

Abstract
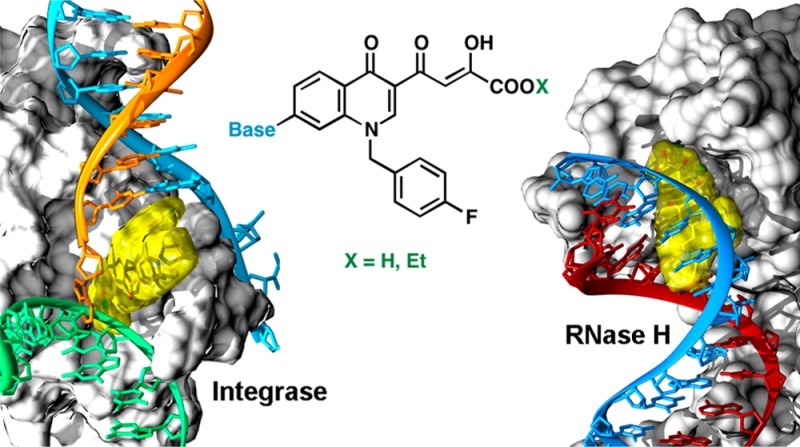
A series of antiviral basic quinolinonyl diketo acid derivatives were developed as inhibitors of HIV-1 IN. Compounds 12d,f,i inhibited HIV-1 IN with IC50 values below 100 nM for strand transfer and showed a 2 order of magnitude selectivity over 3′-processing. These strand transfer selective inhibitors also inhibited HIV-1 RNase H with low micromolar potencies. Molecular modeling studies based on both the HIV-1 IN and RNase H catalytic core domains provided new structural insights for the future development of these compounds as dual HIV-1 IN and RNase H inhibitors.
Introduction
HIV/AIDS remains one of the most important global health challenges, especially in sub-Saharan Africa.1,2 Highly active antiretroviral therapy (HAART),3 the standard of care for HIV/AIDS, comprises a multitarget regimen combining antiviral drugs with orthogonal mechanisms of action, thus increasing the genetic barrier against resistance selection when compared to monotherapy. Nevertheless, treatment adherence resides primarily on treatment tolerance and simplicity of administration, which remains a challenge with multipill HAART cocktails.4 A single compound capable of inhibiting simultaneously two viral targets could represent a therapeutic alternative. Multitarget inhibitors may alleviate dosing complexity, drug–drug interactions, and toxicities.5 In the field of medicinal chemistry, the design of active dual inhibitors against HIV reverse transcriptase (RT) and integrase (IN) is subject of great interest.6 These inhibitors act on the catalytic sites of the IN enzyme and the ribonuclease H (RNase H) domain of HIV RT. IN contains three catalytic carboxylate residues, D64, D116, and E152, forming the DDE motif that coordinates two magnesium atoms of the IN catalytic site. Many HIV-1 IN inhibitors with metal-complexing properties have been reported.7 These inhibitors are referred to as strand transfer IN inhibitors (INSTIs). Three INSTIs, elvitegravir (EVG, 1), raltegravir (RAL, 2), and dolutegravir (DTG, 3) have already been approved by the Food and Drug Administration (Figure 1).8,9
Figure 1.

Anti HIV-1 agents targeting IN (1–3) and RNase H (4–6).
RT is another important HIV-1 enzyme and the target of many anti-HIV drugs. This enzyme has RNA- and DNA-dependent DNA polymerase, strand displacement, strand transfer, and RNase H activities.10 RNase H activity, which degrades RNA from RNA–DNA hybrid molecules, is required at several steps during reverse transcription and essential for virus replication. The crystal and NMR structures of isolated HIV RNase H domain are similar to that of the RNase H domain in the context of the full-length HIV-RT protein.11
These structures also showed that the folding of the HIV-1 RNase H catalytic core domain (CCD) is similar to that of HIV-1 IN and, consequently, the catalytic sites of the two enzymes share a similar geometry. Indeed, also RNase H features the DDE catalytic motif (comprising D443, E478, and D498 residues) chelating two magnesium ions, although a fourth carboxylate residue (D549) is required for catalysis.12 Similar structural characteristics including three aspartate residues and two magnesium ions at a distance of 3.57 Å from each other were shown in the DNA polymerase active site of RT in complex with DNA primer template and an incoming nucleotide.12
Some diketo acid inhibitors of HIV-1 IN have shown activity on RNase H,13,14 whereas DNA aptamers used as inhibitors of RNase H have also been employed to inhibit HIV-1 IN.15 Tropolone (5),16 madurahydroxylactone (6),17 and 2-hydroxyquinoline-1,3(2H,4H)-dione (4)18 were recently described to be able to inhibit both enzymes (Figure 1).
Therefore, concurrent inhibition of HIV-1 RNase H and IN by compounds with metal-chelating activity could represent an interesting approach to alleviate some of the problems linked to multipill combination therapies.
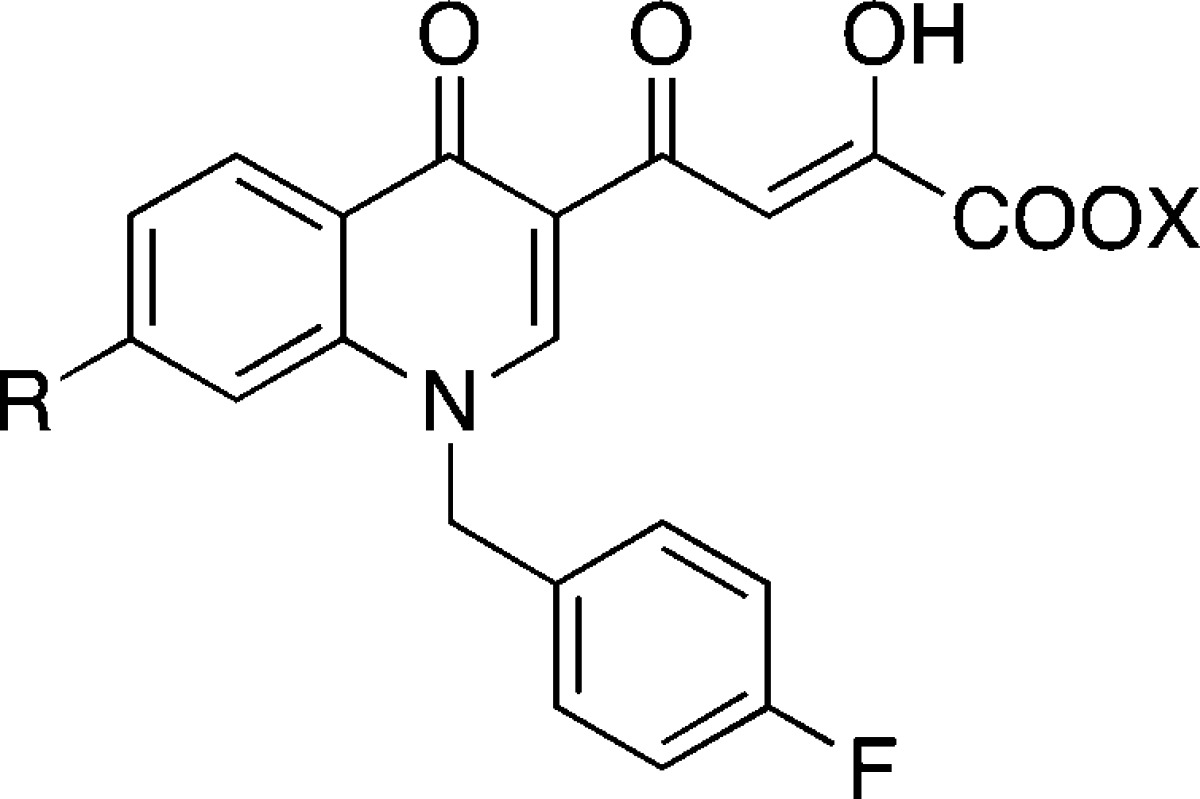
The work of our research group commenced in the context of diketo acids (DKAs) compounds as HIV-1 IN inhibitors.19 The design of quinolinone derivatives characterized by one DKA moiety on position 3 of the quinolinone ring led to HIV-1 IN inhibitors with selectivity for strand transfer versus 3′-processing. Moreover, inserting a functional group with basic properties on position 7 of the quinolinone ring generated derivative 7 (Figure 2), which inhibited ST with nanomolar IC50 value and submicromolar antiviral activity.19b These results prompted us to investigate the position 7 of the quinolinone ring of newly designed DKAs, keeping unchanged the skeleton of the quinolinonyl diketo acid inhibitors, including the p-fluoro benzyl group linked to the quinolone nitrogen.
Figure 2.

1-Pyrrolidinyl substituted quinolinonyl DKA (7) and general structure of “base” substituted quinolinonyl DKAs.
Because of the similarities of the IN and RNase H catalytic sites and reports on dual activities of a number of compounds against IN/RNase H, we decide to test our newly designed quinolonyl DKAs against both enzymes.19b
Herein we describe the synthesis and biological studies of a series of DKA derivatives that present a p-fluoro benzyl group on quinolone nitrogen and a variable “base-like” functional group in position 7 of the quinolone ring as inhibitors of HIV-1 IN. A comparative evaluation of the activity against RNase H function of the HIV RT of the newly synthesized compounds was also performed (Figure 2).
Results and Discussion
Chemistry
Synthesis of derivatives 11a–g,i and 12a–g,i is outlined in Scheme 1. 3-Acetyl-4(1H)-quinolinone 8 were prepared by reaction of 3-fluoroaniline with ethyl orthoformate and ethyl acetoacetate, which were thermally condensed in the presence of an inert heating medium (dowtherm A) under argon atmosphere according to the Yoshizawa procedure.20 Then 8 was alkylated with 4-fluorobenzyl bromide in alkaline medium (K2CO3) to give the N-1 substituted quinolinone 9.
Scheme 1. Synthetic Route to Quinolinonyl DKAs 11a–g,i and 12a–g,i.

Reagents and conditions: (i) triethyl orthoformate, ethyl acetoacetate, Dowtherm A, 95–254 °C, 8 h, 41% yield; (ii) 4-fluorobenzyl bromide, K2CO3, DMF, 100 °C, 1 h, 29% yield; (iii) proper base, Et3N, DMF, microwave, 100 W, 153 °C, 10 min, 14–78% yield; (iv) N,N-dimethylamine aq soln 40%, 100 W, 175 °C, 10 min, 52% yield; (v) CCl4, CH2Cl2, PPh3, reflux, 24 h, 48% yield; (vi) diethyl oxalate, C2H5ONa, THF, room temp, 2 h, 76–99% yield; (vii) 1 N NaOH, THF/CH3OH, room temp, 40 min, 30–100% yield.
Derivatives 10a–f and 10h were obtained in few minutes with good yields by substituting the fluorine atom of 9 with the appropriate base in the presence of NEt3 under microwave irradiation. Using the same microwave assisted technique, we obtained 10g from 9 using N,N-dimethylamine (aq soln 40%) as base. 10h was converted to the desired chlorinated derivative 10i by reaction with PPh3 in CCl4 and CH2Cl2 using a modified procedure of a patent of Otsuka Pharmaceutical.21 Derivatives 10a–f and 10i were then condensed with ethyl oxalate, and the resulting esters 11a–g,i were hydrolyzed to afford the required acids 12a–g,i (Scheme 1). Chemical, physical, and analytical data of intermediates 8, 9, and 10a–i, together with the spectroscopic data, are reported in Supporting Information. Data for final products 11a–g,i and 12a–g,i are listed in the Experimental Section.
Evaluation of Biological Activities
All newly synthesized compounds 11a–g,i and 12a–g,i were tested in vitro for their ability to inhibit RNase H activity of recombinant RT, and on recombinant IN to evaluate their potencies against its 3′-processing and strand transfer functions. IN activities were determined using gel-based assays and the corresponding IC50 values were generated from the dose–response curve and are summarized in Table 1.
Table 1. Cytotoxicity, Antiviral, Anti-IN, and Anti-RNase H Activities of Compounds 11a–g,i and 12a–g,i.
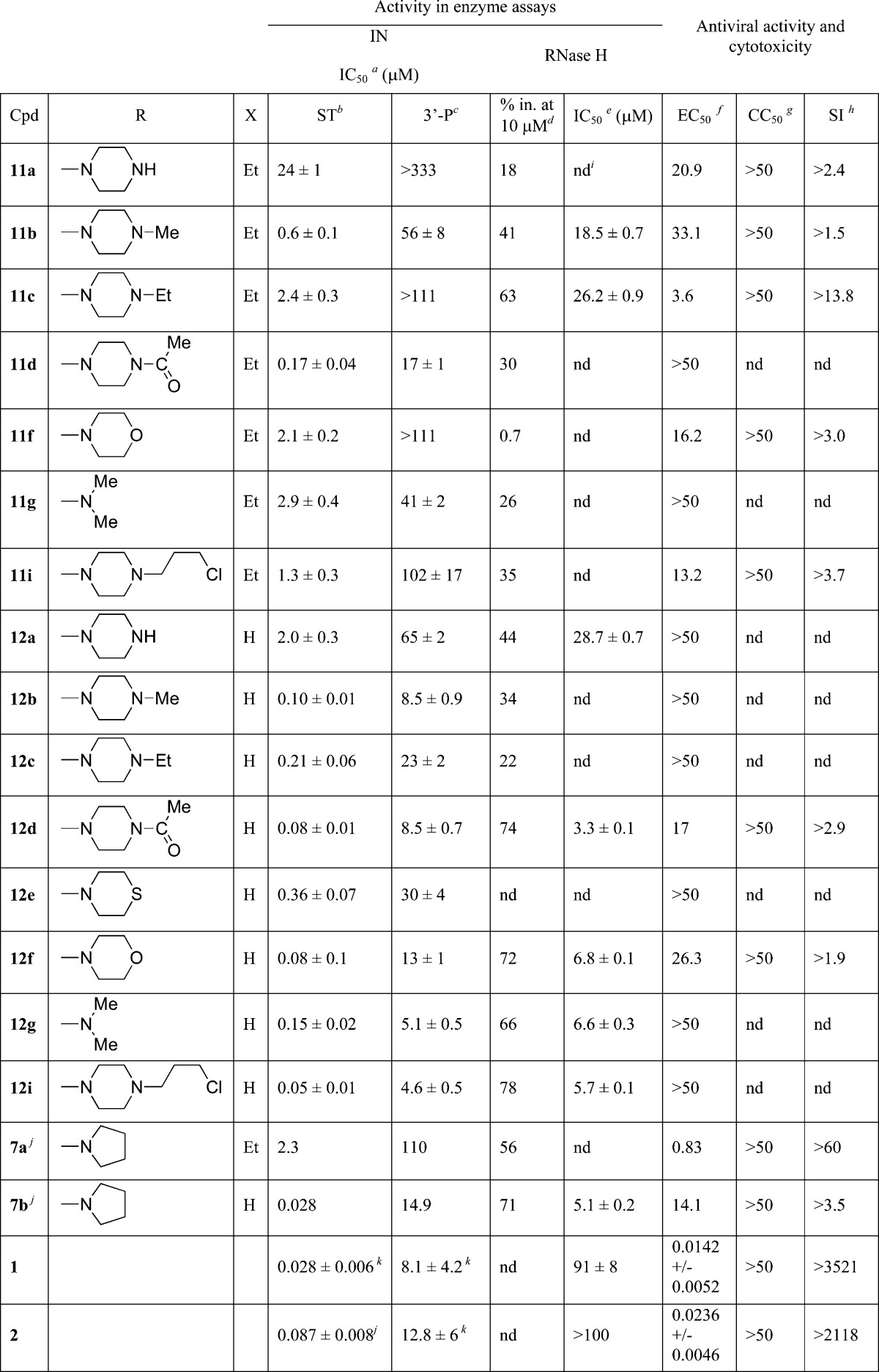
Inhibitory concentration 50% (μM) determined against rIN from gel-based assays and expressed as mean ± SD from at least three independent experiments.
ST: strand transfer.
3′-P: 3′-processing.
Percentage of inhibition of RT-associated RNase H activity at fixed concentration of inhibitor 10 μM (%).
Inhibitory concentration 50% against HIV-1 RT-associated RNase H activity determined from dose response curves (μM).
Effective concentration 50% in HIV-1 infected HeLa cells (μM).
Cytotoxic concentration 50% in HeLa cells (μM).
Selectivity index (CC50/EC50).
nd: not determined.
Reference (19b).
Reference (22).
Anti-IN Activities
The newly synthesized DKAs inhibited IN with nano- to submicromolar potencies for strand transfer and IC50 values ranging from 2.5 to of 333 μM for 3′-processing. All compounds exhibited some selectivity for strand transfer versus 3′-processing, with half of the compounds showing a selectivity of approximately 2 orders of magnitude or greater (Table 1). Carboxylic acid-containing compounds were always more potent than their corresponding ester counterparts.
Three compounds (12d,f,i) inhibited strand transfer with IC50 values below 100 nM with a 100-fold selectivity over the 3′-processing step. Compounds 12d,f showed an antiviral EC50 of 17 and 26.3 μM, respectively. Compound 12i was unable to block replication, possibly indicating reduced uptake. The least active acid-containing compound was 12a (IC50 2.0 μM), in which the pyrrolidine of the lead was replaced by a piperazine moiety. This compound was the only one containing an unsubstituted heteroatom on the 7-substituent. In fact, when the quinolinone ring was substituted with piperazine bioisosters, like morpholine (12f) and thiomorpholine (12e), inhibition of strand transfer activity increased of 1–2 orders of magnitude (IC50 0.08 and 0.36 μM, respectively). The same trend was observed when the piperazine hydrogen was replaced by alkyl or acyl chain. Among the 4-substituted piperazine derivatives, increased potency was achieved when the N-4 of the piperazine ring was substituted with acetyl (12d) or 3-chloropropyl (12i) chains (IC50 0.08 and 0.05 μM, respectively). Substitution with methyl (12b) or ethyl (12c) chains lead to submicromolar activity (IC50 0.10 and 0.21 μM, respectively), indicating that these modifications were not optimal for strand transfer inhibition. Cumulatively, these results demonstrate that quinolinone derivatives with piperazine bioisoster substitutions are potent INSTIs with antiviral activity.
Inhibition of HIV-1 RNase H Activity
The newly synthesized quinolinone DKA also inhibited the RNase H function of HIV-1 RT. RNase H inhibition was first calculated as percentage of inhibition at a single concentration of 10 μM, then for the most potent compounds the IC50 values were determined (Table 1). Interestingly, 4-methylpiperazine (11b) and 4-ethylpiperazine (11c) ester derivatives were more active than the corresponding acids (12b and 12c, respectively) when tested against RNase H (41% and 63% inhibition (not shown) and IC50 18.5 and 26.2 μM, respectively, versus 34% and 22% of the corresponding acids, Table 1). Five derivatives, including the lead compound 7b, inhibited RNase H at micromolar concentration showing IC50s in the range 3.3–6.8 μM: the 4-acetyl-1-piperazine (12d), the morpholine (12f), the dimethylamine (12g), and the 4-(3-chloropropyl)-1-piperazine (12i) acid derivatives. As representative examples, compounds 7b, 12f, and 12i were also tested against the polymerase function of RT and were found inactive up to 40 μM, suggesting that the observed inhibition is RNase H-mediated and not an indirect inhibition of the polymerase function of RT. RAL (2), EVG (1), and DTG (3, data not shown) were also tested for their ability to inhibit RNase H and found to be inactive, demonstrating that not all potent INSTIs can efficiently inhibit this enzyme in vitro (Table 1). These results demonstrate that in general the best INSTIs (7b and 12d,f,i) were also the most potent compounds to inhibit RNase H (Table 1).
Cell-Based Assays
All compounds including the previously reported 7a, 7b, RAL, and EVG were tested for antiviral activity and cytotoxicity using HeLa-CD4-LTR-β-gal cells infected by HIV-1(IIIB). EC50 and CC50 values for compounds 11a–g,i and 12a–g,i are reported in Table 1. In this assay, 7b exhibited an EC50 value of 14 μM, which is sensibly higher than the originally reported EC50 value of 0.17 μM in an HTLV-IIIB-based infection assay.19b Among the tested compounds, derivatives 11a–c,f,g,i and 12d,f,g show EC50 < 50 μM. In particular, 11c and 11g are active in the micromolar range (EC50 = 3.6 and 2.5 μM, respectively), with selectivity indices (SI) of 14 and 20, respectively. All active compounds were characterized by low cytotoxicity against the same HeLa-CD4-LTR-β-gal cells, showing CC50 values >50 μM.
Molecular Modeling
To better rationalize the structure–activity relationship (SAR) data obtained for this new series of DKAs, molecular modeling studies were performed. Given the inhibitory activity of these compounds, docking studies were undertaken to detect the structural features responsible for the binding at the active sites of both the aforementioned enzymes.
Several X-ray structures of HIV-1 IN CCD have been reported,23−38 but unfortunately none of them were cocrystallized with nucleic acids. On the other hand, a number of crystal structures of the intasome (comprising an IN tetramer assembled on a pair of viral DNA ends) from the prototype foamy virus (PFV) have recently been resolved.39−42 These pioneering studies have demonstrated that two Mg2+ ions are chelated by residues of the DDE motif (D64, D116, and E152 in HIV-1) inside the active site. Furthermore, both cations seem to be required for both IN catalytic activity and INSTI inhibition. Additionally, it has been proven that an induced-fit mechanism of inhibition for INSTIs occurs through displacement of the terminal 3′-adenosine of the viral DNA from the active site. Given these recent advances, we decided to revise our previous theoretical model37 by constructing a new homology model of the HIV IN CCD/viral DNA complex, in its INSTI-inhibited form, starting from the structure of full-length IN from PFV in complex with raltegravir (PDB code 3OYA).41 This model was constructed using Prime v3.0 (Schrodinger)43 based on the sequence alignment suggested by Tang and co-workers.44 This alignment identified conserved regions and gave the best overlap between the catalytic DDE motives of the PFV and HIV enzymes. Moreover, original nucleotides were mutated in order to obtain the HIV-1 retroviral DNA sequence. The model was then subjected to the Prime refinement protocol and finally used for docking studies. This new theoretical model was first used to perform docking simulations of raltegravir using Glide v. 5.7 (Schrödinger).45 Interestingly, this inhibitor adopts a binding mode similar to that shown in the parent crystal structure with its chelating oxygens oriented toward both active-site Mg2+ cations and its halobenzyl group fitting into the narrow pocket created by displacement of the terminal adenosine on the 3′-end of the viral DNA. These results prompted the docking calculations on our reference compound 7b, which is an efficient dual INSTI and RNase H inhibitor (Table 1). In the lowest energy conformation (GlideScore = −9.24), this compound occupies the DNA/IN interface with the DKA moiety chelating both the Mg+ ions (Figure 3a), similarly to the binding mode adopted by raltegravir (Figure 3b).
Figure 3.
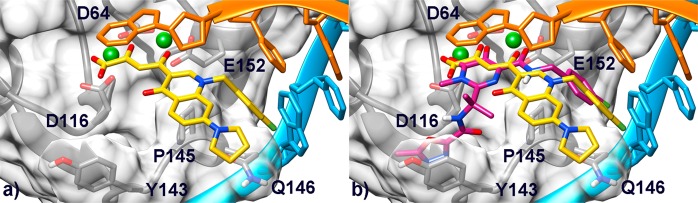
(a) Binding mode of compound 7b (yellow) within the HIV-1 IN/DNA model. The CCD is depicted as transparent light-gray surface and ribbons. Amino acid side chains involved in ligand binding are represented as sticks. The noncleaved (cyan) and processed (orange) viral DNA strands are shown as ribbon and sticks. Mg2+ metal ions are represented as green spheres. (b) Overlay of compound 7b and raltegravir (magenta) in the IN binding pocket.
Interestingly, the predicted conformation of the DKA moiety strongly resembles that of a close analogue crystallized in its unbound conformation,46 suggesting that for these set of compounds, the unbound conformation would also account for the bioactive one. Moreover, chelation by the carboxylate group should explain the higher inhibitory activity displayed by acidic compounds compared to the corresponding esters despite sharing the same predicted binding position in the HIV-1 IN binding site (compare docking pose of 7b in Figure 3a with that of 7a in Figure S1a in the Supporting Information).
In addition, the quinolinone ring is involved in a parallel-displaced π–π interaction with the terminal adenosine of the reactive DNA strand, further stabilizing the ligand in the IN active site. Moreover, the p-F-benzyl group is embedded in a hydrophobic cleft created by residues P145 and Q146, forming a well-oriented π-stacking interaction with the penultimate cytidine residue of the processed DNA strand. This interaction pattern (chelation of the Mg2+ cations and insertion in the DNA/enzyme cleft) is also established by raltegravir in the PFV IN catalytic site39,41 and in our HIV-1 IN model. Nevertheless, it is worth mentioning that raltegravir also places its terminal oxadiazole ring in a cleft, which cannot be explored by our current set of derivatives. In contrast, our INSTIs feature a basic substituent in position 7 of the quinolinone ring (−R substituent, Table 1), which points outward the catalytic site, as further demonstrated by docking of compound 12i (Figure S1b in the Supporting Information), which is the most potent ST inhibitor among the newly synthesized derivatives.
To explain the strand transfer-selective inhibiting properties of our compounds, the predicted INSTI/HIV-1 IN CCD/viral DNA complex was superimposed with the crystal structure of the precatalytic target capture complex (TCC) from PFV IN (PDB code 3OS1, Figure 4).40
Figure 4.
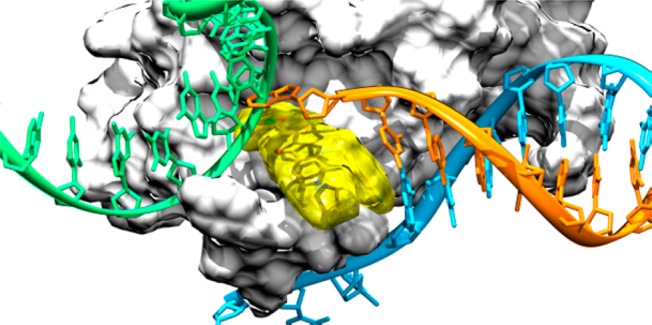
Superimposition between our model of the INSTI/HIV-1 IN CCD/viral DNA ternary complex and the precatalytic TCC from PFV (PDB code 3OS1). IN enzymes are represented as light-gray surfaces. The host DNA (green) and the nonprocessed (cyan) and processed (orange) viral DNA strands are shown as ribbon and sticks. Compound 7b is depicted as yellow transparent surface and sticks.
This superimposition suggests that our ligands, similarly to raltegravir, would sterically hamper host DNA binding by complexing the Mg2+ cofactors and displacing the terminal 3′-adenosine of the viral DNA, thus accounting for the selective strand transfer integration step inhibition. On the other hand, the presence of the viral DNA in the inhibited complex might also explain why the 3′-processing step is not efficiently inhibited by the presented ligands.
To explain at molecular level the reasons for the RNase H inhibition, this enzyme was also considered in our theoretical studies. Several crystal structures of the full-length wild-type RT or of its isolated RNase H CCD from HIV-1 in complex with RNase H inhibitors have been reported.47−50 A superposition of all of these structures on Cα atoms shows that folding, loops shape, and the side chain conformations of the majority of residues within the catalytic site are highly conserved (Figure S2 in the Supporting Information). On the basis of such an investigation, we selected the high resolution (2.09 Å) crystal structure of the full-length RT structure in complex with a RNase H pyrimidinol carboxylic acid inhibitor and nevirapine (PDB code 3QIP)49 to carry out docking studies with Glide. The latter structure is made up by the RNase H domain in complex with an inhibitor, which is structurally related to our new series of DKAs. Because this structure is cocrystallized with Mn2+ ions, we replaced them with the most physiologically relevant Mg2+ cations, also used in the biochemical assays. Docking of compound 7b within the RNase H active site shows that, as happened for IN, also in this case our reference compound places its DKA branch so as to chelate the two catalytic Mg2+ cations (Figure 5a). Remarkably, the DKA moiety again adopts an orientation similar to the unbound conformation of a closed analogue.46 In addition to the chelation of the metal ions, the DKA branch H-bonds with the side chain of N474. Furthermore, compound 7b establishes favorable lipophilic interactions with the Cβ and Cγ carbons of the Q475 through its quinolonyl nucleus. Additionally, the p-F-benzyl group forms favorable contacts with the lipophilic residues Q500 and W535.
Figure 5.
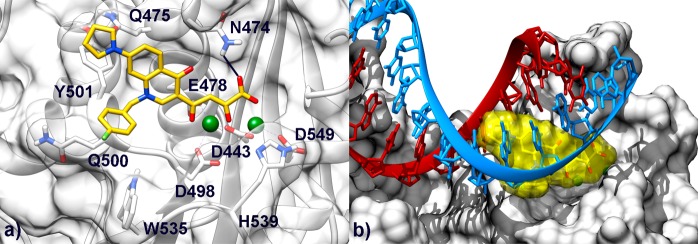
(a) Binding mode of compound 7b (yellow) in the HIV-1 RNase H active site. The active site is shown as transparent white surface and ribbons. Amino acid side chains important for ligand binding are represented as sticks. Mg2+ metal ions are depicted as green spheres. (b) Superimposition between our model of the INSTI/RNase H complex and the crystal structure of the full-length HIV-1 RT (p66 subunit) in complex with an RNA:DNA hybrid (PDB code 4B3O). RNase H residues are represented as white surfaces. The DNA (red) and RNA (light blue) strands are shown as ribbon and sticks. Compound 7b is depicted as yellow transparent surface and sticks.
Finally, the pyrrolidine ring is able to take additional contacts with the Q475 and Y501 residues side chains. Docking of compound 7a (Figure S3a in the Supporting Information) shows that, as observed for IN, ester and acidic derivatives feature analogous predicted binding modes. Thus, also in the case of RNase H, the generally higher potency displayed by the acidic compounds might be ascribed to a more tight coordination of the metal ions by the DKA branch. Finally, also the newly synthesized derivative 12i, which displays interesting RNase H inhibitory properties, is predicted to bind similarly to 7b (Figure S3b in the Supporting Information), thus suggesting that our INSTIs would all share the same binding position to the RNase H catalytic site.
To better explain the RNase H inhibitory activity of our compounds, we superimposed the predicted 7b/RNase H complex with the recently solved crystal structure of the full length HIV-1 RT in complex with a RNA:DNA hybrid51 (PDB code 4B3O) (Figure 5b). This superimposition suggests that binding of compound 7b would sterically hamper the hosting of the RNA:DNA hybrid at the RNase H catalytic site, thus preventing its hydrolysis.
Finally, to provide new hints for future lead optimization studies, we compared the binding pose of our reference compound 7b with that of two more potent inhibitors, namely a naphthyridinone ligand (IC50 ≈ 0.11 μM)48 and a pyrimidinol carboxylic acid (IC50 = 0.23 ± 0.01 μM)49 that were cocrystallized with the HIV-1 RT enzyme at the RNase H active site (PDB codes 3LP0(48) and 3QIP,49 respectively). From this comparison, it turned out that, similarly to 7b, these inhibitors chelate both the two Mg2+ cations, although adopting a slightly different coordination geometry (Figure S4 in the Supporting Information). On the other hand, due to size and structural differences, the considered compounds and our inhibitors establish different interaction patterns at the RNase H active site. In particular, the pyrimidinol carboxylic acid derivative occupies through its 2-phenyl indole substituent a lipophilic pocket shaped by W535, P537, and A538 residues. This additional cleft might be thus explored by our next-generation DKAs to achieve an enhanced RNase H inhibitory profile to obtain dual IN/RNase H inhibitors.
Conclusions
We have designed, synthesized, and evaluated a series of novel quinolinonyl diketo acid derivatives endowed with a “base-like” moiety for their inhibition of HIV-1 IN. The compounds were also tested against the RNase H function of the RT. We identified nanomolar INSTIs that also exhibited low micromolar RNase H inhibition and antiviral properties. Our molecular modeling study also provided new insights for the future development of dual inhibitors of HIV-1 IN and RNase H.
Experimental Section
Chemistry. General
Melting points were determined with a Büchi 530 capillary apparatus and are uncorrected. Compounds purity were always >95% determined by high pressure liquid chromatography (HPLC). HPLC analysis were carried out with a Shimadzu LC-10AD VP CTO-10AC VP. Column used was generally Discovery Bio Wide Pore C18 (10 cm × 4.6 mm, 3 μm). Infrared (IR) spectra were recorded on a PerkinElmer Spectrum-One spectrophotometer. 1H NMR spectra were recorded on a Bruker AC 400 spectrometer. Merck silica gel 60 F254 plates were used for analytical TLC. Developed plates were visualized by UV light. Column chromatography was performed on silica gel (Merck; 70–230 mesh). Concentration of solution after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of approximately 20 Torr. Analytical results agreed to within ±0.40% of the theoretical values. Dimethyl sulfoxide-d6 99.9% (code 44,139-2) and deuterochloroform 98.8% (code 41,675-4) of isotopic purity (Aldrich) were used. Solvents were reagent grade and, when necessary, were purified and dried by standard methods. Organic solutions were dried over anhydrous sodium sulfate (Merck).
Microwave Irradiation Experiments
Microwave reactions were conducted using a CEM Discover system unit (CEM. Corp., Matthews, NC). The machine consists of a continuous focused microwave-power delivery system with operator selectable power output from 0 to 300 W. The temperature of the contents of the vessel was monitored using a calibrated infrared temperature control mounted under the reaction vessel. All experiments were performed using a stirring option whereby the contents of the vessel are stirred by means of a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the vessel.
3-Acetyl-7-fluoroquinolin-4(1H)-one (8)
Triethyl orthoformate (4.0 g, 27 mmol), ethyl acetoacetate (3.5 g, 27 mmol), 3-fluoroaniline (3.0 g, 27 mmol), and Dowtherm A (5.6 mL) were charged in a three-necked flask equipped with a condenser and a Dean-Stark apparatus. This mixture was stirred under argon atmosphere while the temperature was increased to 95 °C in 1 h then gradually to 162 °C in a further hour. Then the mixture was stirred at this temperature for 6 h. After this time, the resulting solution was added in portions during 3 h into 42 mL of Dowtherm A stirred in a three-necked flask equipped with thermometer and a Dean-Stark apparatus and heated at 253–254 °C. After addition, the mixture was heated at the same temperature for 2 h. Then the mixture was cooled at 90 °C, treated with 2-propanol (10 mL), cooled at 30 °C, filtered, and washed with 2-propanol and light petroleum ether to give pure derivatives 8 (41% yield). Chemical and physical data of derivative 8 are reported in the Supporting Information. For spectroscopic data see ref (19b).
3-Acetyl-7-fluoro-1-[(4-fluorophenyl)methyl]quinolin-4(1H)-one (9)
A mixture of derivative 8 (0.23 g, 1.1 mmol), 4-fluorophenylmethyl bromide (610 mg, 3.3 mmol), and anhydrous K2CO3 (210 mg, 1.5 mmol) in dry DMF (10 mL) was stirred at 100 °C for 1 h. After cooling, water was added (40 mL) and the precipitate that formed was filtered, washed with water and light petroleum ether in turn, and then dried under IR lamp to provide pure product 9 (29% yield). Chemical and physical data of derivatives 9 are reported in the Supporting Information. For spectroscopic data, see ref (19b).
General Procedure for the Synthesis of 7-(Base)-4(1H)-quinolinones (10a–f,h)
A mixture of 9 (2.7 g, 8.5 mmol), proper amine (25.6 mmol), and TEA (0.8 g, 7.7 mmol) in dry DMF (40 mL) was irradiated with microwave at 153 °C for 10 min (applied potency 100 W) in an open vessel equipped with a condenser. After cooling, the reaction mixture was diluted with water (100 mL) and treated with 1 N HCl until pH 7. The solid that formed was collected by filtration. Purification of crude product was performed by column chromatography on silica gel (10:1 chloroform/ethanol as eluent) to give of pure 10a–f,h (14–78% yield). For spectroscopic, chemical, and physical data of derivatives 10a–f,h, see the Supporting Information.
3-Acetyl-7-(dimethylamino)-1-[(4-fluorophenyl)methyl]quinolin-4(1H)-one (10g)
A mixture of dimethylamine in H2O (40%, 1.3 mmol) and quinolinone 9 (0.45 mmol) was irradiated with microwave at 175 °C, 100 W, for 10 min. After cooling, the reaction mixture was diluted with water. The formed precipitate was collected by filtration thorugh a sintered glass septum and washed with water and diethyl ether. Purification of crude product was performed by column chromatography on silica gel (chloroform/ethanol 5:1 as eluent) to give pure product 10g (52% yield). For spectroscopic, chemical, and physical data of derivatives 10g, see the Supporting Information.
3-Acetyl-7-[4-(3-chloropropyl)piperazin-1-yl]-1-[(4-fluorophenyl)methyl]quinolin-4(1H)-one (10i)
A suspension of 10h (500 mg, 1.14 mmol) in 3 mL of CCl4 and 3 mL of CH2Cl2 was added triphenylphosphine (300 mg, 1.14 mmol). The mixture was heated at reflux for 24 h. The reaction was monitored with TLC (10:1 chloroform/methanol as eluent). After this time, the reaction was cooled at 25 °C, and the precipitate was filtered. The recovered organic solution was evaporated to afford 400 mg of crude product. Purification of crude product was performed by column chromatography on silica gel (10:1 chloroform/methanol as eluent) to give of pure 10i (48% yield). For spectroscopic, chemical, and physical data of derivatives 10i, see Supporting Information.
General Procedure for the Synthesis of Diketo Esters 11a–g,i
Freshly prepared sodium ethoxide (390 mg, 5.5 mmol) was added into a well stirred mixture of the appropriate acetyl derivative 10a–g,i (2.7 mmol) and diethyl oxalate (790 mg, 5.4 mmol) in anhydrous THF (2.7 mL) under nitrogen atmosphere. The mixture was stirred at room temperature for 2 h, and then was poured into n-hexane (50 mL). The collected precipitate was vigorously stirred for 30 min in 1 N HCl (50 mL). The yellow solid that formed was filtered, washed with water, and dried under IR lamp to afford the pure diketo esters 11a–g,i. Yield (%), melting point (°C), recrystallization solvent, IR, and 1H NMR are reported for each of the following compounds.
4-[1-[(4-Fluorophenyl)methyl]-7-(piperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11a)
Yield 98%; 150–152 °C; washed with 2-propanol; IR ν 3347 (OH enol), 1711 (C=O ester), 1666 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 1.31 (t, 3H, CH3CH2), 3.45–3.55 (m, 4H, piperazine H), 3.25–3.35 (m, 4H, piperazine H), 4.31 (q, 4H, CH2CH3), 5.71 (s, 2H, CH2 benzyl), 6.86 (s, 1H, quinolinone C8-H), 7.22–7.31 (m, 3H, benzene H and quinolinone C6-H), 7.43–7.52 (m, 2H, benzene H), 8.00 (s, 1H, CH enol), 8.10 (d, 1H, quinolinone C5-H), 9.00 (s, 1H, quinolinone C2-H). Anal. (C26H26FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-methylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11b)
Yield 99%; 96–99 °C; washed with 2-propanol; IR ν 3381 (OH enol), 1713 (C=O ester), 1605 (ketone) cm–1. 1H NMR (DMSO-d6) δ 1.02 (s, 3H, CH3 piperazine), 1.22 (t, 3H, CH3CH2), 2.25–2.53 (m, 4H, piperazine H), 3.23–3.31 (m, 4H, piperazine H), 4.10 (q, 4H, CH2CH3), 5.54 (s, 2H, CH2 benzyl), 6.72 (s, 1H, quinolinone C8-H), 7.05 (d, 1H, quinolinone C6-H), 7.11–7.20 (m, 2H, benzene H), 7.23–7.31 (m, 2H, benzene H), 8.07 (d, 1H, quinolinone C5-H), 8.55 (s, 1H, CH enol), 8.61 (s, 1H, quinolinone C2-H). Anal. (C27H28FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-ethylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11c)
Yield 99%; 200 °C (dec); washed with 2-propanol; IR ν 3388 (OH enol), 1711 (C=O ester), 1606 (ketone) cm–1. 1H NMR (DMSO-d6) δ 1.17 (t, 3H, J = 5.5 Hz, CH3CH2 piperazine), 1.21 (t, 3H, CH3CH2), 2.19 (q, 3H, J = 5.5 Hz, CH2CH3 piperazine), 2.30–2.40 (m, 4H, piperazine H), 3.15–3.30 (m, 4H, piperazine H), 4.11 (q, 4H, CH2CH3), 5.55 (s, 2H, CH2 benzyl), 6.72 (s, 1H, quinolinone C8-H), 7.01–7.07 (m, 2H, CH enol and quinolinone C6-H), 7.18–7.20 (m, 2H, benzene H), 7.22–7.30 (m, 2H, benzene H), 8.07 (d, 1H, quinolinone C5-H), 8.54 (s, 1H, quinolinone C2-H). Anal. (C28H30FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-acetylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11d)
Yield 91%; 178–180 °C; washed with 2-propanol; IR ν 3409 (OH enol), 1716 (C=O ester), 1604 (ketone) cm–1. 1H NMR (DMSO-d6) δ 1.24 (t, 3H, CH3CH2), 2.03 (s, 3H, CH3 acetylpiperazine), 3.16–3.35 (m, 4H, piperazine H), 3.45–3.60 (m, 4H, piperazine H), 4.11 (q, 4H, CH2CH3), 5.56 (s, 2H, CH2 benzyl), 6.75 (s, 1H, quinolinone C8-H), 7.01 (s, 1H, CH enol), 7.05 (d, 1H, quinolinone C6-H), 7.18–7.22 (m, 2H, benzene H), 7.28–7.32 (m, 2H, benzene H), 8.09 (d, 1H, quinolinone C5-H), 8.53 (s, 1H, quinolinone C2-H). Anal. (C28H28FN3O6) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(thiomorpholin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11e)
Yield 99%; 170 °C; washed with 2-propanol; IR ν 3388 (OH enol), 1711 (C=O ester), 1606 (ketone) cm–1. 1H NMR (DMSO-d6) δ 1.21 (t, 3H, CH3CH2), 3.16–3.30 (m, 4H, thiomorpholine H), 3.65–3.82 (m, 4H, thiomorpholine H), 4.11 (q, 4H, CH2CH3), 5.56 (s, 2H, CH2 benzyl), 6.77 (s, 1H, quinolinone C8-H), 7.08–7.37 (m, 6H, CH enol, quinolinone C6-H and benzene H), 8.10 (d, 1H, quinolinone C5-H), 8.60 (s, 1H, quinolinone C2-H). Anal. (C26H25FN2O5S) C, H, F, N, S.
4-[1-[(4-Fluorophenyl)methyl]-7-(morpholin-4-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11f)
Yield 76%; 175–177 °C; trituration on diethyl ether; IR ν 3388 (OH enol), 1711 (C=O ester), 1606 (ketone) cm–1. 1H NMR (DMSO-d6) δ 1.21 (t, 3H, CH3CH2), 3.14–3.32 (m, 4H, morpholine H), 3.62–3.80 (m, 4H, morpholine H), 4.11 (q, 4H, CH2CH3), 5.56 (s, 2H, CH2 benzyl), 6.77 (s, 1H, quinolinone C8-H), 7.08–7.37 (m, 6H, CH enol, quinolinone C6-H and benzene H), 8.10 (d, 1H, quinolinone, C5-H), 8.60 (s, 1H, quinolinone C2-H). Anal. (C26H25FN2O6) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(dimethylamino)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11g)
Yield 98%; 150 °C (dec); washed with 2-propanol; IR. ν 3345 (OH enol), 1710 (C=O ester), 1636 (ketone) cm–1. 1H NMR (DMF-d7) δ 1.30 (t, 3H, CH3CH2), 3.00 (s, 6H, CH3 dimethylamine), 4.19 (q, 4H, CH2CH3), 5.65 (s, 2H, CH2 benzyl), 6.52 (s, 1H, quinolinone C8-H), 6.88 (s, 1H, quinolinone C6-H), 7.18 (s, 1H, CH enol), 7.21–7.25 (m, 2H, benzene H), 7.45–7.48 (m, 2H, benzene H), 8.19 (d, 1H, quinolinone C5-H), 8.70 (s, 1H, quinolinone C2-H). Anal. (C24H23FN2O5) C, H, F, N.
4-[1-(4-[(4-Fluorophenyl)methyl]-7-[N-(3-chloroprop-1-yl)-piperazin-1-yl]-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid Ethyl Ester (11i)
77%; 175–177 °C; washed with acetone; IR ν 3488 (OH enol), 1723 (C=O ester), 1617 (ketone) cm–1. 1H NMR (DMF-d7) δ 1.37 (t, 3H, CH3CH2), 1.98 (q, 2H, J = 5.5 Hz, ClCH2CH2CH2N piperazine), 2.48–2.54 (m, 6H, piperazine H and ClCH2CH2CH2N piperazine), 3.38–3.39 (m, 4H, piperazine H), 3.73–3.76 (q, 4H, CH2CH3), 4.35–4.39 (q, 2H, ClCH2CH2CH2N piperazine), 5.87 (s, 2H, CH2 benzyl), 6.98 (s, 1H, quinolinone C8-H), 7.22–7.29 (m, 3H, quinolinone C6-H and benzene H), 7.58–7.62 (m, 2H, benzene H), 8.10 (m, 2H, CH enol and quinolinone C5-H), 9.14 (s, 1H, quinolinone C2-H). Anal. (C29H31ClFN3O5) C, H, Cl, F, N.
General Procedure for the Synthesis of Diketo Acids 12a–g,i
A mixture of 1 N NaOH (6.5 mL) and the appropriate ester 11a–g,i (1.3 mmol) in 1:1 THF/methanol (12 mL) was stirred at room temperature for 40 min and then poured onto crushed ice. The aqueous layer was separated and treated with 1 N HCl until pH 3 was reached, and the yellow solid that formed was collected by filtration, then washed with water, hot dry ethanol, and light petroleum ether to afford pure acids 12a–g,i. Yield (%), melting point (°C), recrystallization solvent, IR, 1H NMR are reported for each of the following compounds.
4-[1-[(4-Fluorophenyl)methyl]-7-(piperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12a)
Yield 98%; 224 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1700 (C=O acid), 1649 (C=O keton) cm–1. 1H NMR (DMSO-d6) δ 3.30–3.37 (m, 4H, piperazine H), 3.41–3.50 (m, 4H, piperazine H), 5.64 (s, 2H, CH2 benzyl), 6.82 (s, 1H, quinolinone C8-H), 7.12–7.20 (m, 3H, benzene H and quinolinone C6-H), 7.37–7.41 (m, 2H, benzene H), 8.08 (d, 1H, quinolinone C5-H), 8.42 (s, 1H, CH enol), 8.75 (s, 1H, C2-H quinolinone), 15.00 (br s, 2H, OH). Anal. (C24H22FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-methylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12b)
Yield 99%; 224 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1700 (C=O acid), 1599 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 2.38 (s, 3H, CH3 piperazine), 2.61–2.75 (m, 4H, piperazine H), 3.21–3.35 (m, 4H, piperazine H), 5.67 (s, 2H, CH2 benzyl), 6.81 (s, 1H, quinolinone C8-H), 7.29–7.08 (m, 3H, benzene H, quinolinone C6-H), 7.31–7.48 (m, 2H, benzene H); 7.69 (br s, 1H, CH enol), 8.07 (d, 1H, quinolinone C5-H), 8.93 (s, 1H, C2-H quinolinone), 15.00 (br s, 2H, OH). Anal. (C25H24FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-ethylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12c)
Yield 99%; > 240 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1700 (C=O acid), 1578 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 1.06 (t, 3H, J = 5.5 Hz, CH3CH2 piperazine), 2.22 (q, 3H, J = 5.5 Hz, CH2CH3 piperazine), 2.28–2.50 (m, 4H, piperazine H), 2.70–3.85 (m, 4H, piperazine H), 5.65 (s, 2H, CH2 benzyl), 6.78 (s, 1H, quinolinone C8-H), 7.12 (s, 1H, quinolinone C6-H), 7.10–7.20 (m, 2H, benzene H), 7.37 (m, 2H, benzene H), 7.69 (br s, 1H, CH enol), 8.06 (d, 1H, quinolinone C5-H), 8.90 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C26H26FN3O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(N-acetylpiperazin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12d)
Yield 90%; 195–197 °C; washed with acetonitrile; IR ν 3392–2000 (OH), 1716 (C=O acid), 1599 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 2.05 (s, 3H, CH3 acetylpiperazine), 3.25–3.42 (m, 4H, piperazine H), 3.50–3.69 (m, 4H, piperazine H), 5.71 (s, 2H, CH2 benzyl), 6.81 (s, 1H, quinolinone C8-H), 7.17–7.28 (m, 3H, benzene H and quinolinone C6-H), 7.30–7.50 (m, 2H, benzene H), 7.97 (br s, 1H, CH enol), 8.10 (d, 1H, quinolinone C5-H), 9.01 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C26H24FN3O6) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(thiomorpholin-1-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12e)
Yield 30%; 190 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1711 (C=O acid), 1606 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 2.43–2.60 (m, 4H, thiomorpholine H), 3.56–3.75 (m, 4H, thiomorpholine H), 5.58 (s, 2H, CH2 benzyl), 6.66 (s, 1H, quinolinone C8-H), 7.10–7.16 (m, 4H, CH enol, quinolinone C6-H and benzene H), 7.32–7.35 (m, 2H, benzene H), 7.22 (d, 1H, quinolinone C5-H), 8.78 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C24H21FN2O5S) C, H, F, N, S.
4-[1-[(4-Fluorophenyl)methyl]-7-(morpholin-4-yl)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12f)
Yield 99%; 187–189 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1726 (C=O acid), 1600 (C=O ketone) cm–1. 1H NMR (DMSO-d6) δ 3.17–3.34 (m, 4H, morpholine H), 3.61–3.80 (m, 4H, morpholine H), 5.70 (s, 2H, CH2 benzyl), 6.83 (s, 1H, quinolinone C8-H), 7.10–7.29 (m, 3H, quinolinone C6-H and benzene H), 7.32–7.48 (m, 2H, benzene H), 7.99 (br s, 1H, CH enol), 8.10 (d, 1H, quinolinone C5-H), 9.02 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C24H21FN2O6) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-(dimethylamino)-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12g)
Yield 98%; 203–205 °C; washed with 2-propanol; IR ν 3500–2000 (OH), 1735 (C=O acid), 1620 (C=O ketone) cm–1. 1H NMR (DMF-d7) δ 3.22 (s, 6H, CH3 dimethylamine), 6.00 (s, 2H, CH2 benzyl), 6.82 (s, 1H, quinolinone C8-H), 7.14 (s, 1H, quinolinone C6-H), 7.42–7.46 (m, 2H, benzene H), 7.70–7.85 (m, 2H, benzene H), 8.36 (d, 1H, quinolinone C5-H), 8.41 (s, 1H, CH enol), 9.27 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C22H19FN2O5) C, H, F, N.
4-[1-[(4-Fluorophenyl)methyl]-7-[N-(3-chloroprop-1-yl)piperazin-1-yl]-4-(1H)-quinolinon-3-yl]-2-hydroxy-4-oxo-2-butenoic Acid (12i)
Yield 100%; 175–177 °C; washed with THF; IR ν 3410–2000 (OH), 1717 (C=O ester), 1655 (ketone) cm–1. 1H NMR (DMF-d7) δ 2.18 (q, 2H, J = 6.0 Hz, ClCH2CH2CH2N piperazine), 2.73–2.79 (m, 6H, piperazine H and ClCH2CH2CH2N piperazine), 3.50–3.65 (m, 4H, piperazine H), 3.92 (q, 2H, J = 6.0 Hz, ClCH2CH2CH2N piperazine), 6.03 (s, 2H, CH2 benzyl), 7.16 (s, 1H, quinolinone C8-H), 7.38–7.45 (m, 3H, quinolinone C6-H and benzene H), 7.74–7.78 (m, 2H, benzene H), 8.37–8.39 (m, 2H, CH enol and quinolinone C5-H), 9.27 (s, 1H, quinolinone C2-H), 15.00 (br s, 2H, OH). Anal. (C27H27ClFN3O5) C, H, Cl, F, N.
Biological Methods
HIV-1 IN Inhibition
HIV-1 IN gel-based assays were carried out as previously published.52
HIV-1 RT RNase H Inhibition
IC50 values were determined as previously reported53 using an 18-nt 3′-fluorescein-labeled RNA annealed to a complementary 18-nt 5′-dabsyl-labeled DNA. To a 96-well plate was added 1 μL of each inhibitor (in DMSO), followed by 10 μL of the appropriate RT (15–80 ng/mL) in reaction buffer. Hydrolysis was initiated by adding 10 μL of RNA/DNA hybrid (2.5 μM). Final assay conditions were 50 mM Tris-HCl, pH 8.0, 60 mM KCl, 10 mM MgCl2, 1% DMSO, 150–800 ng of RT, 250 nM substrate, and increasing concentrations of inhibitor. Wells containing only DMSO were used as negative control. Plates were incubated at 37 °C in a Spectramax Gemini EM fluorescence spectrometer for 10 min, and fluorescence (λex = 485 nm; λem = 520 nm) was measured at 1 min intervals such that linear initial rates could be measured in the presence (vi) and absence (v0) of inhibitor. Percent inhibition was calculated as 100(v0 – vi)/v0, and plotted against log[I]. IC50 values were determined using Prism5 (GraphPad Software). All assays were performed in triplicate.
HIV-1 RT Polymerase Inhibition
HIV-1 RT polymerase assays were carried out as previously reported.13b
HIV-1 Replication Inhibition
Compounds antiviral activity was determined in a cell-based assay according to the procedure described previously54 and modified as follows. HeLa-CD4-LTR-β-gal cells were maintained in DMEM with 10% serum and 0.5 mg/mL G418. The day prior experimentation, 96-well plates were prepared to contain 10000 cells per well in 100 μL of DMEM medium complemented with 10% serum. On day one, each drug was serial diluted directly on cells following a 3-fold dilution over 6 points and each well was then filled to 200 μL with either fresh medium or concentrated viral supernatant (HIV-1(IIIB), Advanced Biotechnologies Inc.). The highest compound concentration tested was 50 μM. On day two, cells were washed three times with PBS before adding 200 μL of a solution containing 50 mM Tris-HCl pH 7.5, 100 mM β-mercaptoethanol, 0.05% Triton X100, and 5 mM 4-methyl-umbelliferyl-β-d-galactopyranoside (4-MUG, Sigma). On day three, sealed plates were read in a SpectraMax GEMINI-XS (Molecular Devices) with λex/em = 360/460 nm.
Cellular Toxicity
Similarly to the antiviral assays, plates were prepared with 10,000 HeLa-CD4-LTR-β-gal cells per well and a serial dilution of compounds in 100 μL. After 24 h of culture, 100 μL of ATPlite reagent (PerkinElmer) was added to each well. After 5 min at room temperature, plates’ luminescence was quantified using an EnVision multilabel reader (PerkinElmer) according to manufacturer’s instructions.
Homology Modeling
A novel homology model of HIV-1 IN CCD/viral DNA complex was built using Schrodinger Prime (version 3.0)43 software accessible through the Maestro interface.55 To construct the model, the crystal structure of full-length IN from PFV in complex with raltegravir41 (PDB code 3OYA) was used as template. The sequence alignment used for the homology modeling was based on the secondary structure alignment of the template and the HIV-1 IN CCD (PDB code 1QS4)26 on the Cα positions of the active-site residues Asp64, Asp116, and Glu152 (DDE motif) to identify the structure conserved regions according to the scheme suggested by Tang and co-workers.44 Prior to model building, bases of the 19-mer PFV DNA were mutated in order to obtain the HIV-1 retroviral DNA sequence (GenBank: AAC37875.1). The newly built model was the subjected to Prime refinement protocol in order to optimize the active site loop and amino acidic side chains conformation.
Ligands Setup
The 3D structures of all of the compounds were generated with the Maestro Build Panel55 and were then submitted to Polak–Ribiere conjugate gradient minimization (0.0005 kJ/(Å mol) convergence) using MacroModel (version 9.9).56 DKAs were modeled in their enolic tautomeric form because it has been clearly established that such compounds mainly exist in this form in solution and also given the influence of the two metal cations in the binding site.57
Proteins Setup
The HIV-1 CCD/DNA model and the RNase H crystal structure (PDB code 3QIP) were prepared for docking calculations using the “Protein Preparation Wizard” panel of Schrödinger 2010 molecular modeling package.55 In particular, using the “preprocess and analyze structure” tool, the bond orders and disulfide bonds were assigned, all the hydrogen atoms were added, and all the water molecules in a distance greater than 5 Å from any heterogroup were deleted; in addition, both Mn2+ ions in the RNase H crystal structure were replaced with Mg2+ cations. Using Epik 2.0, a prediction of the side chains heterogroups ionization and tautomeric states was performed.58 An optimization of the hydrogen-bonding network was performed using the “H-bond assignment” tool. Finally, using the “impref utility”, the positions of the hydrogen atoms were optimized by keeping all the heavy atoms in place.
Docking Calculations
Docking studies at both the HIV-1 IN and RNase H active sites were carried out with Glide v. 5.7 (Schrödinger).45 Glide is a grid-based ligand docking with energetics approach and searches for favorable interactions between ligands and receptors. The shape and properties of the receptor are represented on a grid by different sets of fields that provide progressively more accurate scoring of the ligand pose. These fields are generated as preprocessing steps in the calculation and hence need to be computed only once for each receptor. For the grid generation of both the receptors, a box centered on the Mg2+ cations was created. This box gives a more precise measure of the effective size of the search space. However, ligands can move outside this box during grid minimization. The Cartesian coordinates of the outer box, X, Y, and Z length were set to 25 Å. The conformational space of the ligand is defined by Glide by several lowest-energy poses that are subjected to a Monte Carlo procedure that examines nearby torsional minima. This procedure is needed in some cases to properly orient peripheral groups and occasionally alters internal torsion angles. The default value (1.00) for the van der Waals radii scaling factor was chosen, which means no scaling for the nonpolar atoms was performed (no flexibility was simulated for the receptor). In the present study, the standard precision (SP) mode of GlideScore function was used to score the obtained binding poses. The force field used for the docking was the OPLS-2005.59
All of the pictures were rendered with the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.60
Acknowledgments
We thank the Italian MIUR for financial support, ISS 40H4, PRIN 2010-2011 (2010W2KM5L_002). R. Di Santo and R. Costi thank the FP7 CHAARM project for support. This work was also supported by the NIH Intramural Research Program, Center for Cancer Research, National Cancer Institute, and by NIH grants from the AIDS Intramural Targeted Program (IATAP).
Glossary
Abbreviations Used
- HAART
highly active antiretroviral therapy
- IN
integrase
- RT
reverse transcriptase
- RNase H
ribonuclease H
- INSTI
strand transfer IN inhibitor
- DKA
diketo acid
- SI
selectivity index
- SAR
structure–activity relationship
- CCD
catalytic core domain
- PFV
prototype foamy virus
- TCC
target capture complex
- IR
infrared
Supporting Information Available
Analyses of compounds 8, 9, 10a–i, 11a–g,i, 12a–g,i and molecular modeling. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contribution of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dual Epidemic Threatens Africa; BBC News, November2, 2007; http://news.bbc.co.uk/2/hi/africa/7074298.stm.
- Chun T. W.; Fauci A. S. Latent reservoirs of HIV: obstacles to the eradication of virus. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10958–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeni P. Update on HAART in HIV. J. Hepatol. 2006, 44, S100–S103. [DOI] [PubMed] [Google Scholar]
- a Chesney M. Adherence to HAART regimens. AIDS Patient Care STDS 2003, 17, 169–177. [DOI] [PubMed] [Google Scholar]; b Conway B. The role of adherence to antiretroviral therapy in the management of HIV infection. JAIDS, J. Acquired Immune Defic. Syndr. 2007, 45, S14–S18. [DOI] [PubMed] [Google Scholar]
- a Negredo E.; Bonjoch A.; Clotet B. Benefits and concerns of simplification strategies in HIV-infected patients. J. Antimicrob. Chemother. 2006, 58, 235–242. [DOI] [PubMed] [Google Scholar]; b Barreiro P.; Garcia-Benayas T.; Soriano V.; Gallant J. Simplification of antiretroviral treatment—how to sustain success, reduce toxicity and ensure adherence avoiding PI use. AIDS Rev. 2002, 4, 233–241. [PubMed] [Google Scholar]
- a Wang Z.; Bennett E. M.; Wilson D. J.; Salomon C.; Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J. Med. Chem. 2007, 50, 3416–3419. [DOI] [PubMed] [Google Scholar]; b Wang Z.; Vince R. Synthesis of pyrimidine and quinolone conjugates as a scaffold for dual inhibitors of HIV reverse transcriptase and integrase. Bioorg. Med. Chem. Lett. 2008, 18, 1293–1296. [DOI] [PubMed] [Google Scholar]; c Wang Z.; Vince R. Design and synthesis of dual inhibitors of HIV reverse transcriptase and integrase: introducing a diketoacid functionality into delavirdine. Bioorg. Med. Chem. 2008, 16, 3587–3595. [DOI] [PubMed] [Google Scholar]
- a Makhija M. T. Designing HIV integrase inhibitors—shooting the last arrow. Curr. Med. Chem. 2006, 13, 2429–2441. [DOI] [PubMed] [Google Scholar]; b Lataillade M.; Kozal M. J. The hunt for HIV-1 integrase inhibitors. AIDS Patient Care STDS 2006, 20, 489–501. [DOI] [PubMed] [Google Scholar]; c Dayam R.; Deng J.; Neamati N. HIV-1 integrase inhibitors: 2003–2004 update. Med. Res. Rev. 2006, 26, 271–309. [DOI] [PubMed] [Google Scholar]; d Pommier Y.; Johnson A. A.; Marchand C. Integrase inhibitors to treat HIV/AIDS. Nature Rev. Drug Discovery 2005, 4, 236–248. [DOI] [PubMed] [Google Scholar]; e Johnson A. A.; Marchand C.; Pommier Y. HIV-1 integrase inhibitors: a decade of research and two drugs in clinical trial. Curr. Top. Med. Chem. 2004, 4, 1059–1077. [DOI] [PubMed] [Google Scholar]; f Maurin C.; Bailly F.; Cotelle P. Structure–activity relationships of HIV-1 integrase inhibitors—enzyme–ligand interactions. Curr. Med. Chem. 2003, 10, 1795–1810. [DOI] [PubMed] [Google Scholar]; g Cotelle P. Patented HIV-1 integrase inhibitors (1998–2005). Recent Pat. Antiinfect. Drug Discovery 2006, 1, 1–15. [DOI] [PubMed] [Google Scholar]
- a Grinsztejn B.; Nguyen B. Y.; Katlama C.; Gatell J. M.; Lazzarin A.; Vittecoq D.; Gonzalez C. J.; Chen J.; Harvey C. M.; Isaacs R. D. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 2007, 369, 1261–1269. [DOI] [PubMed] [Google Scholar]; b Markowitz M.; Morales-Ramires J. O.; Nguyen B. Y.; Kovacs C. M.; Steigbigel R. T.; Cooper D. A.; Liporace R.; Schwartz R.; Isaacs R.; Gilde L. R.; Wenning L.; Zhao J.; Teppler H. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naïve HIV-1-infected individuals. JAIDS, J. Acquired Immune Defic. Syndr. 2006, 43, 509–515. [DOI] [PubMed] [Google Scholar]
- Min S.; Sloan L.; Dejesus E.; Hawkins T.; McCurdy L.; Song I.; Stroder R.; Chen S.; Underwood M.; Fujiwara T.; Piscitelli S.; Lalezari J. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS 2011, 25, 1737–1745. [DOI] [PubMed] [Google Scholar]
- a Freed E. O.; Martin M. A.. HIVs and their replication. In Fields Virology; Knipe D., Howley P., Eds.; Lippincott Williams & Wilkins: Philadelphia, 2001; pp 1971–2042. [Google Scholar]; b Telesnitsky A.; Goff S. P.. Reverse transcriptase and the generation of viral DNA. In Retroviruses; Coffin J. M., Hughes S. H., Varmus H. E., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, 1997; pp 121–160. [PubMed] [Google Scholar]
- a Jacobo-Molina A.; Ding J.; Nanni R. G.; Clark A. D. Jr.; Lu X.; Tantillo C.; Williams R. L.; Kamer G.; Ferris A. L.; Clark P.; Hizi A.; Hughes S. H.; Arnold E. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huang H.; Chopra R.; Verdine G. L.; Harrison S. C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- a Nowotny M.; Gaidamakov S. A.; Crouch R. J.; Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 2005, 121, 1005–1016. [DOI] [PubMed] [Google Scholar]; b Kaushik N.; Rege N.; Yadav P. N.; Sarafianos S. G.; Modak M. J.; Pandey V. N. Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry 1996, 35, 11536–11546. [DOI] [PubMed] [Google Scholar]; b Patel H. P.; Jacobo-Molina A.; Ding J.; Tantillo C.; Clark A. D.; Raag R.; Nanni R. G.; Hughes S. H.; Arnold E. Insights into DNA polymerization mechanisms from structure and function analysis of HIV-1 reverse transcriptase. Biochemistry 1995, 34, 5351–5363. [DOI] [PubMed] [Google Scholar]
- a Shaw-Reid C. A.; Munshi V.; Graham P.; Wolfe A.; Witmer M.; Danzeisen R.; Olsen D. B.; Carroll S. S.; Embrey M.; Wai J. S.; Miller M. D.; Cole J. L.; Hazuda D. J. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 2003, 278, 2777–2780. [DOI] [PubMed] [Google Scholar]; b Tramontano E.; Esposito F.; Badas R.; Di Santo R.; Costi R.; La Colla P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 2005, 65, 117–124. [DOI] [PubMed] [Google Scholar]
- Di Santo R. Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. Curr. Med. Chem. 2011, 18, 3335–3342. [DOI] [PubMed] [Google Scholar]
- de Soultrait V.; Lozach P.; Altmeyer R.; Tarrago-Litvak L.; Litvak S.; Andreola M. L. DNA aptamers derivated from HIV-1 RNase H inhibitors are strong anti-integrase agents. J. Mol. Biol. 2002, 324, 195–203. [DOI] [PubMed] [Google Scholar]
- a Didierjean J.; Isel C.; Querré F.; Mouscadet J. F.; Aubertin A. M.; Valnot J. Y.; Piettre S. R.; Marquet R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob. Agents Chemother. 2005, 49, 4884–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Budihas S.; Gorshkova I.; Gaidamakov S.; Wamiru A.; Bona M.; Parniak M.; Crouch R.; McMahon J.; Beutler J.; Le Grice S. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005, 33, 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Semenova E. A.; Johnson A. A.; Marchand C.; Davis D. A.; Yarchoan R.; Pommier Y. Preferential inhibition of the magnesium-dependent strand transfer reaction of HIV-1 integrase by alpha-hydroxytropolones. Mol. Pharmacol. 2006, 69, 1454–1460. [DOI] [PubMed] [Google Scholar]
- Marchand C.; Beutler J. A.; Wamiru A.; Budihas S.; Mollmann U.; Heinisch L.; Mellors J. W.; Le Grice S. F.; Pommier Y. Madurahydroxylactone derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and RNase H. Antimicrob. Agents Chemother. 2008, 52, 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billamboz M.; Bailly F.; Lion C.; Touati N.; Vezin H.; Calmels C.; Andreola M.-L.; Christ F.; Debyser Z.; Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J. Med. Chem. 2011, 54, 1812–1824. [DOI] [PubMed] [Google Scholar]
- a Di Santo R.; Costi R.; Roux A.; Artico M.; Lavecchia A.; Marinelli L.; Novellino E.; Palmisano L.; Andreotti M.; Amici R.; Galluzzo C. M.; Nencioni L.; Palamara A. T.; Pommier Y.; Marchand C. Novel bifunctional quinolonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, biological activities, and mechanism of action. J. Med. Chem. 2006, 49, 1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Di Santo R.; Costi R.; Roux A.; Miele G.; Cuzzucoli Crucitti G.; Iacovo A.; Rosi F.; Lavecchia A.; Marinelli L.; Di Giovanni C.; Novellino E.; Palmisano L.; Andreotti M.; Amici R.; Galluzzo C. M.; Nencioni L.; Palamara A. T.; Pommier Y.; Marchand C. Novel quinolinonyl diketo acid derivatives as HIV-1 integrase inhibitors: design, synthesis, and biological activities. J. Med. Chem. 2008, 51, 4744–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa H.Process for producing 5,7-dichloro-4-hydroxy quinoline. PCT Int. Appl. WO 9523787, 1995;; CAN 124:29619, 1995.
- Yamashita H.; Matsubara J.; Oshima K. Kuroda H.; Ito N.; Miyamura S.; Shimizu S.; Tanaka T.; Takahashi H.. Process for producing piperazine-substituted benzothiophenes for treatment of mental disorders. PCT Int. Appl. WO 112464, 2006.
- Marinello J.; Marchand C.; Mott B. T.; Bain A.; Thomas C. J.; Pommier Y. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 2008, 47, 9345–9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyda F.; Hickman A. B.; Jenkins T. M.; Engelman A.; Craigie R.; Davies D. R. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Dyda F.; Hickman A. B.; Jenkins T. M.; Craigie R.; Davies D. R. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan S.; Guilloteau J. P.; Zhou-Liu Q.; Clément-Mella C.; Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J. Mol. Biol. 1998, 282, 359–368. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Craigie R.; Cohen G. H.; Fujiwara T.; Yoshinaga T.; Fujishita T.; Sugimoto H.; Endo T.; Murai H.; Davies D. R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 13040–13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald J.; Le V.; Butler S. L.; Bushman F. D.; Choe S. The mobility of an HIV-1 integrase active site loop is correlated with catalytic activity. Biochemistry 1999, 38, 8892–8898. [DOI] [PubMed] [Google Scholar]
- Chen J. C.; Krucinski J.; Miercke L. J.; Finer-Moore J. S.; Tang A. H.; Leavitt A. D.; Stroud R. M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni V.; Greenwald J.; Rhodes D.; Hwang Y.; Kwiatkowski W.; Bushman F. D.; Siegel J. S.; Choe S. Identification of a small-molecule binding site at the dimer interface of the HIV integrase catalytic domain. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001, 57, 536–544. [DOI] [PubMed] [Google Scholar]
- Wang J. Y.; Ling H.; Yang W.; Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001, 20, 7333–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Ambrosio A. L.; Rahman S.; Ellenberger T.; Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17308–17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielens J.; Headey S. J.; Jeevarajah D.; Rhodes D. I.; Deadman J.; Chalmers D. K.; Scanlon M. J.; Parker M. W. Crystal structure of the HIV-1 integrase core domain in complex with sucrose reveals details of an allosteric inhibitory binding site. FEBS Lett. 2010, 584, 1455–1462. [DOI] [PubMed] [Google Scholar]
- Christ F.; Voet A.; Marchand A.; Nicolet S.; Desimmie B. A.; Marchand D.; Bardiot D.; Van der Veken N. J.; Van Remoortel B.; Strelkov S. V.; De Maeyer M.; Chaltin P.; Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nature Chem. Biol. 2010, 6, 442–448. [DOI] [PubMed] [Google Scholar]
- Rhodes D. I.; Peat T. S.; Vandegraaff N.; Jeevarajah D.; Newman J.; Martyn J.; Coates J. A.; Ede N. J.; Rea P.; Deadman J. J. Crystal structures of novel allosteric peptide inhibitors of HIV integrase identify new interactions at the LEDGF binding site. ChemBioChem 2011, 12, 2311–2315. [DOI] [PubMed] [Google Scholar]
- Rhodes D. I.; Peat T. S.; Vandegraaff N.; Jeevarajah D.; Le G.; Jones E. D.; Smith J. A.; Coates J. A.; Winfield L. J.; Thienthong N.; Newman J.; Lucent D.; Ryan J. H.; Savage G. P.; Francis C. L.; Deadman J. J. Structural basis for a new mechanism of inhibition of HIV-1 integrase identified by fragment screening and structure-based design. Antiviral Chem. Chemother. 2011, 21, 155–168. [DOI] [PubMed] [Google Scholar]
- Wielens J.; Headey S. J.; Deadman J. J.; Rhodes D. I.; Le G. T.; Parker M. W.; Chalmers D. K.; Scanlon M. J. Fragment-based design of ligands targeting a novel site on the integrase enzyme of human immunodeficiency virus 1. ChemMedChem 2011, 6, 258–261. [DOI] [PubMed] [Google Scholar]
- Peat T. S.; Rhodes D. I.; Vandegraaff N.; Le G.; Smith J. A.; Clark L. J.; Jones E. D.; Coates J. A.; Thienthong N.; Newman J.; Dolezal O.; Mulder R.; Ryan J. H.; Savage G. P.; Francis C. L.; Deadman J. J. Small molecule inhibitors of the LEDGF site of human immunodeficiency virus integrase identified by fragment screening and structure based design. PLoS One 2012, 7, e40147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielens J.; Headey S. J.; Rhodes D. I.; Mulder R. J.; Dolezal O.; Deadman J. J.; Newman J.; Chalmers D. K.; Parker M. W.; Peat T. S.; Scanlon M. J. Parallel screening of low molecular weight fragment libraries: do differences in methodology affect hit identification?. J. Biomol. Screening 2013, 18, 147–159. [DOI] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens G. N.; Hare S.; Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 2010, 468, 326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Vos A. M.; Clayton R. F.; Thuring J. W.; Cummings M. D.; Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Smith S. J.; Métifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S. H.; Cherepanov P. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol. Pharmacol. 2011, 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime, version 3.0; Schrodinger, LLC: New York, 2011.
- Tang J.; Maddali K.; Pommier Y.; Sham Y. Y.; Wang Z. Scaffold rearrangement of dihydroxypyrimidine inhibitors of HIV integrase: docking model revisited. Bioorg. Med. Chem. Lett. 2010, 20, 3275–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glide, version 5.7; Schrödinger, LLC: New York, 2009.
- Vandurm P.; Cauvin C.; Guiguen A.; Georges B.; Le Van K.; Martinelli V.; Cardona C.; Mbemba G.; Mouscadet J. F.; Hevesi L.; Van Lint C.; Wouters J. Structural and theoretical studies of [6-bromo-1-(4-fluorophenylmethyl)-4(1H)-quinolinon-3-yl)]-4-hydroxy-2-oxo-3-butenoic acid as HIV-1 integrase inhibitor. Bioorg. Med. Chem. Lett. 2009, 19, 4806–4809. [DOI] [PubMed] [Google Scholar]
- Himmel D. M.; Maegley K. A.; Pauly T. A.; Bauman J. D.; Das K.; Dharia C.; Clark A. D. Jr.; Ryan K.; Hickey M. J.; Love R. A.; Hughes S. H.; Bergqvist S.; Arnold E. Structure of HIV-1 reverse transcriptase with the inhibitor β-thujaplicinol bound at the RNase H active site. Structure 2009, 17, 1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H.-P.; Yan Y.; Prasad G. S.; Smith R. F.; Daniels C. L.; Abeywickrema P. D.; Reid J. C.; Loughran H. M.; Kornienko M.; Sharma S.; Grobler J. A.; Xu B.; Sardana V.; Allison T. J.; Williams P. D.; Darke P. L.; Hazuda D. J.; Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 2010, 84, 7625–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdon E. B.; Liu Q.; Leavitt S. A.; Balakrishnan M.; Perry J. K.; Lancaster-Moyer C.; Kutty N.; Liu X.; Squires N. H.; Watkins W. J.; Kirschberg T. A. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxyquinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 2011, 55, 2905–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S.; Himmel D. M.; Jiang J. K.; Wojtak K.; Bauman J. D.; Rausch J. W.; Wilson J. A.; Beutler J. A.; Thomas C. J.; Arnold E.; Le Grice S. F. Synthesis, activity, and structural analysis of novel α-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 2011, 54, 4462–4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapkouski M.; Tian L.; Miller J. T.; Le Grice S. F.; Yang W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nature Struct. Mol. Biol. 2013, 20, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métifiot M.; Maddali K.; Johnson B. C.; Hare S.; Smith S. J.; Zhao X. Z.; Marchand C.; Burke T. R. Jr.; Hughes S. H.; Cherepanov P.; Pommier Y. Activities, crystal structures, and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 2013, 8, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S.; Wendeler M.; Rausch J. W.; Beilhartz G.; Gotte M.; O’Keefe B. R.; Bermingham A.; Beutler J. A.; Liu S.; Zhuang X.; Le Grice S. F. Structure–activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métifiot M.; Faure A.; Guyonnet-Duperat V.; Bellecave P.; Litvak S.; Ventura M.; Andréola M. L. Cellular uptake of ODNs in HIV-1 human-infected cells: a role for viral particles in DNA delivery?. Oligonucleotides 2007, 17, 151–165. [DOI] [PubMed] [Google Scholar]
- Maestro, version 9.2; Schrodinger, LLC: New York, 2011.
- MacroModel, version 9.9; Schrodinger, LLC: New York, 2011.
- Maurin C.; Bailly F.; Buisine E.; Vezin H.; Mbemba G.; Mouscadet J. F.; Cotelle P. Spectroscopic studies of diketoacids-metal interactions. A probing tool for the pharmacophoric intermetallic distance in the HIV-1 integrase active site. J. Med. Chem. 2004, 47, 5583–5586. [DOI] [PubMed] [Google Scholar]
- Epik, version 2.2; Schrodinger, LLC: New York, 2011.
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.