

Summary
Mycobacterium tuberculosis, the bacterium that causes tuberculosis (TB), is an intracellular pathogen of mononuclear phagocytes. Although M. tuberculosis has traditionally been thought to survive and replicate in macrophages, recent work in our laboratory and others has revealed that M. tuberculosis infects multiple subsets of mononuclear phagocytes in vivo and in vitro. In experimental animals, M. tuberculosis infects no fewer than five distinct cell subsets in the lungs, including resident alveolar macrophages and 4 types of cells that recruited to the lungs in response to inflammatory signals: neutrophils, monocytes, interstitial macrophages, and dendritic cells. A characteristic of the adaptive immune response in TB is that it is delayed for several weeks following infection, and we have determined that this delay is due to prolonged residence of the bacteria in lung phagocytes prior to acquisition of the bacteria by dendritic cells. Among the mechanisms used by M. tuberculosis to delay acquisition by dendritic cells is to inhibit apoptosis of alveolar macrophages and neutrophils, which sequester the bacteria and prevent their acquisition by dendritic cells in the early stages of infection. We hypothesize that each infected cell subset makes a distinct contribution to the overall biology of M. tuberculosis and allows the bacteria to evade elimination by T-cell responses and to avoid rapid killing by antimycobacterial drugs.
Keywords: tuberculosis, monocyte, macrophage, dendritic cell, antigen presentation, cytokine
Introduction
Mycobacterium tuberculosis, the bacteria that cause tuberculosis (TB), can establish life-long, chronic infection in the face of seemingly appropriate immune responses. This extraordinary persistence may be explained by the fact that M. tuberculosis resides predominantly inside cells of the immune system itself. Since the 1920s, when Florence Sabin and her colleagues described tubercle bacilli in mononuclear phagocytes (1, 2), there has been considerable interest in understanding the roles of these cells in TB pathogenesis and immunity. Until recently, it was believed that macrophages were the sole cells harboring M. tuberculosis. In particular, since the bacteria enter the body via the lung alveoli, it has been widely believed that alveolar macrophages are the predominant infected cells in TB, despite the lack of direct evidence to support that belief. As we describe in this review, M. tuberculosis can infect many subsets of mononuclear cells in vivo, and recent findings indicate that these cell subsets have different functional roles in TB.
An important advance in understanding the phenotypic diversity of infected cells in TB was the development of flow cytometry detection of infected cells isolated from the lungs of mice after aerosol infection with a strain of M. tuberculosis expressing a FACS-optimized variant of green fluorescent protein (GFP) (3). Although the technique is not suited for detection of infected cells prior to 10-14 days after low-dose aerosol infection [<100 colony-forming units (cfu) per mouse], it allowed the phenotypic identification and quantitation of infected cells during the late innate immune and the early adaptive immune stages of TB (days 14-17 and 19-28 post infection, respectively). The initial studies used a simplified phenotypying scheme using the integrins CD11c and CD11b to distinguish subsets of lung mononuclear cells: alveolar macrophages (AM) are CD11c+CD11b-/lo; recruited interstitial macrophages (RIM) are CD11cintCD11bint; myeloid dendritic cells (DC) are CD11c+CD11bhi; and monocytes are CD11c-CD11bhi (3, 4). In addition, this approach allowed identification of polymorphonuclear cells, neutrophils in particular, as CD11c-CD11bhiGr-1hi. With the exception of alveolar macrophages, the other mononuclear cell subsets are massively recruited to the lungs following M. tuberculosis infection, increasing in absolute numbers by 20- to 30-fold compared with uninfected lungs (3, 5). Examination of the distribution of M. tuberculosis in these cell subsets on day 14 post infection revealed a surprising result: going against the conventional dogma that M. tuberculosis resides mainly in alveolar macrophages, GFP-expressing bacteria were equally distributed in AMs, myeloid DCs, and neutrophils (3). With increasing time after infection, recruited interstitial macrophages became a prominent infected cell subset, outnumbering alveolar macrophages, which only constituted a minor subset of the population of infected cells. Neutrophils were also a predominant population of infected cells, albeit transiently, and their contribution decreased markedly after 19 days of infection. Monocytes represented a constant, but minor, fraction of the infected cell population throughout the infection. Most surprising was the finding that, by 21 days post-infection, DC, not macrophages, accounted for the largest fraction of the infected cells (Table 1, Fig. 1). Since expression of surface markers alone are insufficient to identify a myeloid cell subset with complete confidence, additional functional criteria were used to identify DCs: they did not require IFN-γ responsiveness to express high levels of surface MHC class II, unlike macrophages, they migrated in response to CCR7 agonists, and they were not depleted from lungs by bronchioalveolar lavage, unlike alveolar macrophages. Subsequent studies in our laboratory have revealed CD103 expression on a subset of these cells.
Table 1.
Percentage of cells in each lung myeloid cell subset that contain GFP-expressing M. tuberculosis.
| Day post infection | Alveolar macrophages | Interstitial macrophages | Myeloid dendritic cells | Monocytes | Neutrophils |
|---|---|---|---|---|---|
| 14 | 0.8 | 0.6 | 1. 9 | 0.1 | 1.3 |
| 19 | 2.1 | 4.9 | 10.1 | 1.2 | 9.1 |
| 21 | 4.1 | 7.9 | 16.2 | 2.8 | 12.9 |
| 28 | 2.8 | 6.8 | 15.6 | 1.9 | 3.5 |
Adapted from Journal of Immunology 2007;179:2509-2519.
Fig. 1. Time course and distribution of M. tuberculosis by lung cell subset.
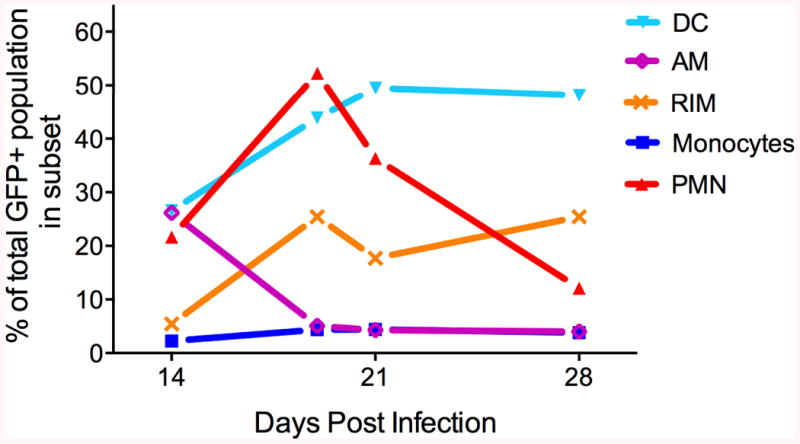
The graph shows the distribution of the population of M. tuberculosis-infected cells in the lungs, according to the proportion of the bacterial population in each myeloid cell subset, and shows the progression over time after infection. DC, dendritic cells; AM, alveolar macrophages; RIM, recruited interstitial macrophages; PMN, neutrophils.
These studies demonstrated that rather than residing only in macrophages as previously thought, M. tuberculosis infects diverse myeloid cell subsets in the lungs and that the dominant infected cell population varies at specific stages of infection. Several models can accommodate these observations. First, M. tuberculosis may orchestrate an inflammatory response that recruits distinct types of myeloid cells during infection, and then exploits cell-specific environments at various stage of infection. Second, M. tuberculosis may be readily ingested by any phagocyte, and distinct cell subsets simply vary in their capacity to kill and dispose of the ingested bacteria. Third, cells from distinct subsets may cooperate with one another to optimize the host response to M. tuberculosis infection, but even under optimal conditions, the bacteria can persist within these cells. Finally, distinct subsets of myeloid cells may vary in their capacity to be recognized by and/or activate antigen-specific CD4+ and CD8+ T cells and their effector functions, i.e. cytokine production or cytotoxic activity, and these differences correlate with the bacterial burden in each myeloid subset.
In the following sections, we describe studies that address specific aspects of each mononuclear cell subset in TB and present a chronological model that integrates those subsets. While most data have been generated in mice, we also refer to data from TB patients as well as other animal models, whether they confirm or contrast with the results obtained in mice.
Lung mononuclear cell phenotypes, subsets, and origins
The definition of mononuclear cell subsets continues to evolve, and until recently, most studies focused on cells isolated from secondary lymphoid organs under homeostatic conditions. Recognizing that phenotypes defined under those circumstances were not applicable to cells in peripheral tissues during infection or inflammatory responses has broadened the understanding of mononuclear cell populations and their regulation (6-9). In this review, we discuss the role of monocytes, alveolar macrophages, recruited interstitial macrophages and lung dendritic cells in TB. Venturing beyond mononuclear cells, we will also briefly describe the importance of neutrophils in M. tuberculosis infection.
Monocytes
Unlike alveolar macrophages whose populations are likely established at birth and replenished by local self-renewal (11, 12), monocytes develop from progenitors in the bone marrow and retain the capacity to differentiate in the periphery and supply tissues with macrophages and DCs, especially during infection or inflammation (13).
Monocyte subsets
Monocytes are pleomorphic in mice as well as in humans. In humans, the two main subsets of monocytes are defined by their expression of CD14, a co-receptor for bacterial lipopolysaccharide, or CD16, the FcγRIII receptor for the Fc domain of IgG (13). These subpopulations are also characterized by the differential expression of chemokine receptors, especially CCR2 and CX3CR1. The more abundant subset in humans, termed ‘classic monocytes’ are CD14+ and express high levels of CCR2 and low levels of CX3CR1. The less abundant ‘non-classic monocytes’ are CD16+, and express the reverse pattern of these chemokine receptors. While CD14 and CD16 are not used to identify monocytes in mice, the differential expression of CCR2 and CX3CR1 also defines the two subsets, which are present at equal frequencies in the blood of mice. Additional markers expressed by murine monocytes include CD115 (macrophage colony-stimulating factor receptor) and the integrin CD11b, which are common to all monocyte subsets, and Ly6C, which is expressed at high levels by CCR2+ monocytes. The expansion of Ly6Chi CCR2+ monocytes in response to inflammation or infection led to their designation as ‘inflammatory monocytes’ (13). Alternatively, expression of CX3CL1 by endothelial or epithelial cells promotes blood vessel patrolling, entry, and tissue retention of monocytes expressing CX3CR1 (14-16), leading to these cells being considered as patrolling, or resident monocytes. Finally, there may be flux between monocyte subsets, as Ly6C and CX3CR1 are inversely expressed during the maturation process and adoptive transfer experiments suggest that Ly6Chi CCR2+ cells can give rise to CX3CR1+ monocytes (17).
Role of inflammatory monocytes in TB
The relative importance of monocyte subsets in human TB remains to be defined. Monocyte chemoattractant protein 1 (MCP-1) (also called CCL2) the predominant ligand for CCR2, is produced by mononuclear cells infected by M. tuberculosis and can be detected in alveolar fluid of TB patients (18). Additionally, increased susceptibility to pulmonary TB in humans is associated with a promoter polymorphism that leads to overproduction of MCP-1/CCL2 (19), and de-sensitization of CCR2 has been proposed to explain this observation (13). The roles of CCR2 in mice infected with M. tuberculosis have been investigated in more depth. We found that CCR2-deficient mice die early following infection, with high bacterial loads in their lungs (20). In that study, CCR2-/- mice exhibited an early defect in macrophage recruitment to the lungs and a later defect in recruitment of DC. Further investigation confirmed the importance of CCR2 for optimal accumulation of macrophages and DC in the lungs in TB (21). An independent study subsequently reported that CCR2-/- mice exhibit delayed recruitment of cells to the lungs after a low-dose aerosol inoculum of M. tuberculosis but are capable of controlling the infection (22), while yet another study that also used a low dose aerosol inoculum revealed a higher bacterial burden in the CCR2-/- mice compared with controls (23). Therefore, the weight of evidence indicates that CCR2-dependent trafficking of mononuclear cells plays an important role in immune responses to M. tuberculosis. Despite these findings, cell transfer studies suggested that CCR2 was not itself required for monocyte recruitment to the lungs of M. tuberculosis-infected mice (5). The solution to this apparent paradox was resolved by finding that CCR2, while not required for cell recruitment to the site of infection, is responsible for egress of Ly6Chi inflammatory monocytes from the bone marrow into the blood (24). We and others have reported the importance of CCR2 or its ligand CCL2 for antigen-specific activation of T cells in the lung draining lymph node in TB (21, 25). Using mixed bone marrow chimeric mice, we established that CCR2 expression is required on myeloid cells, and not on T cells. Recently, an in vivo depletion approach using mice expressing the simian diphtheria toxin receptor under control of the CCR2 promoter showed that inflammatory monocytes are required for transport of M. tuberculosis to the lung draining lymph node and priming of antigen-specific CD4+ T cells (26). This study also revealed that M. tuberculosis-infected monocytes are not responsible for directly priming T cells. Instead, they differentiate into DCs and transport the bacteria to local lymph nodes and must cooperate with resident lymph node cells to prime CD4+ T cells (27).
Although there is considerable evidence for the importance of monocytes in TB, the essential mechanistic contributions of monocytes in TB are less clear. In particular, the functions that monocytes accomplish as monocytes versus the functions that they provide after differentiation into tissue macrophages or DC have not been distinguished. Following TB initiated by low-dose aerosol infection, a small fraction (2-3%) of monocytes in the lungs is infected with M. tuberculosis (Fig. 1). It is notable that the fraction of monocytes containing bacteria in the lungs is low but nearly constant during the innate and adaptive immune phases of infection. Whether this reflects a population of monocytes that become infected shortly after entry into the lungs, and then remains stable, or whether infection of monocytes is dynamic and continuous has not been determined.
Type I interferon and monocytes
Monocyte recruitment to the lungs and the functions of monocytes in the lungs in TB are conditioned by cytokines that induce or modulate production of CCR2 ligands and/or affect local monocyte differentiation. In particular, type I interferons (IFNs) regulate monocyte recruitment during chronic inflammation (28). In mice unable to respond to type I IFN (IFNAR-/-), accumulation of monocytes in the lungs is reduced after infection with M. tuberculosis (29). Conversely, potent induction of type I IFN production by poly-IC treatment of M. tuberculosis-infected mice enhances CCR2-dependent recruitment of CD11b+ Gr-1int monocytes, which then become infected, and contribute to higher bacterial burdens and accelerated mortality (23). A similar imbalance in monocyte recruitment may explain the hypervirulence of certain M. tuberculosis strains that induce large amounts of type I IFN (30). Type I IFN and M. tuberculosis may also impact monocyte differentiation, as in vitro studies have revealed that the bacteria divert human cells from a type I IFN-induced DC fate to a more pathogen-permissive, macrophage phenotype (31). Finally, illustrative of the complexity of the type I IFN-monocyte-M. tuberculosis triangle, type I IFN can directly affect the effector functions of inflammatory monocytes during TB, by suppressing their production of protective interleukins 1α and 1β (32, 33).
Other monocyte subsets
The roles of monocyte subsets other than inflammatory monocytes in TB are even less well understood. While Ly6Chi CCR2+ inflammatory monocytes can give rise to CX3CR1+ monocytes, the significance and mechanism of this transition in TB have not been determined. The only published study indicates that CX3CR1 is dispensable for the control of M. tuberculosis (34), but it is not known whether this is attributable to compensation by CCR2-expressing cells. Humans and mice have other minor monocyte subsets (13), such as CD14+CD16+ or Ly6Cint cells, but their contributions to the control or pathogenesis of TB are unknown.
Alveolar macrophages: initial encounters
The term macrophage applies to cells with lineage and tissue specificities (7). Recognition of the relationship between M. tuberculosis and macrophages has a long history. Whether they were described as ‘reticulo-endothelial cells’, ‘histiocytes’, or by the more generic term of ‘phagocytes’, macrophages were the first host cells to be studied for their interactions with M. tuberculosis (1, 2). Early studies were facilitated by the ease with which macrophages could be identified, isolated, and cultured from tissues (35). Macrophages serve more than one role in TB, as they are important innate and effector cells in the immune response to M. tuberculosis, and they are dominant targets and resident cells for the bacteria.
Alveolar macrophages (AMs) reside in the airspaces of the lungs. AMs make up a large fraction (∼90-95%) of the cells in bronchoalveolar lavage fluid, they are present in uninfected lungs, they are phagocytic, and have a marked capacity for self-renewal (37). AMs are widely believed to be the first phagocytes to encounter M. tuberculosis in vivo, but despite considerable effort, we have been unable to identify a primary publication that provides direct evidence for this textbook-grade tenet (36). Despite recent evidence that AMs, like other tissue-resident macrophages, are derived from the yolk sac during development (11, 12), there is also evidence that these cells can arise from monocytes (7, 38): monocytes transferred into M. tuberculosis infected mice contribute to the CD11bloCD11c+ AM compartment in the lungs (5). Whatever the origin of these cells, in the lungs of M. tuberculosis-infected mice, AMs are distinguishable from recruited interstitial macrophages by their higher expression of CD11c, MHC class II, and costimulatory molecules such as CD80 and CD86. Consistent with their location and phagocytic ability, AMs harbor M. tuberculosis after aerosol infection. Although earlier time points have not yet been studied due to limitations of the technology available, AMs represented approximately 30% of the lung cells with intracellular M. tuberculosis 14 days post-infection (3). At later time points, newly recruited mononuclear cells (recruited interstitial macrophages and DCs) outnumber AMs as the dominant cells harboring intracellular M. tuberculosis (3); these findings were confirmed and extended in an independent investigation (39). In the latter study, cytomegalovirus expressing GFP was used to label lung leukocytes prior to M. tuberculosis infection, and showed that in uninfected mice AM account for 97% of the total GFP+ leukocytes. After 4 weeks of M. tuberculosis infection, the proportion of lung leukocytes represented by AMs decreased to ∼68%, accompanied by an increase in DCs (from 2.8% to 27%). Although this reciprocal increase in DCs and decrease in AMs could reflect a phenotypic conversion of AMs to DC-like cells, the authors calculated that a phenotypic shift could account for no more than ∼3% of the increase in DCs, thus confirming other reports that inflammatory monocytes are the major source of lung myeloid DCs (20, 40).
AMs and innate immunity
Since AMs are likely to encounter bacteria early after aerosol infection and since growth of bacteria in the lungs 10-14 days post-infection coincides with spread of bacteria to other myeloid cells, AMs appear to provide a favorable niche for M. tuberculosis replication. Mice depleted of AMs with clodronate-containing liposomes before infection with M. tuberculosis, AM depletion showed enhanced survival together with increased recruitment of neutrophils and altered cytokine expression, with an increased Th1 response (41). However, these results should be interpreted cautiously in light of the subsequent finding that intrapulmonary administration of clodronate liposomes causes a rapid influx of myeloid DC (10). In vitro studies with human alveolar macrophages have revealed that they respond to exposure to M. tuberculosis and other microbial stimuli in a manner similar to that of other macrophage populations: they produce TNF and other proinflammatory and immunoregulatory cytokines (42), they support intracellular survival and replication of M. tuberculosis, and they undergo apoptosis when infected with an attenuated strain of M. tuberculosis.
One especially innovative study used human AMs to confirm the fundamental observation first made in cultured murine macrophages by Armstrong and Hart in the 1970s (43), that live virulent mycobacteria reside in phagosomes that fail to fully mature and acquire the properties of lysosomes. Mwandumba and colleagues (44) took advantage of the loading of AM lysosomes with indigestible carbon particles acquired by frequent inhalation of wood smoke to determine that, in AM isolated from subjects with active TB, lysosomal cargo is not delivered to mycobacterial phagosomes.
An additional speculative role for AMs in TB is the elimination of the bacteria, even without detectable involvement of adaptive immune responses. This is based on the frequent observation that ∼50% of close contacts of an active case of TB remain uninfected (as detected by T-cell responses to mycobacterial antigens in skin tests or in vitro assays) (45). Since it is widely believed that AMs are the first cells to encounter M. tuberculosis after the bacteria are inhaled, this epidemiological observation has been used to suggest that certain individuals have highly effective innate immunity to TB and that they possess AM with the intrinsic ability to kill M. tuberculosis. While this is an attractive hypothesis, there is currently no direct evidence to support it, since virulent M. tuberculosis replicate intracellularly in alveolar macrophages from a large fraction of subjects (46).
Since alveolar macrophages are likely to encounter inhaled M. tuberculosis early in infection and since the bacteria are found in diverse populations of newly recruited leukocytes 10-14 days later, M. tuberculosis must spread from alveolar macrophages to these other cells. Spread from alveolar macrophages is likely prolonged by the well-established ability of M. tuberculosis to inhibit apoptosis of the cells it infects (46-48). Once the earliest infected cells die by apoptosis or necrosis, the bacteria are released to the extracellular space for uptake by previously uninfected cells (49). It is likely through this process that M. tuberculosis is accessed by neutrophils, and subsequently by dendritic cells (47). Thus, the long delay in initiation of T-cell responses after M. tuberculosis infection of humans (50) and mice (27, 51) is due at least in part by the ability of the bacteria to dwell in and prevent apoptosis of alveolar macrophages before spreading to DC, which eventually migrate to local lymph nodes and initiate T-cell responses.
Although AMs themselves cannot initiate T-cell responses, strong evidence indicates that they participate with DCs in this process. In particular, AMs isolated from M. tuberculosis infected Alox5-/- mice that lack 5-lipoxygenase and cannot synthesize the lipoxin A4 (LXA4), led to superior priming of naive CD8+ T cells when given intratracheally to naive wildtype mice (52). The authors attributed the observed superior T-cell priming to enhanced cross-priming and suggested that the lack of LXA4 results in increased apoptosis (53) of the Alox5-/- AMs and enhanced acquisition of bacterial antigens by DCs, leading to efficient cross-priming of T cells. The authors inferred that one of the ways M. tuberculosis manipulates the cells it infects is by preventing apoptosis of infected cells and subsequent acquisition of antigens by DCs for cross-presentation, eventually resulting in poor T-cell priming (52). While apoptosis of infected cells leads to generation of apoptotic vesicles that cross-prime CD8+ T cells leading to protective immunity (54), apoptosis can also promote cell-to-cell spread of bacteria (49).
AMs and adaptive immunity
The contribution of AMs to adaptive immune responses in TB is poorly understood, but it might be minimal, since AMs are in a distinct anatomic compartment that potentially restrict their contact with effector T cells, most of which are in the lung interstitium. Nevertheless, AMs express moderate-to-high levels of MHC class II and costimulatory molecules, indicating that they should be able to be recognized by and stimulate effector T cells.
One fascinating observation indicates that, at least in the first week of M. tuberculosis infection, antigen-specific CD4+ T cells may not contact or recognize infected alveolar macrophages. When Th1-differentiated T-cell receptor transgenic CD4+ T cells specific for ESAT-6, a secreted bacterial protein, were transferred into mice prior to infection with M. tuberculosis, they had a significant protective effect, manifest as lower lung bacterial burdens (55). However, even though the T cells were transferred prior to infection and had populated the lungs, their antimycobacterial effects were only manifest after the first week of infection. Although there are several possible mechanisms that account for the inability of T cells to provide protection during the first week after infection, it is likely that bacteria are sequestered in AMs and therefore inaccessible to effector T cells. These results potentially have very important implications for development of TB vaccines: if vaccine-induced T cells cannot access the infected cells until at least a week after M. tuberculosis exposure, they will not be able to prevent infection.
Recruited interstitial macrophages
Phenotype, origins, and differentiation of recruited macrophages
Macrophages with a distinct phenotype (CD11clowCD11bhigh) from that of AMs are present at low frequencies in the lungs of uninfected mice and increase ∼40-fold in response to aerosol infection with M. tuberculosis (3). Since these cells are (i) not found in bronchoalveolar lavage fluid, (ii) derived from bone marrow precursors (56, 57), and (iii) depend on the presence of CCR2 for their accumulation in the lungs (20, 21), we term them recruited interstitial macrophages (RIMs). RIMs are distinguished from other lung mononuclear cells by their lack of expression of CD11c (DCs), CD115 or Ly6C (monocytes), or Ly6G (granulocytes). RIMs also upregulate MHC class II and costimulatory molecules, such as CD40 and CD86, as they enter the lung tissues (4), although not to the same extent as DCs (3). It is generally thought that during infection, these macrophages differentiate from monocytes, in particular Ly6Chi CCR2+ inflammatory monocytes (see preceding section on monocytes), although CX3CR1+ monocytes can give rise to similar tissue macrophages under inflammatory conditions (7).
The factors and mechanisms responsible for differentiation of inflammatory monocytes to RIMs in vivo are incompletely understood. Nevertheless, IFNs play a complex role in the accumulation of RIMs in TB. Both type I and type II IFNs influence the recruitment of monocyte precursors (23, 29) while differentiation into RIM appears to be negatively regulated by type I IFNs (IFN-α/β) (29) and is independent of type II (IFN-γ)(5, 29).
M. tuberculosis infection of RIMs_in vivo
Since the population of RIMs in uninfected lungs is small, RIMs are unlikely to be major initial targets of inhaled M. tuberculosis. However, once they are recruited in response to infection, they become infected and represent one of the major subsets of cells harboring M. tuberculosis: by 19 days post infection, RIMs harbor a much larger fraction of the bacterial population than do alveolar macrophages (3, 29, 39) (Fig. 1). Unlike neutrophils, whose prevalence as infected cells is transient, RIMs represent a large and stable fraction of the cells harboring M. tuberculosis in the lungs. Whether this reflects an inability of RIMs to kill intracellular M. tuberculosis, even in the presence of adaptive immune responses, or whether it is a dynamic state in which individual cells kill M. tuberculosis and other cells become infected at an equivalent rate, remains to be defined. Moreover, whether RIMs represent a single homogeneous population or are composed of distinct subsets needs to be determined; the latter state is suggested by monocyte transfer studies that indicated that IFN-γ-dependent production of nitric oxide (iNOS) in the lungs of M. tuberculosis-infected mice is attributable to a distinct population of cells (5).
M. tuberculosis interactions with macrophages studied in vitro
Many types of primary (e.g. M-CSF-differentiated from bone marrow or blood, resident or thioglycolate-induced from peritoneum, alveolar) or immortalized (e.g. RAW264.7, J774.1, THP-1) human and mouse macrophages are used to study host-bacteria interactions in TB. As professional phagocytes, macrophages possess impressive armamentaria for killing internalized bacteria, including low pH, proteolytic enzymes, reactive oxygen and nitrogen intermediates, and antimicrobial peptides such as cathelicidin. Expression and activation of these antimicrobial factors is tightly regulated and closely tied to macrophage activation, in particular through Toll-like receptors and IFN-γ Using bone marrow chimeric mice, we confirmed in vivo the in vitro observation (58, 59) that hematopoietic cells, including macrophages, must be responsive to IFN-γ to restrict growth of M. tuberculosis (56). We have only a limited understanding of the mechanisms induced by IFN-γ that mediate control of M. tuberculosis by macrophages in vivo, as only three IFN-γ-responsive genes, Nos2 and Irgm1 (also termed LRG-47) in mice (60, 61), and cathelicidin in humans (62) have been directly implicated in the control of M. tuberculosis. While well established in mice, the importance of nitric oxide in the control of M. tuberculosis human macrophages remains unsettled (63). In vitro, Irgm1/LRG47 regulates autophagy, a process in which cells digest their own macromolecules and organelles in an effort to kill intracellular M. tuberculosis (64). Autophagy-dependent control of M. tuberculosis in macrophages is also regulated by Toll-like receptor 4 (TLR4) signaling and by sensing of bacterial DNA (65, 66). In addition to killing intracellular organisms, macrophages also process and present bacterial antigenic peptides to CD4+ T cells via MHC class II. However, as revealed by our work (67-69) and that of others (70-72), antigen presentation and microbicidal mechanisms are selectively blocked by M. tuberculosis, allowing the bacteria to persist. More recently, we and others established that this inhibition was at least partially dependent on prolonged signaling through TLR2 (67, 73-75). This exploitation of innate immune signaling by M. tuberculosis may be significant, since macrophages are equipped with a variety of surface, cytosolic and vesicular sensors, i.e. TLRs, NOD-like receptors (NLRs) and RIG-I like receptors (RLRs) (76), suggesting that innate sensing may have complex roles in the context of TB. While in vivo data on the overall importance of NOD2 for control of M. tuberculosis is inconsistent (77, 78), NOD2 is required for the production of inflammatory cytokines and control of intracellular bacteria in human macrophages (79). In mice, NOD2 has been implicated in the production of type I IFN by macrophages after sensing bacterial peptides that have translocated from phagosomes into the cytosol (80). However, more recent findings challenged this view and attributed induction of type I IFN to the sensing of mycobacterial DNA by STING, another cytosolic sensor, independently of NOD or TLR signaling (81). Interestingly, STING is also the DNA sensor involved in regulation of autophagy in M. tuberculosis-infected macrophages (65).
M. tuberculosis and macrophage death
As a highly successful pathogen, M. tuberculosis manipulates intracellular trafficking and cellular responses to IFN-γ and PAMPs to its long-term advantage. As a successful intracellular pathogen, it is not surprising that M. tuberculosis also manipulates programmed cell death or apoptosis, to preserve its cellular niche. M. tuberculosis employs multiple mechanisms to inhibit apoptosis, including the nuoG gene, which encodes a component of the bacterial type I NADH-dehydrogenase complex (47, 48), and a mechanism of protein secretion, secA2, which is required for secretion of bacterial superoxide dismutase (85).
Alternatively, macrophage death by necrosis is observed in multiple mycobacterial infection contexts, including in human lung granulomas (86) and is considered a hallmark of infection by virulent mycobacteria (87, 88). M. tuberculosis-driven macrophage necrosis requires the mycobacterial type VII secretion system ESX-1 and its substrate ESAT-6, permeabilizing the phagosomal compartment in which M. tuberculosis resides, with subsequent activation of the NLRP3 inflammasome (89). Although it involves inflammasome activation, M. tuberculosis-induced macrophage necrosis occurs independently of caspase 1 and cathepsin B in both mouse and human macrophages, thus distinguishing it from pyroptosis and pyronecrosis respectively (89-91).
These results exemplify the extent of M. tuberculosis' ability to manipulate macrophages, allowing sustained bacterial replication by inhibiting apoptosis and promoting cell-to-cell spread by necrosis. Although these mechanisms have all been described in macrophages, M. tuberculosis resides in other cell types, with which it may interact in distinct ways to alter their functions and optimize the opportunities to survive, replicate, and be transmitted.
Dendritic cells: bridges to adaptive immunity
Classical dendritic cells (cDCs) are professional antigen-presenting cells located in secondary lymphoid organs and in peripheral tissues. Following exposure to a pathogen, cDCs internalize and transport the pathogen (and/or its soluble antigens) to local draining lymph nodes, where they present antigens to naive T cells and initiate adaptive immune responses. Precursors for DCs originate in the bone marrow, circulate in the blood, and populate tissues as resident DCs. This tissue homing occurs in both steady state as well as during infection and is driven by chemokines and their receptors as well as by cytokines and growth factors. CCR2+Ly6Chi inflammatory monocytes migrate to sites of infection and inflammation and differentiate into monocyte-derived DCs. These appear to provide similar functions as cDCs: acquire antigens in peripheral tissues, respond to proinflammatory cues by maturing and migrating to local lymph node, where they contribute to priming naive T lymphocytes. Ly6Chi monocytes also traffic to peripheral tissues in steady state, but under these conditions, they remain as monocytes, rather than to differentiate into DCs or macrophages (8).
While macrophages have been studied extensively with respect to their roles in M. tuberculosis infection and immunity, less attention has been paid to the roles of DCs in TB. However, recent studies revealed that mouse (3) and human (92) DCs harbor M. tuberculosis in the lungs and draining lymph nodes. Indeed, 3-4 weeks after aerosol infection with the virulent H37Rv strain of M. tuberculosis, CD11chiCD11bhi DCs represent the most prevalent subset of infected cells in the lungs and in the local draining lymph nodes (3). Moreover, DC acquisition of M. tuberculosis in the lungs is clearly the rate-limiting step in activation of antigen-specific CD4+ T-cell responses in TB (27, 47, 93). However, DC transport of M. tuberculosis from the lungs to the lymph node appears to be inefficient, and may even cease after the early stages of infection, as the population of infected DCs appears to reach a steady state (3). Since DCs are traditionally considered to be efficient antigen-presenting cells, they should be readily recognized by and trigger activation of antigen-specific CD4+ effector T cells. Although data from bone marrow chimeric mice prepared with a 1:1 mix of MHC II-/- and MHC II+/+ donor cells provides clear evidence that MHC II+/+ lung DCs are recognized by CD4+ T cells (57), a large number of lung DCs remain infected despite development of adaptive immune responses. This observation suggests either that T-cell recognition of infected DCs is inefficient or infrequent or that DCs cannot respond effectively to the effector mechanisms provided by CD4+ T cells to express mycobactericidal activity. Persuasive evidence for the former mechanism has been reported, indicating that activation of CD4+ effector T cells occurs with low frequencies in the lungs of M. tuberculosis-infected mice (94, 95), and that CD4+ effector T-cell activation is remarkably lower than is observed in influenza (96). While these results indicate that DCs in the lungs may present antigen less well than they do in other infections, they do not exclude the possibility that other mechanisms contribute to the inability of T-cell responses to eliminate M. tuberculosis.
DCs and their functions during M. tuberculosis infection
A notable characteristic of TB (and certain other persistent infections) is the long interval between initiation of infection and the development of adaptive immune responses. In humans, the appearance of measurable adaptive immune responses requires an average of 6 weeks after infection (50). In mice, a minimum of 11-14 days is required from the time of aerosol infection to the first detectable T-cell response, using sensitive techniques (27, 51). Since this gives the bacteria the opportunity to expand their population ∼40,000-fold before the initial appearance of effector T cells in the lungs (27), this is undoubtedly one of the most important features of TB pathogenesis. As in other infections or administration of antigen, M. tuberculosis-specific T-cell responses are initiated in the local draining lymph nodes. Since M. tuberculosis enters and proliferates in the lungs, and since most of the known immunodominant T-cell antigens of M. tuberculosis are bacterial secreted proteins, it was logical to assume that lung DC transport soluble M. tuberculosis antigens to the lung-draining mediastinal lymph nodes to prime antigen-specific T cells. Therefore, it was unexpected to find that DC must transport intact M. tuberculosis bacteria from the lungs to the mediastinal lymph nodes to prime naive T cells (27). This initiation of adaptive immunity involves transport of bacteria to the mediastinal lymph nodes by DCs, as ablation of DCs in M. tuberculosis infected mice results in further delayed adaptive immunity and enhanced mortality (97). CD11bhi DCs are the major subset that harbors bacteria in the lungs and mediastinal lymph node, and are responsible for transport of the bacteria (3). Migration of M. tuberculosis-infected DC is driven by chemokine receptors, as plt mice that are deficient for the CCR7 ligands CCL19 and CCL21 contain far fewer DCs as well as bacteria (∼95% less than the wild type mice) (3) and show delayed activation of M. tuberculosis-specific CD4+ T cells in their mediastinal lymph nodes (27). Similar results have been reported in CCR7-/- mice (98). CCR2-/- mice also show delayed and reduced priming of M. tuberculosis-specific CD4+ T cells following M. tuberculosis infection, which eventually results in poor control of bacterial growth (20, 23), indicating that CCR2+ inflammatory monocytes are the likely precursors of the relevant lung DC in TB. In support of this, Samstein and colleagues (26) used a combination of CCR2-RFP reporter mice and CCR2-deleter mice to show that CCR2+ inflammatory monocytes that acquire a DC phenotype are crucial for transporting M. tuberculosis to the lung-draining lymph nodes for T-cell priming. Moreover, blocking responsiveness by treatment of M. tuberculosis-infected DC with pertussis toxin prior to intratracheal transfer prevents their migration to the local lymph node, resulting in the lack of appearance of DC and of M. tuberculosis in the mediastinal lymph node (93).
By mechanisms that are not yet well defined, M. tuberculosis-infected cells are impaired in their ability to activate T cells (reviewed in 99). The findings that DCs transport M. tuberculosis from the lungs to the lymph node and that DCs harbor M. tuberculosis in the lymph node, together with evidence that infected DCs are poor at activating antigen-specific CD4+ T cells (3) presented a conundrum, since antigen-specific CD4+ (and CD8+) T-cell responses are successfully generated in M. tuberculosis-infected humans and mice. The solution to the conundrum required either (i) refuting the evidence that M. tuberculosis infection inhibits T-cell activation, (ii) refuting the evidence that T-cell responses are generated in response to M. tuberculosis infection, or (iii) discovery of a novel mechanism. Although longstanding evidence from multiple studies shows that M. tuberculosis-infected macrophages or DCs activate CD4+ T cells poorly, the evidence was limited to in vitro studies until recently. Using adoptive transfer of inflammatory monocytes that differentiate into DC during M. tuberculosis infection, Samstein and colleagues (26) found that antigen-specific CD4+ T cells were not primed if the recipient mice lacked MHC class II. This implied that migratory DC must collaborate with one or more resident DCs to successfully prime CD4+ T cells in the context of TB, thus providing the first in vivo evidence that M. tuberculosis interferes with antigen presentation by the cells that it infects. Consistent with these findings, we found that intratracheal transfer of bone marrow-derived DCs (BMDCs) could lead to priming of M. tuberculosis antigen 85B (Ag85B)-specific CD4+ T cells, and that optimal priming required expression of MHC class II by both donor and recipient cells (93). These results are consistent with the findings of Samstein and colleagues (26) and indicate that, in the context of TB, infected migratory DCs are indeed poor antigen-presenting cells. Furthermore, they suggest that infected cells have a mechanism to transfer antigen to uninfected cells so that uninfected cells could prime CD4+ T cells. Consistent with this model, we found that cultured DCs and macrophages release multiple M. tuberculosis protein antigens into the extracellular medium. The extracellular mycobacterial antigens are not present as a consequence of extracellular bacteria, are full-length and unprocessed, are not associated with exosomes or apoptotic vesicles, and can be taken up, processed, and presented to CD4+ T cells by uninfected DCs (93). Most importantly, this form of antigen transfer occurs without transfer of the pathogen itself, either in vitro or in vivo, thus providing a mechanism for host cells to bypass the inhibitory effects of M. tuberculosis on antigen presentation and allow for effective priming of antigen-specific CD4+ T cells.
In addition to presenting antigens to T cells, DCs contribute to the cytokine environment that determines the effector phenotype of CD4+ T cells. Considerable evidence indicates that Th1-polarized CD4+ T cells are essential for control of M. tuberculosis infection, although the precise effector mechanisms they provide are uncertain (100). Infection of cultured DC+ results in secretion of IL-12p70, which drives Th1 differentiation. In addition, infected DCs produce large quantities of the IL-12p40 subunit, which is shared by IL-12 and IL-23. M. tuberculosis induction of IL-12p40 requires TLR9 signaling (101) and, following systemic infection with M. tuberculosis, mice lacking IL-12p40 show poor control of bacterial growth, low IFNγ levels, and poor recruitment of lymphocytes (102). The IL-12p40 homodimer also promotes migration of DCs from lungs to the local lymph node, a defect that is restored upon providing exogenous IL-12p40 homodimer (103). During mycobacterial infection in vivo, CD11b+ DCs are the predominant source of IL-12p40 (104). Notably, uninfected CD11b+ DC are the source of IL-12p40 in vivo during mycobacterial infection (104). In vitro studies show that, in response to M. tuberculosis infection, DCs also produce other cytokines such as IL-23, IL-1α and IL-1β, IL-6, TNF, and IL-10 that contribute to immunity (105, 106). IL-23 is required for full differentiation of Th17 effector T cells following M. tuberculosis infection. The exact role of Th17 cells in M. tuberculosis protective immunity remains unclear (107, 108), but there is evidence that their roles are bacterial strain-dependent (109). IL-10 production by DCs has been reported in vitro and in some situations, IL10-/- mice show enhanced inflammation (110), while in others, IL-10-/- mice have lower bacterial burdens (111), indicating that control of M. tuberculosis requires a fine balance with inflammation. IL-1α and IL-1β are both produced by CD11b+ DC in vivo during M. tuberculosis infection, and both are required for effective control of the bacterial growth in the lungs (32). Together, these studies indicate that DCs, as well as macrophages, are important targets of M. tuberculosis, and that these cell types make unique and shared contributions to the response to infection.
Beyond mononuclear cells: neutrophils in TB
Given the historical focus on macrophages, neutrophils have only recently received attention in TB. In humans, a low normal neutrophil count is independently associated with the risk of acquiring M. tuberculosis infection after exposure to an active case, and neutrophil antimicrobial peptides contributed to killing M. tuberculosis in vitro (112). A neutrophil-associated transcriptional signature has also been detected in the blood of patients with active pulmonary TB (84), implying that infection with M. tuberculosis imposes functional differences on neutrophils, aside from stimulating their proliferation and release from the bone marrow. Finally, in sputum, bronchoalveolar lavage fluid, and pulmonary TB cavity contents, neutrophils are a dominant cell type, implying that neutrophilic inflammation can be a manifestation of failed immunity to M. tuberculosis in humans (113).
Studies in M. tuberculosis-infected mice have revealed evidence for multiple roles for neutrophils. Consistent with the findings in sputum and lung samples that revealed a preponderance of neutrophils in the context of failed immunity, distinct strains of inbred mice that succumb to M. tuberculosis infection show a higher influx of neutrophils (114). Large numbers of neutrophils are often associated with necrotic lesions, increased pathology, and severe disease in M. tuberculosis-infected mice (56, 115). Beyond the association of neutrophils with failed immunity in TB, there is strong evidence that neutrophils contribute to the pathology and lethality in these contexts (115-117).
Contrasting with the detrimental roles of neutrophils in TB, there is also evidence that during the early stages of infection in mice, they contribute to the initiation of adaptive immune responses. Depletion of neutrophils in M. tuberculosis-infected mice leads to delayed DC transport of bacteria from the lungs to the local lymph node, further delaying priming of CD4+ T cells (118). That study also provided evidence that DCs that ingested M. tuberculosis-infected neutrophils are more efficient in migrating in response to CCR7 ligands than are DCs that ingested bacteria directly, and showed that neutrophils attract mature DCs by secreting one or more CCR7 ligands. Likewise, in zebrafish, neutrophils kill virulent M. marinum, using reactive oxygen species, after the bacteria have been released by dying macrophages (119). Taken together, these studies imply beneficial and detrimental roles for neutrophils in TB. Broader investigation of the factors that regulate and determine the specific contributions of neutrophils in protective immunity and pathology in TB is clearly warranted, to optimize host responses and decrease the morbidity and transmission of TB.
Outstanding questions and future directions
In this review, we present evidence that M. tuberculosis inhabits cells with diverse mononuclear cell phenotypes in the lungs. Since individual subsets of monocytes, macrophages, and DCs exhibit distinct functional properties, we speculate that cells in each of these subsets differ in their responses to infection with M. tuberculosis. Since the outcome of infection with M. tuberculosis varies widely, from lifelong asymptomatic latent infection, to potentially lethal transmissable disease, the diversity of infected cells, and a skewing toward dominance by one subset or another, may dictate the ultimate outcome of infection. For example, does earlier acquisition of M. tuberculosis by lung DCs result in earlier initiation of T-cell responses and superior control of infection? While modulation of cell trafficking at the onset of infection would be difficult to approach therapeutically, if distinct cell subsets differ in the intracellular environment they provide to M. tuberculosis, they may also differ in their ability to sequester the bacteria from recognition by T cells and/or by antituberculous drugs during the chronic stages of infection. Since the potential to modulate macrophage subsets for therapeutic benefit is gaining momentum for certain cancers, the possibility that a similar approach could shorten the course of antituberculous chemotherapy should also be considered. Likewise, since some TB patients suffer from lung pathology due to inflammatory responses, identification and selective modulation of specific mononuclear cell subsets might prevent long term pulmonary morbidity from scarring or lung destruction in TB.
The growing knowledge of the mediators and mechanisms of mononuclear cell trafficking in TB enables novel studies of the dynamics of cells in granulomas, which are initiated by aggregates of infected and uninfected macrophages and dendritic cells, joined later by B and T lymphocytes. While granulomas have been considered to be static structures, it is more likely that they are highly dynamic, with cells dying and/or egressing and being replaced through cell recruitment or proliferation. Since there is great interest in development of drugs that target ‘persisters’ (bacteria that survive despite the presence of drugs, without acquiring drug resistance mutations) in TB, more knowledge of cell dynamics in granulomas might enable use of drugs that target host cells and make the bacteria more vulnerable to antituberculous drugs, thereby shortening the long course of chemotherapy currently required to cure TB. As efforts to develop new TB vaccines progress, we suggest that increased understanding of the interactions of M. tuberculosis with distinct phenotypic subsets of mononuclear cells will enable more rational selection of vaccine antigens, adjuvants, and/or vectors, to assure the development of vaccine-induced T cells that can recognize and eliminate the infection. There is no shortage of questions, needs, and opportunities to combat the global problem of TB; the advances in understanding of monocytes, macrophages, and dendritic cells are sure to pave the way for development of better drugs, vaccines, and methods for modulating immunity and pathology in TB.
Fig. 2. Dynamics of M. tuberculosis spread to myeloid cell subsets in the lungs after aerosol infection.
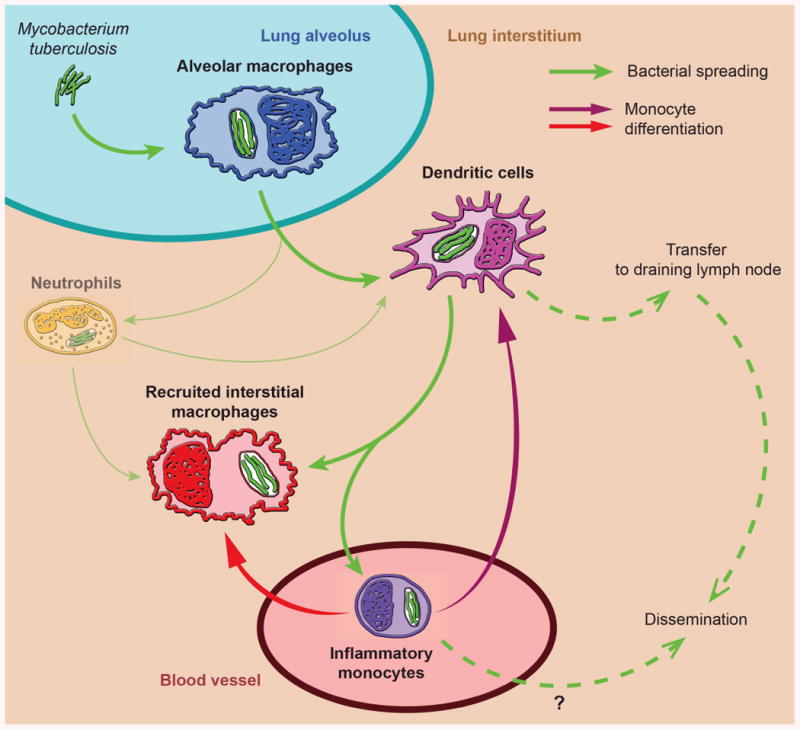
Based on their location, alveolar macrophages are likely to be the initial cells infected by M. tuberculosis after entry into the lungs. After 6-7 days of replication in alveolar macrophages, bacteria are released and ingested by newly recruited neutrophils, recruited interstitial macrophages, and dendritic cells (the latter two are both derived from monocytes). Subsequent rounds of replication in these cells support expansion of the bacterial population and promotes bacterial spread to additional phagocytic cells. Neutrophils are a transiently dominant subset of the infected cells; peaking at 17-19 days post infection; this peak is followed immediately by the peak of infected dendritic cells (see Fig. 1). Dendritic cells transport the bacteria to the local lymph node, where they produce antigens that are transferred to resident lymph node cells, which then prime naive T cells. Dissemination to other lymphoid and nonlymphoid tissues follows, and may be mediated by dendritic cells and/or monocytes.
Acknowledgments
Supported by grants from the National Institutes of Health (AI084041 and AI051242), and the Stony Wold-Herbert Foundation.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Cunningham RS, Sabin FR, Sugiyama S, Kindwali JA. The role of the monocyte in tuberculosis. Vol. 37. Bull Johns Hopkins Hosp; 1925. p. 231. [Google Scholar]
- 2.Sabin FR, Doan CA. The Relation of Monocytes and Clasmatocytes to Early Infection in Rabbits with Bovine Tubercle Bacilli. The Journal of experimental medicine. 1927;46:627–644. doi: 10.1084/jem.46.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf AJ, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. Journal of immunology. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. Journal of immunology. 2003;171:3128–3135. doi: 10.4049/jimmunol.171.6.3128. [DOI] [PubMed] [Google Scholar]
- 5.Skold M, Behar SM. Tuberculosis triggers a tissue-dependent program of differentiation and acquisition of effector functions by circulating monocytes. Journal of immunology. 2008;181:6349–6360. doi: 10.4049/jimmunol.181.9.6349. [DOI] [PubMed] [Google Scholar]
- 6.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jakubzick C, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 10.Jakubzick C, Tacke F, Llodra J, van Rooijen N, Randolph GJ. Modulation of dendritic cell trafficking to and from the airways. Journal of immunology. 2006;176:3578–3584. doi: 10.4049/jimmunol.176.6.3578. [DOI] [PubMed] [Google Scholar]
- 11.Hashimoto D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annual review of immunology. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bazan JF, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 15.Auffray C, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 16.Imai T, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 17.Sunderkotter C, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. Journal of immunology. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 18.Sadek MI, Sada E, Toossi Z, Schwander SK, Rich EA. Chemokines induced by infection of mononuclear phagocytes with mycobacteria and present in lung alveoli during active pulmonary tuberculosis. American journal of respiratory cell and molecular biology. 1998;19:513–521. doi: 10.1165/ajrcmb.19.3.2815. [DOI] [PubMed] [Google Scholar]
- 19.Flores-Villanueva PO, et al. A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. The Journal of experimental medicine. 2005;202:1649–1658. doi: 10.1084/jem.20050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7958–7963. doi: 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters W, et al. CCR2-dependent trafficking of F4/80dim macrophages and CD11cdim/intermediate dendritic cells is crucial for T cell recruitment to lungs infected with Mycobacterium tuberculosis. Journal of immunology. 2004;172:7647–7653. doi: 10.4049/jimmunol.172.12.7647. [DOI] [PubMed] [Google Scholar]
- 22.Scott HM, Flynn JL. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infection and immunity. 2002;70:5946–5954. doi: 10.1128/IAI.70.11.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antonelli LR, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. The Journal of clinical investigation. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nature immunology. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 25.Kipnis A, Basaraba RJ, Orme IM, Cooper AM. Role of chemokine ligand 2 in the protective response to early murine pulmonary tuberculosis. Immunology. 2003;109:547–551. doi: 10.1046/j.1365-2567.2003.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, Pamer EG. Essential yet limited role for CCR2+ inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife. 2013;2:e01086. doi: 10.7554/eLife.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf AJ, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. The Journal of experimental medicine. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee PY, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. The American journal of pathology. 2009;175:2023–2033. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. Journal of immunology. 2012;188:6205–6215. doi: 10.4049/jimmunol.1200255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manca C, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mariotti S, et al. Mycobacterium tuberculosis diverts alpha interferon-induced monocyte differentiation from dendritic cells into immunoprivileged macrophage-like host cells. Infection and immunity. 2004;72:4385–4392. doi: 10.1128/IAI.72.8.4385-4392.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer-Barber KD, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer-Barber KD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014 doi: 10.1038/nature13489. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall JD, et al. The impact of chemokine receptor CX3CR1 deficiency during respiratory infections with Mycobacterium tuberculosis or Francisella tularensis. Clinical and experimental immunology. 2009;156:278–284. doi: 10.1111/j.1365-2249.2009.03882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 36.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nature reviews Molecular cell biology. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 37.Tarling JD, Lin HS, Hsu S. Self-renewal of pulmonary alveolar macrophages: evidence from radiation chimera studies. Journal of leukocyte biology. 1987;42:443–446. [PubMed] [Google Scholar]
- 38.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. Journal of immunology. 2007;179:3488–3494. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 39.Repasy T, et al. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS pathogens. 2013;9:e1003190. doi: 10.1371/journal.ppat.1003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hohl TM, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell host & microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leemans JC, et al. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. Journal of immunology. 2001;166:4604–4611. doi: 10.4049/jimmunol.166.7.4604. [DOI] [PubMed] [Google Scholar]
- 42.Keane J, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infection and immunity. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. The Journal of experimental medicine. 1971;134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mwandumba HC, et al. Mycobacterium tuberculosis resides in nonacidified vacuoles in endocytically competent alveolar macrophages from patients with tuberculosis and HIV infection. Journal of immunology. 2004;172:4592–4598. doi: 10.4049/jimmunol.172.7.4592. [DOI] [PubMed] [Google Scholar]
- 45.Kwan CK, Ernst JD. HIV and tuberculosis: a deadly human syndemic. Clinical microbiology reviews. 2011;24:351–376. doi: 10.1128/CMR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. Journal of immunology. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- 47.Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell host & microbe. 2012;11:81–90. doi: 10.1016/j.chom.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velmurugan K, et al. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS pathogens. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 2009;136:37–49. doi: 10.1016/j.cell.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulsen A. Some clinical features of tuberculosis. 1. Incubation period. Acta tuberculosea Scandinavica. 1950;24:311–346. [PubMed] [Google Scholar]
- 51.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infection and immunity. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nature immunology. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen M, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. The Journal of experimental medicine. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winau F, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–117. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 55.Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. The Journal of experimental medicine. 2008;205:2359–2368. doi: 10.1084/jem.20080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srivastava S, Ernst JD. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. Journal of immunology. 2013;191:1016–1020. doi: 10.4049/jimmunol.1301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. The Journal of experimental medicine. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nathan CF, et al. Activation of human macrophages. Comparison of other cytokines with interferon-gamma. The Journal of experimental medicine. 1984;160:600–605. doi: 10.1084/jem.160.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ehrt S, et al. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. The Journal of experimental medicine. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 62.Fabri M, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Science translational medicine. 2011;3:104ra102. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thoma-Uszynski S, et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 64.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 65.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fortune SM, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. Journal of immunology. 2004;172:6272–6280. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 68.Kincaid EZ, Ernst JD. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. Journal of immunology. 2003;171:2042–2049. doi: 10.4049/jimmunol.171.4.2042. [DOI] [PubMed] [Google Scholar]
- 69.Nagabhushanam V, Solache A, Ting LM, Escaron CJ, Zhang JY, Ernst JD. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. Journal of immunology. 2003;171:4750–4757. doi: 10.4049/jimmunol.171.9.4750. [DOI] [PubMed] [Google Scholar]
- 70.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. Journal of immunology. 2004;173:2660–2668. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 71.Noss EH, et al. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. Journal of immunology. 2001;167:910–918. doi: 10.4049/jimmunol.167.2.910. [DOI] [PubMed] [Google Scholar]
- 72.Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a potential mechanism for immune evasion. Journal of immunology. 2003;171:175–184. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 73.Banaiee N, Kincaid EZ, Buchwald U, Jacobs WR, Jr, Ernst JD. Potent inhibition of macrophage responses to IFN-gamma by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. Journal of immunology. 2006;176:3019–3027. doi: 10.4049/jimmunol.176.5.3019. [DOI] [PubMed] [Google Scholar]
- 74.Kincaid EZ, et al. Codominance of TLR2-dependent and TLR2-independent modulation of MHC class II in Mycobacterium tuberculosis infection in vivo. Journal of immunology. 2007;179:3187–3195. doi: 10.4049/jimmunol.179.5.3187. [DOI] [PubMed] [Google Scholar]
- 75.Pai RK, Pennini ME, Tobian AA, Canaday DH, Boom WH, Harding CV. Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infection and immunity. 2004;72:6603–6614. doi: 10.1128/IAI.72.11.6603-6614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 77.Divangahi M, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. Journal of immunology. 2008;181:7157–7165. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 78.Gandotra S, Jang S, Murray PJ, Salgame P, Ehrt S. Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infection and immunity. 2007;75:5127–5134. doi: 10.1128/IAI.00458-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brooks MN, et al. NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cellular microbiology. 2011;13:402–418. doi: 10.1111/j.1462-5822.2010.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pandey AK, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS pathogens. 2009;5:e1000500. doi: 10.1371/journal.ppat.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell host & microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ottenhoff TH, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PloS one. 2012;7:e45839. doi: 10.1371/journal.pone.0045839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maertzdorf J, et al. Functional correlations of pathogenesis-driven gene expression signatures in tuberculosis. PloS one. 2011;6:e26938. doi: 10.1371/journal.pone.0026938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hinchey J, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. The Journal of clinical investigation. 2007;117:2279–2288. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nature immunology. 2009;10:943–948. doi: 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pan H, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434:767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hsu T, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong KW, Jacobs WR., Jr Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cellular microbiology. 2011;13:1371–1384. doi: 10.1111/j.1462-5822.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee J, Repasy T, Papavinasasundaram K, Sassetti C, Kornfeld H. Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PloS one. 2011;6:e18367. doi: 10.1371/journal.pone.0018367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Welin A, Eklund D, Stendahl O, Lerm M. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PloS one. 2011;6:e20302. doi: 10.1371/journal.pone.0020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tailleux L, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. The Journal of experimental medicine. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Srivastava S, Ernst JD. Cell-to-Cell Transfer of M. tuberculosis Antigens Optimizes CD4 T Cell Priming. Cell host & microbe. 2014;15:741–752. doi: 10.1016/j.chom.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS pathogens. 2011;7:e1002063. doi: 10.1371/journal.ppat.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, Germain RN. Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity. 2011;34:807–819. doi: 10.1016/j.immuni.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Torabi-Parizi P, Vrisekoop N, Kastenmuller W, Gerner MY, Egen JG, Germain RN. Pathogen-related differences in the abundance of presented antigen are reflected in CD4+ T cell dynamic behavior and effector function in the lung. Journal of immunology. 2014;192:1651–1660. doi: 10.4049/jimmunol.1301743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. Journal of immunology. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- 98.Olmos S, Stukes S, Ernst JD. Ectopic activation of Mycobacterium tuberculosis-specific CD4+ T cells in lungs of CCR7-/- mice. Journal of immunology. 2010;184:895–901. doi: 10.4049/jimmunol.0901230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baena A, Porcelli SA. Evasion and subversion of antigen presentation by Mycobacterium tuberculosis. Tissue antigens. 2009;74:189–204. doi: 10.1111/j.1399-0039.2009.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gallegos AM, van Heijst JW, Samstein M, Su X, Pamer EG, Glickman MS. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS pathogens. 2011;7:e1002052. doi: 10.1371/journal.ppat.1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. The Journal of experimental medicine. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. The Journal of experimental medicine. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Khader SA, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. The Journal of experimental medicine. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rothfuchs AG, et al. In situ IL-12/23p40 production during mycobacterial infection is sustained by CD11bhigh dendritic cells localized in tissue sites distinct from those harboring bacilli. Journal of immunology. 2009;182:6915–6925. doi: 10.4049/jimmunol.0900074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cooper AM. Cell-mediated immune responses in tuberculosis. Annual review of immunology. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annual review of immunology. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- 107.Cruz A, et al. Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. Journal of immunology. 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- 108.Khader SA, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. Journal of immunology. 2005;175:788–795. doi: 10.4049/jimmunol.175.2.788. [DOI] [PubMed] [Google Scholar]
- 109.Gopal R, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS pathogens. 2014;10:e1004099. doi: 10.1371/journal.ppat.1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Lee EJ, Orme IM, Gonzalez-Juarrero M. Lack of IL-10 alters inflammatory and immune responses during pulmonary Mycobacterium tuberculosis infection. Tuberculosis. 2009;89:149–157. doi: 10.1016/j.tube.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 111.Redford PS, et al. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. European journal of immunology. 2010;40:2200–2210. doi: 10.1002/eji.201040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martineau AR, et al. Neutrophil-mediated innate immune resistance to mycobacteria. The Journal of clinical investigation. 2007;117:1988–1994. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eum SY, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Eruslanov EB, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infection and immunity. 2005;73:1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. The Journal of experimental medicine. 2011;208:2251–2262. doi: 10.1084/jem.20110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dorhoi A, et al. The adaptor molecule CARD9 is essential for tuberculosis control. The Journal of experimental medicine. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nouailles G, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. The Journal of clinical investigation. 2014;124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. Journal of immunology. 2011;186:7110–7119. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell host & microbe. 2012;12:301–312. doi: 10.1016/j.chom.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]