-6″), 8.00 (dd, J = 1.3 and 7.8 Hz, 1H, H-6), 9.50 (s, 1H, SO2NH), 10.35 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 19.66, 19.70, 39.21, 39.36, 121.29, 122.11, 123.46, 123.70, 124.66, 125.11, 126.40, 126.89, 130.39, 130.51, 130.81, 131.12, 133.36, 134.61, 135.12, 137.11, 138.99, 139.08, 139.18, 158.89. Anal. Calcd for C26H25N5O5S2: C, 56.61; H, 4.57; N, 12.70. Found: C, 56.59; H, 4.53; N, 12.75.
1-(3-Chloro-4-methylphenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2r)
Following the general procedure method C, compound 2r was obtained, from 8r, in 40% yield as a white solid: mp 287–289 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.45 (s, 3H, CH3), 2.95 (s, 3H, SO2CH3), 3.20 (s, 3H, NCH3), 7.10 (dd, J = 1.4 and 7.5 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.50 (dd, J = 2.1 and 8.0 Hz, 1H, H-6′), 7.60 (d, J = 8.0 Hz, 1H, H-5′), 7.65–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.85 (d, J = 2.1 Hz, 1H, H-2′), 8.05 (dd, J = 1.6 and 7.3 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 19.92, 39.12, 39.33, 121.26, 122.14, 123.41, 124.72, 124.90, 125.14, 126.41, 126.69, 130.49, 130.70, 130.82, 132.68, 133.48, 134.63, 134.68, 135.03, 138.10, 138.14, 139.55, 158.72. Anal. Calcd for C25H22ClN5O5S2: C, 52.49; H, 3.88; N, 12.24. Found: C, 52.89; H, 4.93; N, 12.15.
1-[4-Chloro-3-(trifluoromethyl)phenyl]-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2s)
Following the general procedure method C, compound 2s was obtained, from 8s, in 50% yield as a white solid: mp 288–290 °C. 1H NMR (400 MHz, DMSO-d6): δ 2.95 (s, 3H, SO2CH3), 3.20 (s, 3H, NCH3), 7.10–7.20 (m, 3H, H-9, H-3″, and H-5″), 7.65–7.80 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.85 (dd, J = 2.5 and 8.6 Hz, 1H, H-6′), 7.95 (d, J = 8.6 Hz, 1H, H-5′), 8.00 (dd, J = 1.2 and 7.4 Hz, 1H, H-9), 8.25 (d, J = 2.5 Hz, 1H, H-2′), 9.60 (s, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.08, 39.34, 121.25, 122.22, 122.68 (q, JC–F = 272 Hz, CF3), 123.14, 125.17, 125.28, 125.60–125.70 (m, C-2′), 127.11, 128.63 (q, JC–F = 31.4 Hz, C-3′), 130.70, 130.86, 130.89, 131.28 (bs, C-5′), 132.35 (bs, C-4′), 133.60, 133.74, 134.78, 134.93, 138.25, 140.15, 158.58. Anal. Calcd for C25H19ClF3N5O5S2: C, 47.96; H, 3.06; N, 11.19. Found: C, 47.76; H, 3.18; N, 11.02.
1-Cyclohexyl-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxyamide 5,5-Dioxide (2t)
Following the general procedure method C, compound 2t was obtained, from 8t, as a white solid in 40% yield after purification by flash column chromatography eluting with CHCl3/MeOH (95:5): mp 254–256 °C. 1H NMR (400 MHz, DMSO-d6): δ 1.25–1.35 (m, 2H, Cy-CH2), 1.50–1.65 (m, 2H, Cy-CH2), 1.70–1.80 (m, 2H, Cy-CH2), 1.85–1.95 (m, 4H, Cy-CH2), 2.95 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 4.70–4.80 (m, 1H, Cy-CH), 7.15–7.25 (m, 2H, H-3″, and H-5″), 7.70–7.85 (m, 3H, H-8, H-2″, and H-6″), 7.90–8.05 (m, 3H, H-6, H-7, and H-9), 9.60 (s, 1H, SO2NH), 10.10 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 25.05, 25.15, 32.93, 38.99, 39.35, 60.63, 121.22, 122.29, 123.77, 124.90, 125.28, 125.55, 130.20, 130.25, 131.17, 134.15, 134.61, 135.02, 137.59, 159.07. Anal. Calcd for C24H27N5O5S2: C, 54.43; H, 5.14; N, 13.22. Found: C, 54.63; H, 5.34; N, 13.01.
4-Methyl-N-{4-[(Methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (2u)
Following the general procedure method C, compound 2u was obtained from 8u as yellow solid in 60% yield after purification by flash column chromatography, eluting with CHCl3/acetone (6:4): mp 298–299 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.40 (s, 3H, NCH3), 7.15–7.20 (m, 2H, H-3″ and H-5″), 7.70–7.80 (m, 3H, H-8, H-2″, and H-6″), 7.85–7.95 (m, 2H, H-7 and H-9), 8.05 (d, J = 7.7 Hz, 1H, H-6), 9.50 (bs, 1H, SO2NH), 10.40 (s, 1H, CONH), 14.50 (s, 1H, NH). 13C NMR (DMSO-d6): δ 39.22, 39.35, 114.70, 115.50, 121.41, 121.90, 124.30, 124.56, 124.78, 127.13, 130.34, 130.52, 133.78, 134.45, 135.34, 159.10. Anal. Calcd for C18H17N5O4S2: C, 50.10; H, 3.97; N, 16.23. Found: C, 49.90; H, 3.76; N, 16.52.
1-(3-Fluorophenyl)-4-methyl-N-phenyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (3)
The title compound was prepared following the general procedure method C. Starting from compound 9(17) and replacing the N-(4-aminophenyl)methanesulfonamide with the aniline, compound 3 was obtained as a white solid in 70% yield after purification by flash chromatography, eluting with CHCl3/MeOH (95:5): mp 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.25 (s, 3H, NCH3), 7.05 (dd, J = 1.2 and 7.6 Hz, 1H, H-9), 7.10 (t, J = 7.4 Hz, 1H, H-4″), 7.35 (t, J = 7.5 Hz, 2H, H-3″ and H-5″), 7.45 (dd, J = 1.0 and 7.9 Hz, 1H, H-6′), 7.55 (dt, J = 1.8 and 8.5 Hz, 1H, H-4′), 7.65–7.75 (m, 4H, H-7, H-8, H-2″, and H-6″), 7.80–7.85 (m, 2H, H-2′ and H-6′), 8.05 (dd, J = 1.8 and 7.8 Hz, 1H, H-6), 10.30 (s, 1H, NH). 13C NMR (DMSO-d6): δ 39.15, 113.88 (d, JC–F = 25 Hz, C-2′), 117.55 (d, JC–F = 21 Hz, C-4′), 121.12, 122.44 (d, JC–F = 3 Hz, C-6′), 123.36, 124.55, 124.91, 125.17, 126.79, 129.08, 130.61, 130.74, 130.86, 132.28 (d, JC–F = 9.1 Hz, C-5′), 133.47, 138.63, 139.76, 140.51 (d, JC–F = 10.2 Hz, C-1′), 158.88, 162.74 (d, JC–F = 245.3 Hz, C-3′). Anal. Calcd for C23H17FN4O3S: C, 61.60; H, 3.82; N, 12.49. Found: C, 61.72; H, 3.93; N, 12.36.
N-[4-(Amino-2-oxoethoxy)phenyl]-1-(3-fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3c][1,2]benzothiazine-3-carboxamide 5,5-Dioxide (4)
The title compound was prepared following the general procedure method C. Starting from 9(17) and replacing the N-(4-aminophenyl)methanesulfonamide with the 2-(4-aminophenoxy)acetamide,21 compound 4 was obtained as a white solid in 60% yield after purification by flash column chromatography, eluting with CH2Cl2/MeOH (98:2): mp 223–225 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.35 (s, 3H, NCH3), 4.50 (s, 2H, OCH2), 6.90–7.00 (m, 2H, H-3″ and H-5″), 7.05 (d, J = 7.7 Hz, 1H, H-9), 7.40 (bs, 1H, NH), 7.45 (dd, J = 1.8 and 7.3 Hz, 1H, H-4′), 7.50–7.55 (m, 2H, H-8 and NH), 7.65–7.75 (m, 6H, H-7, H-2′, H-5′, H-6′, H-2″, and H-6″), 8.00 (dd, J = 1.4 and 7.7 Hz, 1H, H-6), 10.40 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 39.16, 67.41, 113.88 (d, JC–F = 25 Hz, C-2′), 115.10, 117.53 (d, JC–F = 21 Hz, C-4′), 122.47 (bs, C-6′), 122.63, 123.37, 124.90, 125.16, 126.70, 130.57, 130.72, 130.84, 132.19, 132.28 (d, JC–F = 9 Hz, C-5′), 133.47, 139.86, 140.53 (d, JC–F = 10.3 Hz, C-1′), 154.76, 158.56, 162.73 (d, JC–F = 245.4 Hz, C-3′), 170.42. Anal. Calcd for C25H20FN5O5S: C, 57.58; H, 3.87; N, 13.43. Found: C, 57.72; H, 4.02; N, 13.15.
1-(3-Fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazin-3-amine 5,5-Dioxide (10)
A mixture of the acid intermediate 9(17) (1.48 g, 4.0 mmol) in an excess of SOCl2 (4 mL) was refluxed under stirring for 1 h. The solvent was distilled off, and the residue was washed three times with dry benzene. The corresponding pyrazolobenzothiazine-3-carbonyl chloride was immediately dissolved in acetone (4 mL) and added dropwise to aqueous NaN3 (1 M, 5.6 mL) at 0 °C. After the mixture was stirred at room temperature for 5 min, water was added and the precipitated pinkish solid was filtered. The crude product was immediately suspended into a mixture of toluene/37% HCl (1:1) (20 mL) and heated at 100 °C for 1.5 h. The mixture was then cooled and the solid formed was filtered to give compound 10 (0.95 g, 80%) as a white solid, used as is in the next reaction step: mp 185–187 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, NCH3), 6.25 (s, 2H, NH2), 7.15–7.20 (m, 2H, H-9 and H-4′), 7.25–7.30 (m, 2H, H-2′ and H-5′), 7.50–7.55 (m, 1H, H-6′), 7.65–7.70 (m, 2H, H-7 and H-8), 7.95–8.00 (m, 1H, H-6).
N-[1-(3-Fluorophenyl)-4-methyl-5,5-dioxido-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazin-3-yl]-4-nitrobenzamide (11)
To a stirred solution of amine derivative 10 (0.76 g, 2.3 mmol) in dry pyridine (10 mL), under nitrogen flux, a solution of 4-nitrobenzoyl chloride (0.85 g, 4.6 mmol) in dry pyridine (10 mL) was added dropwise. The mixture was heated at 40 °C for 18 h and then was concentrated to half volume and poured into ice–water, acidified with 2 N HCl (pH 3), and the precipitate formed was filtered under vacuum. The solid was suspended in 20% NaHCO3, filtered, and purified by flash column chromatography, eluting with CH2Cl2/MeOH (95:5) to give compound 11 (0.43 g, 45%) as a white solid: mp 273–275 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.00 (s, 3H, NCH3), 7.15 (d, J = 6.5 Hz, 1H, H-9), 7.25–7.30 (m, 1H, H-4′), 7.40–7.50 (m, 2H, H-2′ and H-5′), 7.55–7.70 (m, 3H, H-7, H-8, and H-6′), 7.90–8.00 (m, 1H, H-6), 8.15–8.25 (m, 2H, H-2″ and H-6″), 8.30–8.40 (m, 2H, H-3″ and H-5″), 11.05 (s, 1H, CONH).
4-Amino-N-[1-(3-fluorophenyl)-4-methyl-5,5-dioxido-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazin-3-yl]benzamide (12)
A stirred solution of nitro derivative 11 (0.40 g, 0.8 mmol) in DMF (50 mL) was hydrogenated over a catalytic amount of Raney nickel at room temperature and atmospheric pressure for 2 h. The mixture was then filtered over Celite. The filtrate was evaporated to dryness and triturated with ethanol to give the amino derivative 12 (0.26 g, 66%) as a white solid: mp 218–220 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, NCH3), 5.80 (s, 2H, NH2), 6.55–6.65 (m, 2H, H-3″ and H-5″), 7.10–7.15 (m, 1H, H-9),7.30–7.35 (m, 1H, H-4′), 7–40–7.50 (m, 2H, H-2′ and H-5′), 7.55–7.70 (m, 3H, H-7, H-8, and H-6′), 7.75–7.80 (m, 2H, H-2″ and H-6″), 7.95–8.00 (m, 1H, H-6), 10.15 (s, 1H, CONH).
N-[1-(3-Fluorophenyl)-4-methyl-5,5-dioxido-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazin-3-yl]-4-(methylsulfonyl)aminobenzamide (5)
Methanesulfonyl choride (0.2 mL, 3 mmol) was added dropwise to a solution of the amino derivative 12 (0.28 g, 0.6 mmol) in dry CH2Cl2 (20 mL) and dry pyridine (2 mL) at 0 °C. The mixture was left overnight at room temperature and then poured into ice/water, acidified with 2 N HCl (pH 3), and extracted with CH2Cl2 (4 times). The combined organic layers were washed with brine (2 times), dried, filtered, and evaporated to dryness. The residue was purified by column chromatography, eluting with CH2Cl2/MeOH (95:5), affording the target compound 5 (0.16 g, 50%) as a white solid: mp 261–263 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.20 (s, 3H, NCH3), 7.15 (dd, J = 2.0 and 7.5 Hz, 1H, H-9), 7.30–7.40 (m, 3H, H-4′, H-3″, and H-5″), 7.45–7.50 (m, 2H, H-2′ and H-5′), 7.60–7.75 (m, 3H, H-7, H-8, and H-6′), 7.95–8.10 (m, 3H, H-6, H-2″, and H-6″), 10.25 (bs, 1H, SO2NH), 10.70 (s, 1H, CONH). 13C NMR (DMSO-d6): δ 36.65, 36.75, 112.84 (d, JC–F = 24.1 Hz, C-2′), 116.65 (d, JC–F = 21.1, C-4′), 118.29, 121.65 (bs, C-6′), 122.20, 123.79, 124.82, 124.96, 127.86, 129.22, 129.92, 130.32, 130.89, 131.24 (d, JC–F = 10 Hz, C-5′), 133.33, 140.48, 140.77 (d, JC–F = 10 Hz, C-1′), 142.61, 162.72 (d, JC–F = 245.1 Hz, C-3′), 166.35. Anal. Calcd for C24H20FN5O5S2: C, 53.23; H, 3.72; N, 12.93. Found: C, 51.43; H, 4.05; N, 12.66.
Computational Methods
Pyrazolobenzothiazine derivative 2a was built using the Schrödinger Maestro Interface42 and then submitted to Polak–Ribiere conjugate gradient minimization [0.0005 kJ/(Å mol) convergence]. The docking of 2a into the PSI of 3SKE was then performed with the same methodology previously described.28
Molecular dynamics (MD) simulation of 2a/NS5B complex was run in explicit solvent, using the TIP4P water model43 in a periodic boundary conditions orthorhombic box. Desmond31 was used to set up and run the MD simulations. The simulated environment was built using the system builder utility, with the structures being neutralized by Na+ and Cl– ions, which were added until a concentration of 0.15 M was reached. The buried regions were solvated using the “solvate pocket” utility. Before the simulations were performed, a series of minimizations and short MD simulations were carried out to relax the model system by means of a relaxation protocol consisting of six stages: (i) minimization with the solute restrained; (ii) minimization without restraints; (iii) simulation (12 ps) in the NVT ensemble using a Berendsen thermostat (10 K) with non-hydrogen solute atoms restrained; (iv) simulation (12 ps) in the NPT ensemble using a Berendsen thermostat (10 K) and a Berendsen barostat (1 atm) with non-hydrogen solute atoms restrained; (v) simulation (24 ps) in the NPT ensemble using a Berendsen thermostat (300 K) and a Berendsen barostat (1 atm) with non-hydrogen solute atoms restrained; (vi) unrestrained simulation (24 ps) in the NPT ensemble using a Berendsen thermostat (300 K) and a Berendsen barostat (1 atm). At this point, a 10 ns long MD simulation was carried out at a temperature of 300 K in the NPT ensemble using a Nose–Hoover chain thermostat and a Martyna–Tobias–Klein barostat (1.01325 bar). The simulation analysis was carried out by means of the Simulation Interaction Diagram utility44 embedded in the Schrödinger Suite 2013 update 2.
Vital Stains
Nile Red was obtained from Sigma. Monodansylcadaverine and LipidTOX Red Neutral lipid stain was obtained from life technologies. Huh-9-13 cells were seeded in glass bottom 35 mm culture dishes (Mattek) at a density of 25.000 cells/dish in complete DMEM without G418 at 37 °C and 5% CO2. The next day medium was replaced with complete DMEM containing either the proper dilution of the compound to be tested or DMSO. The cells were further incubated for 3 days at 37 °C and 5% CO2, after which the medium was replaced with complete medium without neutral red and either Nile Red (2 μg/mL), monodansylcadaverine (50 μM), or LipidTOX Red Neutral lipid stain (diluted 1/1000). Next, the cells were incubated for an additional 30 min at 37 °C and 5% CO2 after which the cells were imaged using a laser-scanning SP5 confocal microscope (Leica Microsystems) equipped with a DMI 6000 microscope and an Acousto optical beam splitter. For Nile Red the yellow-gold fluorescence was monitored at an excitation wavelength (λex) of 552 nm and an emission wavelength (λem) of 636 nm. The red fluorescence was monitored at λex of 485 nm and λem of 525 nm. Monodansylcadaverine was imaged at λex of 340 nm and λem of 530 nm, and LipidTOX Red Neutral lipid stain was assessed at λex of 577 nm and λem of 609 nm.
Physicochemical and ADME Properties. Solubility
For thermodynamic and kinetic solubility determination, solutions were shaken in a shaker bath at room temperature for 48 h before the filtration. Determinations were performed in triplicate by LC–UV–MS assay. Quantification of the compound was made by comparison with corresponding calibration curves realized with standard solutions in methanol.
Shake Flask Method for Lipophilicity
The octanol/water partitioning experiment was performed in a test tube. The test tube was filled with 2.5 mL of water and 2.5 mL of octanol to form the shake flask setup. The tested compound was dissolved directly in octanol.
The sample was sealed and then agitated to produce good mixing between the phases. Time required to reach the equilibrium was 10 h. An aliquot was obtained from each of the phases and is analyzed, typically by HPLC, to determine the concentration of selected compound in each phase.
The experimental log P was calculate using the following equation:
LC analysis was performed with a Perkin-Elmer (series 200) instrument equipped with an UV detector (Perkin-Elmer 785A, UV/vis detector). Chromatographic analysis was conducted using a Polaris C18-A column (150 mm × 4.6 mm, 5 μm particle size) at a flow rate of 0.8 mL min–1 with a mobile phase composed of 50% ACN/50% H2O.
PAMPA Assay
Donor solution (0.5 mM) was prepared by diluting 1 mM dimethylsulfoxide (DMSO) compound stock solution using phosphate buffer (pH 7.4, 0.025 M). Filters were coated with 5 μL of a 1% (w/v) dodecane solution of phosphatidylcholine or 4 μL of brain polar lipid solution (20 mg/mL 16% CHCl3, 84% dodecane) prepared from CHCl3 solution, 10% w/v, for intestinal permeability and BBB permeability, respectively. Donor solution (150 μL) was added to each well of the filter plate. To each well of the acceptor plate was added 300 μL of solution (50% DMSO in phosphate buffer). The compound was tested in three different plates on different days. The sandwich was incubated for 5 or 22 h at room temperature under gentle shaking. After the incubation time, the plates were separated and samples were taken from both receiver and donor sides and analyzed using LC with UV detection at 254 nm. LC analyses were performed with a Perkin-Elmer (series 200) instrument equipped with an UV detector (Perkin-Elmer 785A, UV/vis detector). Chromatographic separations were conducted using a Polaris C18-A column (150 mm × 4.6 mm, 5 μm particle size) at a flow rate of 0.8 mL min–1 with a mobile phase composed of 50% ACN/50% H2O.
Permeability (Papp) for PAMPA was calculated according to the following equation, obtained from the Wohnsland and Faller45 and Sugano et al.46 equation with some modification in order to obtain permeability values in cm/s,
where VA is the volume in the acceptor well, VD is the volume in the donor well (cm3), A is the “effective area” of the membrane (cm2), t is the incubation time (s), and r is the ratio between drug concentration in the acceptor and equilibrium concentration of the drug in the total volume (VD + VA). Drug concentration was estimated by measuring the peak area.
Membrane retention (%) was calculated according to the following equation:
where r is the ratio between drug concentration in the acceptor and equilibrium concentration and where D, A, and Eq represent the drug concentration in the donor, acceptor, and equilibrium solution, respectively.
Metabolic Stability
The compound in DMSO solution was incubated at 37 °C for 60 min in 125 mM phosphate buffer (pH 7.4), 5 μL of human liver microsomal protein (0.2 mg/mL), in the presence of a NADPH-generating system at a final volume of 0.5 mL (compound final concentration, 50 μM); DMSO did not exceed 2% (final solution). The reaction was stopped by cooling in ice and adding 1.0 mL of acetonitrile. The reaction mixtures were then centrifuged, and the parent drug and metabolites were subsequently determined by LC–UV–MS. Chromatographic analyses were performed with an Agilent 1100 LC/MSD VL system (G1946C) (Agilent Technologies, Palo Alto, CA) consisting of a vacuum solvent degassing unit, a binary high-pressure gradient pump, an 1100 series UV detector, and an 1100 MSD model VL benchtop mass spectrometer. Chromatographic separation was obtained using a Varian Polaris C18-A column (150 mm × 4.6 mm, 5 μm particle size) and gradient elution, eluent A being ACN and eluent B consisting of water. The analysis started with 2% of eluent A, which was rapidly increased to 70% in 12 min, then slowly increased to 98% in 20 min. The flow rate was 0.8 mL min–1, and injection volume was 20 μL. The Agilent 1100 series mass spectra detection (MSD) single-quadrupole instrument was equipped with the orthogonal spray API-ES (Agilent Technologies, Palo Alto, CA). Nitrogen was used as nebulizing and drying gas. The pressure of the nebulizing gas, the flow of the drying gas, the capillary voltage, the fragmentor voltage, and the vaporization temperature were set at 40 psi, 9 L/min, 3000 V, 70 V, and 350 °C, respectively. UV detection was monitored at 280 nm. The LC–ESI-MS determination was performed by operating the MSD in the negative ion mode. Spectra were acquired over the scan range m/z 100–1500 using a step size of 0.1 μm. The percentage of compound not metabolized was calculated by comparison with reference solutions.
Acknowledgments
This work was supported by the UMDNJ Foundation and National Institutes of Health Research Grant CA153147 to N.K.-B. We thank Roberto Bianconi, Stijn Delmotte, Katrien Geerts, and Inge Vliegen for excellent technical assistance. We are grateful to Els Vanstreels for assisting with fluorescence microscopy.
Glossary
Abbreviations Used
- RBV
ribavirin
- pegIFN-α
pegylated interferon α
- SVR
sustained virological response
- DAA
direct-aging antiviral
- NS5B
nonstructural 5B
- NI
nucleoside inhibitor
- NNI
non-nucleoside inhibitor
- TSI
thumb site I
- TSII
thumb site II
- PSI
palm site I
- PSII
palm site II
- PSIII
palm site III
- SI
selectivity index
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- EMCV
encephalomyocarditis virus
- IRES
internal ribosome entry site
- neo
neomycin phosphotransferase
- MDC
monodansylcadaverine
- PAMPA
parallel artificial membrane permeability
- MR
membrane retention
- HLM
human liver microsome
Supporting Information Available
Biochemical and biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [DOI] [PubMed] [Google Scholar]
- Tran T. T. Overview of epidemiology, diagnosis, and disease progression associated with hepatitis C. Am. J. Managed Care 2012, 18, S335–S339. [PubMed] [Google Scholar]
- Fauvelle C.; Lepiller Q.; Felmlee D. J.; Fofana I.; Habersetzer F.; Stoll-Keller F.; Baumert T. F.; Fafi-Kremer S. Hepatitis C virus vaccines—progress and perspectives. Microb. Pathog. 2013, 58, 66–72. [DOI] [PubMed] [Google Scholar]
- Berman K.; Kwo P. Y. Boceprevir, an NS3 protease inhibitor of HCV. Clin. Liver Dis. 2009, 13, 429–439. [DOI] [PubMed] [Google Scholar]
- Kwong A. D.; Kauffman R. S.; Hurter P.; Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011, 29, 993–1003. [DOI] [PubMed] [Google Scholar]
- Ferenci P.; Reddy K. R. Impact of HCV protease-inhibitor-based triple therapy for chronic HCV genotype 1 infection. Antiviral Ther. 2011, 16, 1187–1201. [DOI] [PubMed] [Google Scholar]
- Manns M. P.; von Hahn T. Novel therapies for hepatitis C—One pill fits all?. Nat. Rev. Drug Discovery 2013, 12, 595–610. [DOI] [PubMed] [Google Scholar]
- Poordad F.; Lawitz E.; Kowdley K. V.; Cohen D. E.; Podsadecki T.; Siggelkow S.; Heckaman M.; Larsen L.; Menon R.; Koev G.; Tripathi R.; Pilot-Matias T.; Bernstein B. Exploratory study of oral combination antiviral therapy for hepatitis C. N. Engl. J. Med. 2013, 368, 45–53. [DOI] [PubMed] [Google Scholar]
- Liang T. J.; Ghany M. G. Current and future therapies for hepatitis C virus infection. N. Engl. J. Med. 2013, 368, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HCV Drugs in Development, October 1, 2013; HCV Advocate, San Francisco, CA. http://www.hcvadvocate.org/hepatitis/hepC/Quick_Ref_Guide.pdf (accessed October 31, 2013).
- Behrens S. E.; Tomei L.; De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 2012, 55, 2481–2531. [DOI] [PubMed] [Google Scholar]
- Barreca M. L.; Iraci N.; Manfroni G.; Cecchetti V. Allosteric inhibition of the hepatitis C virus NS5B polymerase: in silico strategies for drug discovery and development. Future Med. Chem. 2011, 3, 1027–1055. [DOI] [PubMed] [Google Scholar]
- Li H.; Shi S. T. Non-nucleoside inhibitors of hepatitis C virus polymerase: current progress and future challenges. Future Med. Chem. 2010, 2, 121–141. [DOI] [PubMed] [Google Scholar]
- Manfroni G.; Meschini F.; Barreca M. L.; Leyssen P.; Samuele A.; Iraci N.; Sabatini S.; Massari S.; Maga G.; Neyts J.; Cecchetti V. Pyridobenzothiazole derivatives as new chemotype targeting the HCV NS5B polymerase. Bioorg. Med. Chem. 2012, 20, 866–876. [DOI] [PubMed] [Google Scholar]
- Manfroni G.; Cannalire R.; Barreca M. L.; Kaushik-Basu N.; Leyssen P.; Winquist J.; Iraci N.; Manvar D.; Paeshuyse J.; Guhamazumder R.; Basu A.; Sabatini S.; Tabarrini O.; Danielson U. H.; Neyts J.; Cecchetti V. The versatile nature of the 6-aminoquinolone scaffold: identification of submicromolar hepatitis C virus NS5B inhibitors. J. Med. Chem. 2014, 57, 1952–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreca M. L.; Manfroni G.; Leyssen P.; Winquist J.; Kaushik-Basu N.; Paeshuyse J.; Krishnan R.; Iraci N.; Sabatini S.; Tabarrini O.; Basu A.; Danielson U. H.; Neyts J.; Cecchetti V. Structure-based discovery of pyrazolobenzothiazine derivatives as inhibitors of hepatitis C virus replication. J. Med. Chem. 2013, 56, 2270–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A. N.; Tedesco R.; Bambal R.; Chai D.; Concha N. O.; Darcy M. G.; Dhanak D.; Duffy K. J.; Fitch D. M.; Gates A.; Johnston V. K.; Keenan R. M.; Lin-Goerke J.; Liu N.; Sarisky R. T.; Wiggall K. J.; Zimmerman M. N. Substituted benzothiadiazine inhibitors of hepatitis C virus polymerase. Bioorg. Med. Chem. Lett. 2009, 19, 4350–4353. [DOI] [PubMed] [Google Scholar]
- Cecchetti V.; Fravolini A.; Schiaffella F.; De Regis M.; Orzalesi G.; Volpato I. Synthesis and pharmacologic activity of the carboxamides of pyrazolo-1,2-benzothiazine and isoxazolo-1,2-benzothiazinecarboxamides. Farmaco 1983, 38, 35–44. [PubMed] [Google Scholar]
- Waslei J.; Rosenthal G. J.; Sun X.; Strong S.; Qiu J.. Novel Pyrrole Inhibitors of S-Nitrosoglutathione Reductase as Therapeutic Agents. PTC WO 2010/019903 A1, 2010.
- Singh R.; Argade A.; Payan D.; Molineaux S.; Holland S. J.; Clough J.; Keim H.; Bhamipidati S.; Sylvain C.; Li H.; Rossi A. B.. 2,4-Pyrimidine Compounds and Their Uses. U.S. 2005/0209230 A1, 2005.
- Nichols D. B.; Fournet G.; Gurukumar K. R.; Basu A.; Lee J. C.; Sakamoto N.; Kozielski F.; Musmuca I.; Joseph B.; Ragno R.; Kaushik-Basu N. Inhibition of hepatitis C virus NS5B polymerase by S-trityl-l-cysteine derivatives. Eur. J. Med. Chem. 2012, 49, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik-Basu N.; Bopda-Waffo A.; Talele T. T.; Basu A.; Costa P. R.; da Silva A. J.; Sarafianos S. G.; Noel F. Identification and characterization of coumestans as novel HCV NS5B polymerase inhibitors. Nucleic Acids Res. 2008, 36, 1482–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Bopda-Waffo A.; Basu A.; Krishnan R.; Silberstein E.; Taylor D. R.; Talele T. T.; Arora P.; Kaushik-Basu N. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antiviral Chem. Chemother. 2009, 20, 19–36. [DOI] [PubMed] [Google Scholar]
- Nichols D. B.; Leao R. A.; Basu A.; Chudayeu M.; de Moraes Pde F.; Talele T. T.; Costa P. R.; Kaushik-Basu N. Evaluation of coumarin and neoflavone derivatives as HCV NS5B polymerase inhibitors. Chem. Biol. Drug Des. 2013, 81, 607–614. [DOI] [PubMed] [Google Scholar]
- Cheng C. C.; Shipps G. W. Jr.; Yang Z.; Kawahata N.; Lesburg C. A.; Duca J. S.; Bandouveres J.; Bracken J. D.; Jiang C. K.; Agrawal S.; Ferrari E.; Huang H. C. Inhibitors of hepatitis C virus polymerase: synthesis and characterization of novel 2-oxy-6-fluoro-N-((S)-1-hydroxy-3-phenylpropan-2-yl)-benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 2119–2124. [DOI] [PubMed] [Google Scholar]
- Betzi S.; Eydoux C.; Bussetta C.; Blemont M.; Leyssen P.; Debarnot C.; Ben-Rahou M.; Haiech J.; Hibert M.; Gueritte F.; Grierson D. S.; Romette J. L.; Guillemot J. C.; Neyts J.; Alvarez K.; Morelli X.; Dutartre H.; Canard B. Identification of allosteric inhibitors blocking the hepatitis C virus polymerase NS5B in the RNA synthesis initiation step. Antiviral Res. 2009, 84, 48–59. [DOI] [PubMed] [Google Scholar]
- Barreca M. L.; Iraci N.; Manfroni G.; Gaetani R.; Guercini C.; Sabatini S.; Tabarrini O.; Cecchetti V. Accounting for target flexibility and water molecules by docking to ensembles of target structures: the HCV NS5B palm site I inhibitors case study. J. Chem. Inf. Model. 2014, 54, 481–497. [DOI] [PubMed] [Google Scholar]
- Anilkumar G. N.; Lesburg C. A.; Selyutin O.; Rosenblum S. B.; Zeng Q.; Jiang Y.; Chan T. Y.; Pu H.; Vaccaro H.; Wang L.; Bennett F.; Chen K. X.; Duca J.; Gavalas S.; Huang Y.; Pinto P.; Sannigrahi M.; Velazquez F.; Venkatraman S.; Vibulbhan B.; Agrawal S.; Butkiewicz N.; Feld B.; Ferrari E.; He Z.; Jiang C. K.; Palermo R. E.; McMonagle P.; Huang H. C.; Shih N. Y.; Njoroge G.; Kozlowski J. A. II; Novel HCV NS5B polymerase inhibitors: discovery of indole 2-carboxylic acids with C3-heterocycles. Bioorg. Med. Chem. Lett. 2011, 21, 5336–5341. [DOI] [PubMed] [Google Scholar]
- Glide, version 6.0; Schrödinger, LLC, New York, NY, 2013.
- Desmond Molecular Dynamics System, version 3.5; D. E. Shaw Research: New York, NY, 2013.
- Windisch M. P.; Frese M.; Kaul A.; Trippler M.; Lohmann V.; Bartenschlager R. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 2005, 79, 13778–13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuyver L. J.; McBrayer T. R.; Tharnish P. M.; Hassan A. E.; Chu C. K.; Pankiewicz K. W.; Watanabe K. A.; Schinazi R. F.; Otto M. J. Dynamics of subgenomic hepatitis C virus replicon RNA levels in Huh-7 cells after exposure to nucleoside antimetabolites. J. Virol. 2003, 77, 10689–10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrolijk J. M.; Kaul A.; Hansen B. E.; Lohmann V.; Haagmans B. L.; Schalm S. W.; Bartenschlager R. A replicon-based bioassay for the measurement of interferons in patients with chronic hepatitis C. J. Virol. Methods 2003, 110, 201–209. [DOI] [PubMed] [Google Scholar]
- Bartenschlager R.; Lohmann V. Novel cell culture systems for the hepatitis C virus. Antiviral Res. 2001, 52, 1–17. [DOI] [PubMed] [Google Scholar]
- Kansy M.; Senner F.; Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- Bermejo M.; Avdeef A.; Ruiz A.; Nalda R.; Ruell J. A.; Tsinman O.; González I.; Fernández C.; Sánchez G.; Garrigues T. M.; Merino V. PAMPA—a drug absorption in vitro model 7. Comparing rat in situ, Caco-2, and PAMPApermeability of fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [DOI] [PubMed] [Google Scholar]
- Koljonen M.; Rousu K.; Cierny J.; Kaukonen A. M.; Hirvonen J. Transport evaluation of salicylic acid and structurally related compounds across Caco-2 cell monolayers and artificial PAMPA membranes. Eur. J. Pharm. Biopharm. 2008, 70, 531–538. [DOI] [PubMed] [Google Scholar]
- Bassendine M. F.; Sheridan D. A.; Felmlee D. J.; Bridge S. H.; Toms G. L.; Neely R. D. HCV and the hepatic lipid pathway as a potential treatment target. J. Hepatol. 2011, 55, 1428–1440. [DOI] [PubMed] [Google Scholar]
- Rasmussen A. L.; Diamond D. L.; McDermott J. E.; Gao X.; Metz T. O.; Matzke M. M.; Carter V. S.; Belisle S. E.; Korth M. J.; Waters K. M.; Smith R. D.; Katze M. G. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato M. T.; Gomez-Lechon M. J. Drug-induced liver steatosis and phospholipidosis: cell-based assays for early screening of drug candidates. Curr. Drug Metab. 2012, 13, 1160–1173. [DOI] [PubMed] [Google Scholar]
- Maestro, version 9.5; Schrödinger, LLC: New York, NY, 2013.
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Maestro-Desmond Interoperability Tools, version 3.5; Schrödinger: New York, NY, 2013.
- Wohnsland F.; Faller B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 2001, 44, 923–930. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Hamada H.; Machida M.; Ushio H. High throughput prediction of oral absorption: improvement of the composition of the lipid solution used in parallel artificial membrane permeation assay. J. Biomol. Screening 2001, 6, 189–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.