

Summary
Macrophages play essential roles in tissue homeostasis, pathogen elimination, and tissue repair. A defining characteristic of these cells is their ability to efficiently adapt to a variety of abruptly changing and complex environments. This ability is intrinsically linked to a capacity to quickly alter their transcriptome, and this is tightly associated with the epigenomic organization of these cells and, in particular, their enhancer repertoire. Indeed, enhancers are genomic sites that serve as platforms for the integration of signaling pathways with the mechanisms that regulate mRNA transcription. Notably, transcription is pervasive at active enhancers and enhancer RNAs (eRNAs) are tightly coupled to regulated transcription of protein-coding genes. Furthermore, given that each cell type possesses a defining enhancer repertoire, studies on enhancers provide a powerful method to study how specialization of functions among the diverse macrophage subtypes may arise. Here, we review recent studies providing insights into the distinct mechanisms that contribute to the establishment of enhancers and their role in the regulation of transcription in macrophages.
Keywords: epigenetic, macrophage, enhancer, eRNA, transcription, PU.1
Introduction
Macrophages are fundamental effectors of the innate immune system (1). These cells participate in a wide spectrum of biological processes in metazoan organisms, making critical contributions not only to defense against pathogenic agents but also to developmental aspects of the body and the preservation of tissue homeostasis. Further attesting to their physiological importance is the observation that macrophages appear to play an inherent role in essentially all known biomedical diseases including cancer, metabolic disorders, neurological dysfunctions, etc. Therefore, understanding their development and functions and how these are regulated constitute one of the great challenges in modern medical research.
Macrophages are among the most transcriptionally dynamic cell types, capable of rapidly and dramatically altering their transcriptional output to quickly adapt to abruptly changing environments, as it occurs during infections or tissue injuries. Furthermore, each subtype of macrophage fulfills dedicated and specialized functions relative to the other members, as illustrated, for example, by the role of microglia in modulating synaptogenesis (2) and that of spleen macrophages in the phagocytosis of erythrocytes (3). Yet the overall gene expression profile across the different macrophage populations exhibits, nonetheless, a substantial amount of similarities (4). Thus, a subtype ‘specialization’ seems to arise from a particular combination of expression or repression of a relatively small subset of genes. This is rather exceptional given the fact that macrophages develop under very different contexts and sometimes from different precursor cells (5-10). From a transcriptional perspective, such ‘substantial similarities’ versus ‘specialized differences’ is rather intriguing, as macrophages, for a given individual, share the same genome and express to a great extent the same array of transcription factors (TFs).
Epigenomics provides an incredibly efficient paradigm to study how transcriptomes can vary from one cell type to another or across time under changing environments for the same cell type. The term ‘epigenome’ encompasses all the features associated with chromatin parameters, including DNA methylation state, covalent modifications of histone proteins, and the overall 3D architecture of the genome in the nucleus, etc., that participate in the optimization of gene regulation of that given cell (11). Thus, a cell's epigenome is intrinsically linked to the structural and spatial organization of its genome, giving it a unique footprint signature, and is ultimately reflected by the global pattern of gene expression.
One of the great breakthroughs in epigenomics over the past decade has been the development of massively parallel sequencing-based assays that allow quantitative measurements of transcription factor binding, histone modifications, DNA methylation states, and nascent RNA transcripts on a genome wide level. Application of these methods has enabled the correlation of combinations of specific histone modifications with promoters and distal transcriptional regulatory elements termed enhancers. As discussed in further detail below, enhancers were found to greatly outnumber promoters in the genome, and to be the most abundant binding sites for signal-dependent transcription factors (SDTFs), like NF-κB. Although enhancers had long been implicated in gene regulation, the recognition that mammalian genomes contain hundreds of thousands of enhancer-like regions, significantly altered our conception of gene regulation, which had until then been rather promoter-centric. Further of note, the repertoire of enhancers for a given cell type is largely specific to that cell type, implicating that differential gene expression between two different cell types can be attributed potentially to differential configuration of their respective enhancer repertoire (Fig. 1). Thus, these two insights provide strong pillars for the elaboration of a conceptual framework to understand the mechanisms responsible for macrophage transcriptional dynamism, similarities, and differences. This review summarizes recent advances in epigenomics, with a focus on enhancers, and how this paradigm has contributed to furthering our understanding of transcription in macrophages.
Fig. 1. Cell type-specific selection of enhancers.
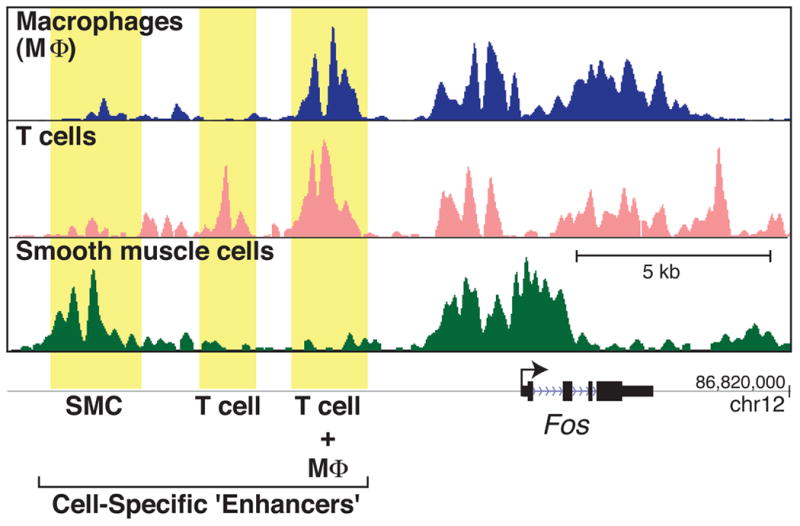
Depiction of H3K4me2 ChIP-seq data obtained from mouse macrophages, T cells, and smooth muscles cells (SMC) in the vicinity of the Fos gene genomic locus. In macrophages, H3K4me2 peaks reveal an enhancer located 5kb upstream of the Fos transcription start site (TSS). This enhancer is also established in T cells but not in SMCs. On the other hand, T cells and SMCs each display a unique enhancer at 7kb and 10kb respectively. H3K4me2 marks are also present at promoters and may extend beyond the TSS at highly transcribed genes, reflecting high levels of transcriptional activity by RNAP II. Note that spacing between H3K4me2 peaks at enhancers is indicative of absence of nucleosome and concords instead with the binding of TFs.
Enhancers possess a defining molecular signature
Enhancers are defined as DNA sequences that ‘enhance’ transcription from promoters. Enhancers can be located upstream, downstream, or within the gene body with respect to the location of the transcriptional start site (TSS) of the protein-coding or non-coding gene that they regulate. Whether or not different positioning relative to the TSS confers different functions to enhancers is not known, however. Functional binding of transcription factors to DNA at enhancers promotes recruitment of general co-activators or co-repressors that typically do not interact by themselves directly with DNA, and the summation of the activity of all the recruited proteins at a given time will determine the ability of the enhancer to modulate transcription. In the canonical sense, enhancers are considered to consist of the set of binding sites required for the binding of the sequence specific TFs. However, the recent findings that enhancers are pervasively transcribed and in at least some cases produce enhancer RNAs (eRNAs) that contribute to enhancer functions require consideration of the minimal functional unit of an enhancer to include its eRNA products. Finally, note that a subset of genes within a cell, often linked to that cell's identity, are associated with clusters of enhancers termed “locus control regions” (LCRs), such as those that control globin gene expression (12). More recently, genomic regions exhibiting high densities of enhancers have been given the term super-enhancers (13, 14).
The molecular features of enhancers include absence of DNA methylation and depletion of nucleosomes at sites bound by TFs. The nucleosomes that are adjacent to transcription factor binding sites are associated with characteristic post-translational modifications (PTMs). With regards to this latter characteristic, nucleosomes at enhancers possess relatively high levels of histone H3 lysine 4 monomethylation (H3K4me1) and low H3K4me3 (15). Note that an opposite ratio of these marks is characteristic of promoters, and that H3K4me2 labels both enhancers and promoters (16) (Fig.1).
Nucleosomal PTMs may function as scaffolds or beacons for factors, sometimes referred to as ‘readers’, that will ensure optimal assembly of TFs-co-activator/repressors. For instance, unmodified H3K4 interacts favorably with repressors proteins, including Dnmt3L, an essential cofactor for de novo DNA methyltransferase Dnmt3a/b, and Uhrf1, a factor required for substrate recognition by the maintenance methyltransferase Dnmt1 (17, 18). Given that DNA methylation represses transcriptional activity (19), H3K4 methylation may interfere with Dnmt3L binding, thus preventing enhancers from being targeted for repression by DNA methylation (20). On the other hand, given the number of proteins that can ‘read’ and interact with H3K4me1, it is possible that this specific PTM plays a more pro-active role in enhancer biology. However, this possibility has yet to be supported experimentally.
The observation that H3K4me1High/H3K4me3Low regions predicts enhancers, coupled with chromatin immunoprecipitation with massively parallel sequencing (ChIP-Seq) has greatly facilitated efforts to identify enhancers genome-wide and has been applied to numerous cell types already.
In mouse macrophages, this approach identifies between 35,000 and 45,000 enhancers (21, 22). In comparison, these numbers exceed that of promoters associated with protein-coding genes, which altogether genome-wide potentially amount to 26,000 in mice, although only fraction of these are active or poised in any given cell type (23). Importantly, macrophage enhancers exhibit strong enrichment for sequences recognized by TFs relevant to macrophage ontology and functions. In particular, the motif recognized by ETS family member PU.1 is present in most macrophage enhancers, and PU.1 binding sites genome-wide, or its cistrome, largely coincide with H3K4me1High/H3K4me3Low regions. Other motifs commonly found within enhancer regions in macrophages include motifs associated with C/EBP family members, AP-1, IRF, and NF-κB factors (21, 22, 24). These co-occurring motifs signify that enhancers provide sites for the integration at the genomic level of TFS regulated by cell intrinsic and/or extrinsic signals, and that interactions between TFS and DNA are more likely to occur at enhancers than at promoters. This feature constitutes an important effector mechanism by which enhancers can in turn participate in the optimal synchronization of transcriptional output in accordance with cellular activity and metabolism. Indeed, a single promoter can be influenced by a variety of different combinations of enhancers that possess different configurations of SDTFs motifs. Given that these latter are intrinsically linked with signaling pathways, this integrating system thus provides great flexibility to tune the expression of a gene according to the particular demand encountered by the cells in a context-dependent manner.
Enhancers are cell type specific transcriptional units that optimize mRNA transcription
Tremendous progress has been achieved over the past few years in our understanding of how the right combination of interacting TFs and co-activators activates an enhancer, and how this in turn translates into whether gene transcription is altered. In particular, recruitment of co-activators that possess histone-modifying enzymatic capabilities appears to be a prevalent feature associated with enhancer activation. Indeed, histone-acetyl-transferases (HATs), including P300, and/or CREB-binding protein (CBP), are present at active enhancers or are quickly recruited to enhancers shortly after they become activated following functional SDTFs binding to DNA (15, 22, 25). These HATS catalyze the acetylation of histone H3 at lysine K27, and H3K27Ac correlates with active enhancers and promoters (16, 26-28). In addition, P300 and CBP, and other HATs including GCN5 and PCAF, catalyze acetylation of H3K9, K14, K18, and H4K5, K8, and K12, which also correlate with active transcription (29-31). Based on these characteristics, genomic regions characterized by high levels of H3K4me1 but low levels of histone acetylation are considered poised, while regions characterized by the combination of high levels of both H3K4me1 and histone acetylation are considered active (12).
Enhancers can be inactivated or repressed by histone tail de-acetylation, histone lysine methylation, and/or H3K4 demethylation. For example, recruitment of NCoR/SMRT co-repressor complexes that contain histone deacetylase 3 (HDAC3) results in active histone tail de-acetylation (32-34). The histone methyltransferase EZH2 present in the polycomb complex trimethylates H3K27, resulting in transcriptional repression (35-37). Removal of the H3K27 trimethylation mark by the histone demethylases JmjC-domain protein JMJD3 and UTX is required for full activation of a large subset of genes that are induced by Toll-like receptor 4 (TLR4) signaling in macrophages (38, 39). As a final example, the histone lysine demethylase LSD1 is capable of demethylating H3K4me1 and H3K4me2, providing a mechanism for the ‘decommissioning’ of enhancers during cellular differentiation (40). These observations provide evidence for a complex and dynamic choreography of TFs, nucleosome remodeling factors, and histone writers, readers, and erasers at enhancers.
The precise molecular consequences of histone acetylation at active enhancers remain incompletely understood. Acetylated lysine residues are recognized by bromodomains present in diverse nuclear proteins including many HATs themselves, various ATP-dependent chromatin remodelers, general transcription factors (GTFs) of the RNA polymerase II (RNAP II) complex, and factors regulating transcriptional pause release (20, 41). In particular, H3 and H4 acetylation promotes interactions with bromodomain-containing 4 protein (BRD4), which then catalyzes recruitment of transcription initiation cofactors forming the Mediator complex and positive transcription elongation factor b (P-TEFb) (42, 43). These latter two elements are rather intriguing, given that they regulate, respectively, pre-initiation complex assembly and elongation of the RNAP II complex downstream of transcriptional start sites (TSS). Particularly, recruitment of Mediator is significant given (i) the positive correlation between gene expression and frequency of promoter-enhancer interactions (44), and (ii) that Mediator directly contributes to enhancer-promoter chromosomal looping (45). Note, however, that though chromosomal looping between an enhancer and a target promoter is considered to be required to mediate enhancer functions, exactly what it entails and how this is achieved molecularly is not completely understood. All in all though, histone acetylation at enhancers likely plays an active, necessary role to allow for the recruitment and control of the RNA polymerase machinery complex at enhancers and promoters of protein-coding genes.
Strong evidence suggests an important role for RNAP II activity at enhancers. RNAP II complexes are enriched at enhancer elements and respond dynamically to signal transduction events (46-49). In addition, early studies reported the detection of RNA transcripts originating from the LCR of the β-globin gene clusters (50-52). More definitive evidence for the pervasiveness of an active role of RNAP II at enhancers was later revealed by a study reporting RNA transcripts derived from ∼2,000 extragenic enhancers in neurons (48). Finally, genome-wide detection of nascent RNA transcripts still associated with RNAP II using global run-on sequencing (GRO-Seq) demonstrated robust expression and regulation of eRNA in macrophages, breast, and prostate cancer cells (47, 49, 53). Thus, enhancer transcription is a widespread phenomenon observed across multiple cell types in different species.
eRNAs modulate transcription of target protein-coding genes
eRNAs are a recently defined species of non-coding RNAs that are derived from active enhancers. All appear to have 5′ cap, but the majority do not seem to be either spliced or polyadenylated. Likely as a result of lack of polyadenylation, eRNAs are relatively unstable and have short half-lives compared to mRNAs and long non-coding RNAs (lncRNAs). Note, however, that the frequency of transcription initiation of eRNAs appears comparable to that of mRNAs (54).
eRNAs are dynamically regulated upon signal transduction events orchestrated by SDTFs including the androgen, estrogen, and peroxisome-proliferator activated receptor γ (PPAR-γ) nuclear receptors and NF-κB (16, 47, 49, 55). In addition, signal-dependent changes in eRNA expression levels are highly correlated with corresponding signal dependent transcriptional changes of nearby genes. Along the same lines, enhancer–promoter interactions positively correlate with eRNA transcription, such that enhancers that interact more with promoters of protein-coding genes through chromatin looping display higher abundance of the associated of eRNA transcripts. Conversely, decreasing eRNA transcription is associated with decreased corresponding mRNA transcription. Indeed, we recently identified that the Rev-Erb nuclear receptors repress gene transcription in mouse macrophages, including Mmp9 and Cx3cr1, by inhibiting eRNA transcription at their respective enhancers (53). These enhancers possess Rev-Erb binding motifs, and binding of Rev-Erbs to these sites presumably recruits the NCoR-HDAC3 complex (56, 57). Thus, in the absence of the Rev-Erb factors, the Mmp9 and Cx3cr1 enhancers become de-repressed, which translates into an aberrant increased transcription of Mmp9 and Cx3cr1 mRNAs.
The correlative nature of the evidence presented above regarding eRNAs and mRNA prevents causative claims as to whether eRNAs actually fulfill relevant biological functions. Although eRNAs per se could conceivably contribute to enhancer activity in promoting gene transcription, their existence could also be a by-product of molecular events associated with RNAP II complex activities and that these events are the actual contributing effectors of enhancers to gene transcription. In that case, it may be that the process of transcription initiation and/or elongation per se at enhancers, or events related to them, are necessary for the activating functions of enhancers. Under this scenario, eRNAs could be considered merely as ‘noise’ from spurious engagement of the RNAP II complex.
Several independent groups have recently addressed these hypotheses experimentally and demonstrated that at least some eRNAs in themselves play a fundamental role in regulating the transcription of associated protein-genes (53, 58-60). For instance, targeted degradation of eRNAs using either small interfering RNA or anti-sense oligonucleotides led to significant reduction of nearby protein-coding genes (53, 58). In addition, in a tethering system in a reporter assay, using actual eRNA sequences as bait increased transcriptional activity of the reporter gene (58, 59). Importantly, inverted control sequences did not provide any potentiating effect. Furthermore, we reported evidence suggesting that different components of an enhancer might contribute differentially to its overall activity (53). By cloning different sizes of genomic fragments corresponding to endogenous enhancer loci active in murine macrophages, we showed that although the ‘core’ element of enhancers, which include lineage-determining transcription factors (LDTFs) binding sites, conferred in themselves potentiating effect in the reporter assay, constructs that also included eRNA sequences promoted even higher transcriptional activity. Importantly, reversing the coding sequence of the eRNA sequence relative to the core abolished this ‘enhancing’ effect. Because this inversion completely changes the sequence of the eRNA product but retains all the putative TF binding sites, these results imply that the sequence of an eRNA is important for its function. In sum, these reports suggest that sequence-specific eRNA transcripts can contribute to enhancer-mediated transcriptional activation of neighboring protein-coding genes.
From a functional standpoint, numerous studies suggest that eRNAs can modulate transcription of protein-coding genes through various epigenomic mechanisms including chromatin remodeling at promoters and chromosomal loopings between enhancers and promoters. With respect to the former, depletion of eRNAs decreased chromatin accessibility at promoters of target genes, leading to reduced RNAP II recruitment and lower levels of mRNA transcripts (61). Exactly what led to these reductions however remains unknown. On the other end, accumulating evidence from independent groups indicates that eRNAs may promote enhancer-promoter chromosomal loopings (59, 60). Indeed, treating human breast cancer cells with estrogen receptor α (ERα) ligand 17β-estradiol induces formation of chromatin looping between activated enhancers and adjacent promoters including NR1P1 and GREB1 (59). Targeted degradation of eRNAs associated with these enhancers reduced enhancer-promoter interactions and decreased transcription of the gene downstream of the corresponding promoter. Interestingly, Li et al. (45) noted that the presence at enhancers of the cohesin complex, which along with the Mediator complex regulates enhancer-promoter chromosomal looping in stem cells, increases as enhancers become activated. Of note, eRNAs can interact with subunits of the cohesin complex, and decreasing eRNAs lowers cohesin recruitment to enhancers. This suggests that interactions of eRNAs with cohesin may promote and/or stabilize the presence of the latter at enhancers (Fig. 2).
Fig. 2. eRNAs contribute to functional chromosomal looping between enhancers and promoters to increase mRNA transcription.
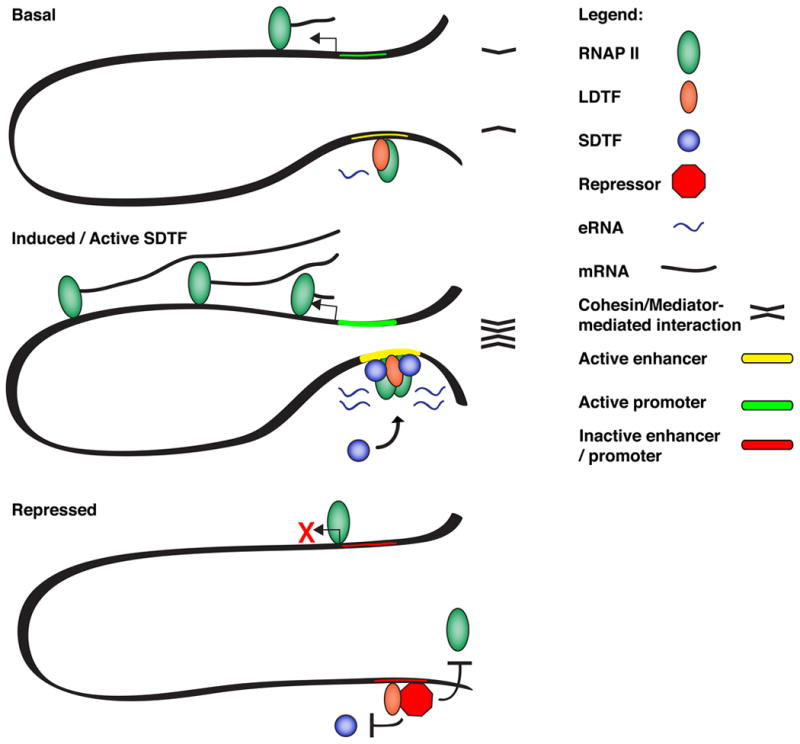
Under basal conditions, low level of constitutive activity promotes RNAP II activity at both a distal enhancer downstream of a gene and at the protein-coding gene. In this context, eRNAs promote enhancer-promoter interactions through cohesin/Mediator complex activity. Strong stimulation of a target SDTF leads to its efficient binding at the enhancer and increased transcription of eRNAs, leading to increase enhancer-promoter interactions strengths and/or frequencies, resulting in more transcription of the protein-coding gene. However, recruitment of a repressor complex at the enhancers interferes with SDTF binding and/or local RNAP II activity. Lower eRNA abundance decreases enhancer-promoter interactions, which correlates with lower mRNA transcription.
Hsieh et al. (60) also reported evidence that eRNAs regulate enhancer-promoter interactions through chromosomal looping. In prostate cancer cells, the androgen receptor (AR) increases transcription of the Kallikrein-related peptidase 2 (KLK2) gene by activating an upstream enhancer known as KLK3. Enhancer RNAs originating from KLK3 immunoprecipitate with both AR and Med1 subunit of the Mediator complex, and KLK3 eRNA silencing decreases the AR-Med1 interaction, resulting in a reduction of both enhancer-promoter interactions and transcription of KLK2. Importantly, eRNAs degradation did not impact the presence of AR or RNAP II at the enhancer, suggesting that eRNA inhibition does not lead to overall collapse of enhancer functional features.
Although the evidence presented above supports the hypothesis that eRNAs promote protein-coding gene transcription by catalyzing enhancer-promoter chromosomal loopings, some discrepancies exist. Indeed, reducing eRNAs by chemically inhibiting elongation of RNAP II with flavopiridol did not decrease estradiol-induced enhancer-promoter loopings between the promoter of either P2RY2 or GREB1 and their corresponding enhancers (62). Note however that as mentioned earlier, it is not clear exactly when or why chromosomal loopings become functional. Thus, there might be a critical aspect to functional enhancer-promoter chromosomal looping that may extend well beyond the sole detection of the occurrence of a physical interaction between an enhancer and a promoter. Therefore, although studies that used targeted degradation of eRNAs do suggest that eRNAs promote enhancer-promoter loopings, their actual enhancing effects on protein-coding gene transcription may very well be downstream of this event. Note also that as mentioned above, eRNAs also promote chromatin accessibility at promoters (61), which hypothetically may be one downstream effect of functional enhancer-promoter looping. In this context, using flavopiridol to assess the role of eRNAs on protein-coding gene transcription becomes problematic because it will also interfere with RNAP II elongation over the gene body. Therefore, this method of eRNA inhibition is less sensitive than targeted eRNA degradation to assess the possible role of eRNAs on mRNA transcription.
Specifying the enhancer repertoire in macrophages: role of lineage-determining factor PU.1
Each cell type possesses a fundamental enhancer repertoire that will give rise to a canonical transcriptional signature in a given context. Lineage-determining transcription factors (LDTFs), or pioneer factors, are a class of TFs that play an essential role in the establishment of the fundamental enhancer atlas characteristic of a given cell type. LDTFs are relatively highly expressed and their interaction with DNA does not seem to be as dynamic and subject to abrupt fluctuations as that of SDTFs, like the RELA/p65 subunit of NF-κB or the SMAD TFs for example. Expression and/or activity of LDTFs seem to be intrinsically linked with fundamental cytokines and factors known to promote the ontology of the associated cell type. Importantly, LDTFs can presumably interact with their DNA recognition elements in the context of closed nucleosomal DNA, decompact chromatin, and reposition nucleosomes (63, 64). Note however that whether this is accomplished in a direct or indirect manner, by recruiting chromatin remodeling enzyme for example, is not completely understood. Nonetheless, LDTFs provide a nuclear effector mechanism used by signals relevant to a cell's lineage to reconfigure the epigenome of a precursor cell such that the transcriptional output of the differentiated cell is optimized to allow it to efficiently carry on the cell's purpose and functions. Numerous LDTFs have been identified, and include FOXA1, GATA family members, among others (65, 66).
Multiple lines of evidence suggest that PU.1 is an essential LDTF for macrophages. At the cellular level for example, macrophages are completely absent in mice deficient for PU.1 (5, 67, 68). Also of note, signaling pathways downstream of CD115, the receptor for cytokines macrophage colony-stimulating factor (M-Csf) and interleukin-34 (IL-34) and whose activity is absolutely required for proper macrophage development and functions in vivo, directly modulate transcription of the Spi1 gene that encodes the PU.1 protein (69). Importantly, however, we provided direct molecular evidence that PU.1 has potent epigenomic remodeling activity genome-wide (21). Using a PU.1-ER fusion protein system to control PU.1 nuclear translocation with tamoxifen, in a context where endogenous PU.1 was knocked out, extensive nucleosome repositioning and chromatin opening at sites bound by PU.1 occurs genome-wide within an hour of tamoxifen treatment. De novo H3K4me1 deposition, which was previously absent at those sites, was gained locally. These regions exhibited enrichment for motifs recognized by other TFs relevant to macrophage differentiation. Importantly, these nascent enhancers localized to genomic regions previously identified as enhancers in an unrelated macrophage primary cell line. Similar events occur following ectopic expression of PU.1 in mouse fibroblast cell line 3T3 (22). Another strong indication of a role for PU.1 as a macrophage LDTF is that its loss in differentiated macrophages results in a decrease of H3K4me1 at several enhancers previously bound by PU.1 (22). On the other hand, note that decreasing the expression levels of nuclear receptors LXRα and β, which are critical regulators of phagocytosis and inflammatory pathway checkpoints in macrophages, does not impact PU.1 binding or H3K4me1 deposition (21). Thus, absence of effects of these SDTFs on chromatin features associated with enhancers in this context illustrates a hierarchical order to the process of enhancer selection and function, in which LDTFs frequently act as initiators of enhancer selection, enabling subsequent binding of SDTFs and conversion of a poised enhancer to an active enhancer. Together, these observations led to the elaboration of a model of macrophage ontology whereby PU.1, acting as a LDTF, specifies a fundamental epigenomic landscape for macrophages that can then be used by SDTFs to integrate a diverse array of signals with gene transcription.
An important point with regards to the establishment of enhancers relates to the molecular mechanisms triggered by PU.1 and other LDTFs that lead to structural changes in local chromatin and culminate with mono- and di-methylation of H3K4. A subset of the family of histone methyltransferases (HMTs) has been demonstrated to catalyze mono- and di-methylation of H3K4 at enhancers in various different cellular types, including mouse embryonic fibroblasts, cancer cell lines, adipocytes, and myocytes (70-72). However, the mechanisms by which these HMTs are recruited to enhancers to methylate H3K4 have not been established. On the other hand, studies of a set of de novo formation of enhancers in macrophages that occurs following TLR4 activation suggests that H3K4me1 and H3K4me2 deposition is linked to enhancer transcription itself. Tracking chromatin state and transcriptional changes associated with the formation of these new enhancers in this particular context indicated the following temporal sequence of events during the selection of an enhancer: (i) SDTFs and PU.1 bind in a collaborative manner to unmarked chromatin and lead to nucleosomal displacement, (ii) subsequently, histone H4K5/8 acetylation and eRNA transcription occurs, which is followed by (iii) progressive mono- and di-methylation at H3K4 by HMTs H3K4 by mixed lineage leukemia 1 (Mll1), Mll2/4, and Mll3. These observations, in addition to positive correlations between enhancer transcription and H3K4 methylation, both in terms of quantity and spatial length of the transcribed region, raised the possibility of a functional relationship between enhancer transcription and H3K4 methylation at de novo enhancers. Consistent with this hypothesis, inhibition of RNAP II initiation or elongation using a variety of different inhibitors reduced local H3K4 methylation at TLR4-induced enhancers. Similar positive correlations also exist between increased transcription and higher H3K4 methylation at pre-existing enhancers and promoters activated downstream of TLR4 signaling. Targeted degradation of eRNAs did not abrogate the enhancer formation process, thus suggesting that the transcription process and not the RNA product is the important effector. These findings are consistent with a sequence of events in which histone acetylation results in the recruitment of the P-TEFb complex via its bromodomain-containing BRD4 component. P-TEFb then phosphorylates the C-terminal domain of RNAP II, which provides docking sites for members of the Mll family of H3K4 methyltransferases. The loading of Mlls to the CTD of Pol II thereby provides an explanation for how they are recruited to enhancers and for the progressive accumulation of H3K4me1 and H3K4me2 during successive rounds of enhancer transcription.
Substantial evidence suggests that selection of signal-dependent enhancers is linked with active transcription and methyltransferase activity. Studies of enhancer methylation during differentiation of adipocytes and muscle cells suggested an alternative mechanism in which Mll3 was recruited to newly formed enhancers by LDTFs themselves and preceded histone methylation (72). It will therefore be of interest to determine whether there are general differences in the mechanisms by which newly selected enhancers acquire their H3K4 enhancer signature depending on the nature of the specific factors involved.
Factors regulating enhancer selection in macrophages
An important question with respect to the ability of PU.1 to establish the macrophage enhancer repertoire pertains to the original mechanisms that lead to the initial selective binding of PU.1 genome-wide. To this end, nucleotide sequences of the DNA are, not surprisingly, critically involved, as PU.1 is more likely to interact with an efficient functional outcome with DNA sequences that match its DNA binding motif than sites that do not (73). Indeed, mutations in the vicinity of potential PU.1 binding sites impair the ability of PU.1 to interact efficiently with DNA and to promote di-methylation deposition at H3K4 histones. On the other hand, mutations in motifs recognized by SDTFs do not impair the establishment of the enhancer as defined by its methylation state.
Given the size of the mouse genome, there are potentially between 650,000 and 1.4 million sites where PU.1 can bind DNA on a spectrum of different stringencies (73, 74). Yet, as mentioned earlier, about only up to 45,000 of those are selected in differentiated macrophages. Thus, a simple DNA sequence – PU-1 interaction model cannot account by itself for the PU.1 cistrome in macrophages. How such stringent selection is achieved is not completely understood, as is the equally important mechanism that prevents PU.1 from forming enhancers at sites irrelevant to macrophage functions. Thus, additional layers of regulations must be at play to optimize PU.1 activity as a LDTF.
PU.1 collaborates with other transcription factors and nucleosomes to establish the macrophage enhancer repertoire
Collaborative activity of PU.1 and C/EBP factors in specifying enhancers in thioglycolate-elicited macrophages
Evidence suggests that a tremendous amount of additional information encoded in DNA, extending well beyond the presence of a PU.1 recognition motif, influences the binding behavior of PU.1 to DNA. Among these, two deserve particular attention: close co-localization of the Pu.1 ETS motif with motifs recognized by other TFs that collaborate with PU.1, including C/EBP and AP-1 factors, and co-occurrence of PU.1 and collaborator motifs within sequences favoring specific nucleosomal placement.
Computational analysis of DNA motifs in the vicinity of PU.1 peaks in thioglycolate-elicited macrophages (TGEMs) revealed strong enrichment for TFs C/EBP and AP-1 (73). ChIP-Seq data also suggests that many of these motifs are biologically relevant, as over 13,000 peaks for C/EBPα or C/EBPβ occur within 100bp of PU.1-bound sites in TGEMs. Thus, about a third of the PU.1 cistrome intersects with C/EBP factors.
We recently provided strong evidence that this genetic information is used in a functional, collaborative manner by PU.1 and C/EBPα to establish functional enhancers. In this study, natural genetic variation between C57BL6/J and BALB/cJ mouse strains was leveraged to determine how genetic variation influences binding patterns of LDTFs. Analyses revealed that a mutation in one strain within regions where a PU.1 peak is detected in the other strain not only interferes with PU.1 binding in the mutant strain but also abrogates nearby C/EBPα binding within the enhancer. A mutation within C/EPBα peaks had similar effects on PU.1 binding. Importantly, both types of mutations negatively affected histone modifications H3K4me2 and H3K27Ac. On the other hand, mutations in SDTF motifs like NF-κB only impaired RELA/p65 binding at this site following TLR4 stimulation but had no effect on the binding behavior of PU.1 or C/EBPα.
Cellular studies also indicated that there is a close synergistic relationship between PU.1 and C/EBP factors. Indeed, while ectopic expression of PU.1 in fibroblastic cell line 3T3 induced upregulation of the myelomonocytic marker Mac-1 in 35-40% of these cells, co-expression of C/EBPα increased the number of Mac-1 positive cells to 90% (75). Interestingly, sole expression of C/EBPα alone could not upregulate Mac-1. Similarly, Hela cells acquire a monocytic-like phenotype and responsiveness to lipopolysaccharide (LPS) only following induced co-expression of both PU.1 and C/EBPα (76).
Although we have until now primarily considered LDTFs and SDTFs to be distinct factors and to operate at different levels of the hierarchy of enhancer selection and function, this distinction is clearly blurred for several classes of SDTFs. NF-κB provides one important example. Although the vast majority of NF-κB binding that occurred following TLR4 activation of macrophages occurred at pre-existing enhancers, consistent with the actions of SDTFs, about 10% of the binding occurred at de novo enhancers. At these locations, PU.1 binding was dependent on NF-κB and NF-κB binding was dependent on PU.1, consistent with NF-κB functioning as a LDTF. Interestingly, C/EBPα also possesses features of both LDTFs and SDTFs. It is indeed necessary to granulopoieis, and its activity is modulated by ERK activity downstream of the receptors for G-Csf (CD114), M-Csf, and FLT3 (CD135), which are all critical regulators of the hematopoietic stem cell system (77-80). Finally, although much less explored, a similar picture is expected for members of the AP-1 family of transcription factors. CD115 also regulates the activity of c-Fos transcription factor, which is a prevalent member of the AP-1 heterodimer that shares, like C/EBP, strong co-localization with the PU.1 binding motif (79). Although hypothetical, activation of c-Fos, and possibly C/EBPα, by CSF-1 receptor signaling would bring PU.1 binding behavior under the immediate influence of CD115, thus coupling a fundamental signaling pathway in macrophages to their fundamental LDTF. This could ensure that the establishment of the macrophage enhancer repertoire is properly synchronized with the physiological environment in which it develops and/or functions.
Nucleosomes mediate collaborative activity of PU.1 with other transcription factors
The data reported by Heinz et al. (73) integrates elegantly with a second important type of information encoded in DNA that influences LDTFs binding pattern, namely sequence specifying nucleosomal placement. Evidence suggests that nucleosome placement within chromatin is not random in all instances, with certain nucleotide sequences increasing the probability that a nucleosome will be located more frequently over a specific region of DNA (81). In addition, a current hypothesis in epigenomics proposes that nucleosomal DNA promotes synergistic binding between TFs, which in turn leads to more efficient displacement or eviction of the nucleosome than if only one transcription factor is present (82-85). Of interest, this hypothesis does not require protein-protein interactions between PU.1 and CEBPα. This concords well with the observation that these two factors seemingly do not interact directly with each other through protein-protein interactions and analyses of DNA sequences at co-bound sites suggest that they cannot physically interact when they are separated by more than 20 nucleotides (86). Although chromatin looping could potentially bring the two factors together, no evidence suggests that this occurs efficiently in macrophages over distances of less than 80 -100 base pairs (bp) (86). Note however that the binding distance between PU.1 and C/EBP cannot be too far apart either, as the collaborative model hypothesis imposes a possible upper limit in the range of a nucleosomal length of nucleotides (i.e. 147 bp) (82, 83, 87).
Recent work provided evidence that the PU.1 binding pattern might be intrinsically linked to some extent with nucleosome location in macrophages in many instances (74) (Fig. 3). Indeed, cell types that do not express PU.1, including embryonic stem cells, neural precursors, and mouse embryonic fibroblasts, exhibit high nucleosomal occupancy at enhancer sites bound by PU.1 in macrophages relative to sites not active in macrophages but that nonetheless possess a PU.1 motif. Interestingly, these regions of the genome displayed a particular set of characteristics with regards to their DNA sequences that appear to promote co-occurrence of both PU.1 binding in macrophages and nucleosomal occupancy in non-expressing PU.1 cells. These DNA parameters are comprised of the inclusion of motifs for TFs known to collaborate with PU.1 and overall G+C content as well as factors previously known to influence nucleosomal placement such as configuration of CG/GG/CC and AT/TA dinucleotides (i.e. nucleosomal container) (81). Another factor, the DNA shape immediately surrounding the ETS core motif recognized by PU.1, influenced only PU.1 binding and not nucleosomal occupancy. Indeed, competitive EMSA revealed that mutating two nucleotides flanking the ETS core motif decreased a competing DNA sequence's ability to pull away PU.1 from binding its site in nuclear extract, but had no effect on nucleosomal arrangements. These results suggest that particular DNA sequences contribute extensively to the specification of the chromatin molecular environment, which in turn directly affect the propensity for PU.1 binding that leads to efficient opening of chromatin, nucleosomal depletion, and enhancer formation in macrophages.
Fig. 3. Collaborative model of epigenomic lineage determination in macrophages.
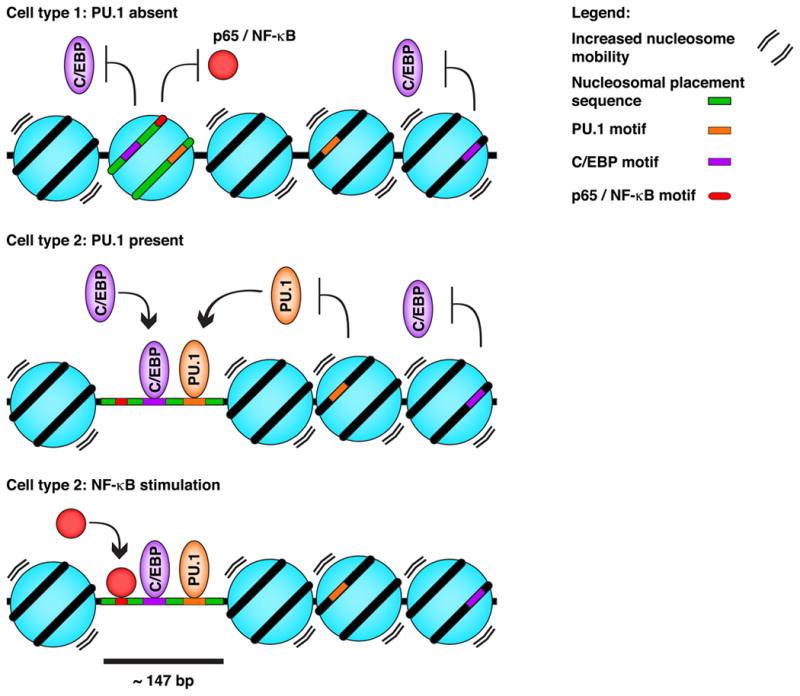
Co-occurrences of binding sequences for PU.1 and collaborative TFs, including C/EBP factors, within a ∼147 nucleotides window are relatively prevalently encoded within larger DNA sequences that promote a more defined nucleosome placement (in green). In cells that do not express PU.1, such specific nucleosome placement may protect these cells from using genomic information not relevant to their functions. Note that neighboring nucleosomes may be more mobile relative to their interaction with DNA (arrowheads). In macrophages however, the same nucleosome is probabilistically more effectively evicted and/or displaced as a result of the collaborative binding activity of PU.1 and C/EBP factors. This then allows for more efficient binding of the NF-κB p65 subunit following TLR4 stimulation, for example.
The biological mechanism(s) promoted by having evolutionary forces selecting DNA sequence features that favor, for a same region of the genome, an enhancer in one cell type and nucleosomal occupancy in another remains speculative. An intuitive, yet very intriguing possibility, however, is that nucleosomal occupancy may efficiently interfere with improper use by SDTFs of information encoded in the genome. Given that SDTFs are typically not expressed in a cell type specific manner and that enrichment for SDTF motifs is a defining characteristic of the majority of all potential enhancer regions genome-wide, nucleosomal occupancy may thus ensure that only regions specified by LDTFs in a given cell type can be used to regulate the transcriptional output (Fig. 3).
Together studies by Heinz et al. (73) and Barozzi et al. (74) offer great support to the epigenomic hypothesis of nucleosome-mediated collaboration between TFs and its application to the macrophage enhancer repertoire. Importantly, evidence from other mammalian cell types also supports this model (87). However, many important aspects remain unknown. For one, the overall binding dynamics and kinetics of the PU.1-C/EBPα-nucleosomal eviction series of events in TGEMs remain to be fully characterized. For instance, it is not clear how stable each event is (i.e. PU.1 binding to DNA), and whether PU.1 and C/EBPα binding must occur sequentially or simultaneously. Whether their impact on nucleosomal eviction is additive or synergistic is not clear either. All in all though, the data presented above suggests that an array of critical information encoded in DNA is used by LDTFs like PU.1 to select enhancers in a cell type-specific manner. It also indicates that a primary function of LDTFs is to displace specific nucleosomes that act as barriers to shield highly potent genomic information that must be used under very specific circumstances.
Roles of composite sequences and ternary molecular complexes
Direct protein-protein interaction between PU.1 and other TFs at the DNA interface is another mechanism by which PU.1 likely promotes the establishment of the macrophage enhancer repertoire. In particular, the ETS-IRF composite element (EICE) 5′-GGAANNGAAA-3′, in which the first four nucleotides correspond to the core ETS motif and the last four are recognized by IRF4 or IRF8, is relatively prevalent among enhancers in macrophages (21). In this context, IRF4 or IRF8 do not bind efficiently by themselves to the IRF-half of the EICE motif due to both intrinsic characteristics of their DNA-binding domain and the presence of an autoinhibitory domain present in their C-terminal. However, upon PU.1 binding to its ETS sequence, Arg222 in the PU.1 ETS domain abandons a hydrogen bond with the DNA backbone in the major groove and, along with Lys223, reaches across the minor groove to favor interactions with hydrophobic residues in IRF4/IRF8 (88). Importantly, this is achieved by PU.1 without it losing affinity for its core 5′-GGAA-3′ motif (88). Thus, ETS-DNA binding energy is redistributed to collaborative protein-protein interactions. Furthermore, phosphorylated PEST domain of PU.1 further potentiates these collaborative interactions by interfering with the autoinhibitory domain of IRF4, resulting in a more stable ternary complex.
The precise contribution of the ternary complex model to the PU.1 cistrome and the establishment of the macrophage enhancer repertoire have yet to be thoroughly investigated. On the other hand, we note that AP-1: IRF composite sequences direct heterodimerization of BATF with IRF4, which is critical to the differentiation of Th17 cells (89). Furthermore, given the evidence demonstrating the important contribution of IRF4 and IRF8 to the development and functions of mononuclear phagocytes (90-92), it is likely that ternary complex mechanisms play an important role in macrophage ontology and functions.
PU.1 concentration as a major determinant of the nucleosome-mediated PU.1-C/EBPα collaborative model
The nucleosome-mediated collaborative model is subject to the influence of numerous parameters (83). Among them, the concentration of LDTFs in the nucleus is predicted to have a substantial impact on their efficiency to promote nucleosomal remodeling, and thus establish enhancers.
The expression level of PU.1 per se correlates with different epigenomic outcomes. In this regard, the role of PU.1 in B cells and macrophage ontology is particularly informative. As is the case for macrophages, B cells also depend on PU.1 to develop. For instance, induction of low activity of PU.1 in the nucleus with the PU.1-ER fusion system described above reprograms hematopoietic progenitors into cells with a B-cell-like phenotype but higher expression leads to macrophage reprogramming (21, 93). Importantly, DNA motifs enriched in the vicinity of PU.1 at enhancers in B cells include EBF/PAX5/E2A, i.e. motifs recognized by TFs important to B-cell development and functions. Note that these motifs are not detected in macrophages. Similarly, B cells can be reprogrammed into macrophages by overexpression of C/EBP factors but will only do so if PU.1 is sufficiently expressed (94).
The key role of PU.1 concentration in specifying macrophage versus B-cell development in hematopoietic progenitors begs the question as to what mechanisms participate in the regulation of PU.1 expression levels. Not surprisingly, many effectors appear to be critically involved. At the transcriptional level, high levels of IRF8 downregulate PU.1 mRNA transcription and promote B-cell development (95). In addition, PAX5, a transcription factor important to B-cell development, can inhibit C/EBP-mediated transcription of the Csf1r gene in B-cell progenitors (96), thus likely desensitizing the cell to macrophage-promoting cytokines M-Csf and/or IL-34 that promote PU.1 expression. Cell cycle regulation also contributes to high PU.1 expression in macrophages (97). Indeed, developing macrophages accumulate high PU.1 levels by lengthening their cell cycle and not by increasing transcription of the Spi1 gene relative to myeloid–lymphoid common progenitors. On the contrary, developing B cells both decrease Spi1 rate of transcription and enter cell division more rapidly.
The development of the macrophage enhancer repertoire is dependent on PU.1 expression level, which is coherent with predictions derived from the nucleosome-mediated collaborative transcription factor model of epigenomics. Importantly, the PU.1 concentration and/or activity level parameter also likely play an important role at other stages of bifurcation as macrophage progenitors develop, including the erythroid-myeloid and the granulopoietic-myelopoietic stages (77, 98, 99). Note, however, that the extent to which PU.1 expression per se directly translates into effective nuclear concentration remains unknown as post-translational modifications of the PU.1 protein may also modulate its activity in certain conditions (100).
Applying and extending the collaborative model to primary tissue macrophages
Complete and efficient macrophage differentiation and/or polarization in vivo likely also require the collaborative activity of PU.1 and a second transcription factor. This collaborative interplay would presumably actually offer great plasticity for the optimization of functions of differentiated macrophages. Indeed, different tissues exhibit very different environmental milieu and requirements to function homeostatically, all of which are subject to fluctuations and perturbations. Together, these impose very different demands on macrophage functions. To efficiently cope with this seemingly infinity of possibilities, PU.1 may provide a developing macrophage with a basic, plastic epigenomic architecture upon which other signals/TFs can act to properly tune in a specific, efficient combination of enhancers. This, in turn, ensures an optimal transcriptional output that will allow the differentiated macrophage to function synergically with a given tissue at a given time and context. Therefore, such collaboration between PU.1 and other TFs offers a simple, yet apparently very efficient, strategy to achieve specialization and provides tremendous plasticity/malleability to the macrophage cell family.
Although a systematic comparison of the epigenomic features and enhancer repertoire of different macrophage family members has yet to be reported, numerous studies have provided insights into the networks of TFs and signaling pathways that potentially contribute to the proper epigenomic configuration of a given macrophage subtype. In particular, work on microglia, peritoneal macrophages, and spleen macrophages has provided valuable information regarding the spectra of signals used by different macrophages to accomplish proper differentiation and functions.
Proper microglia development and functions depend on IRF8 and TGF-β1 signaling
Microglia are the resident macrophages of the central nervous system (CNS). The microglial population exclusively originates during embryogenesis from yolk sac-derived c-KIT+ erythromyeloid progenitors (101, 102). These progenitors seed the developing CNS early in embryogenesis during the primitive stage of hematopoiesis. Although the half-life of microglia is not known, it appears that the population as a whole is maintained throughout life independently of blood-borne, peripheral contribution (5, 7). Interestingly, a recent study suggests that a microglia progenitor whose survival does not require CD115 signaling may also exist in the brain (103).
In spite of some conflicting data, mice lacking IRF8 generally exhibit defects in microglia ontology and functions, with studies reporting a decrease, no change, or increase numbers of microglia in IRF8-/- mice (101, 104, 105). Minten et al. (104) and Horiuchi et al. (105) noted that IRF8-/- displayed reduced morphological complexity, namely a wider and shorter cell body and cruder ramifications. Lack of IRF8 also impairs gene expression in quiescent and activated microglia and negatively affects their phagocytotic abilities under certain conditions (104, 105). Thus, although IRF8 may not be absolutely required for a microglia-like cells to develop and survive in the CNS, it likely plays an important role in properly tuning the transcriptome of these cells, as was proposed in the discussion of the PU.1-IRF8 ternary model.
Although still speculative, it is highly probable that the SMAD TFs also play a critical role in microglia ontology and functions given the essential role that TGF-β signaling plays in microglia biology. A significant decrease in microglia cell number was observed in the CNS of TGF-β1 -/- mice, and those that survived were small, had less elaborate ramifications, and expressed lower levels of CD11b (106). These effects were recently validated (107). Importantly, this latter study offered evidence that addition of TGF-β1 to primary microglia cultured with M-Csf promotes a gene expression signature that approximates better to that of freshly sorted microglia, compared to microglia cultured with M-Csf alone. Thus, TGF-β1 is not only critical to the survival and/or expansion of microglia during development, but also likely plays a role in organizing microglial functions in addition to helping restrain their activity and maintain quiescence (108, 109).
Retinoic acid promotes GATA6 expression, which regulates the development and functions of resident peritoneal macrophages
Although heterogeneous, macrophages from the peritoneal cavity can be broadly divided into two groups in mice based on F4/80 and MHCII expression (110). F4/80LowMHCIIHigh macrophages likely depend on the CCR2High-expressing monocyte subtype to maintain their population. On the other hand, F4/80HighMHCIILow macrophages, which are required for the control of IgA production by B-1 cells, might be derived from progenitors that originate either from the bone marrow in a CCR2-independent manner or that seeded local proliferative niches early during embryogenesis, although this remains to be precisely characterized. F4/80HighMHCIILow macrophages also appear to maintain their population through a self-replenishment mechanism. Importantly, evidence suggests that the F4/80HighMHCIILow macrophage population depends on transcription factor GATA6 to survive in the proper compartmental environment of the peritoneal cavity but not to develop per se. Indeed, absence of GATA6 promotes apoptosis in those resident peritoneal macrophages, and this may be a consequence of decreased expression of acetyl CoA synthesis-promoting enzyme aspartoacylase (111). Importantly, retinoic acid positively modulated GATA6 transcription and removing dietary vitamin A intake in mice decreased GATA6 expression in F4/80HighMHCIILow macrophages. This latter observation provides another elegant example of how a factor in a given tissue can contribute to the differentiation and functions of the resident macrophage population of the same tissue. In addition, it demonstrates how an organism's environment and life habits can potentially modulate gene expression, and possibly the epigenome, of its macrophages. Note that another study also recently confirmed the critical role of GATA6 to the resident macrophages of the peritoneal cavity (112).
SPI-C and LXRα contribute to the development and functions of different subtypes of spleen macrophages
The spleen provides a potent cellular milieu for the generation of immune responses to blood-borne antigens and for filtering the blood of senescent cells or potentially noxious materials. As with the resident macrophages of the peritoneal cavity, the spleen encompasses numerous diverse macrophage populations including the red pulp (RP), marginal zone (MZ), and white pulp (WP) subtypes. The development, survival, and/or functions of these different populations depend on the expression of different TFs. Indeed, RP macrophages fail to develop in mice lacking SPI-C, an ETS transcription factor closely related to PU.1, and this leads to decreased phagocytosis of red blood cells and dysregulated iron homeostasis (3).
MZ macrophages, but not the RP or WP subtypes, critically depend on LXRα (113). Indeed, MZ progenitors, including monocytes, fail to differentiate into MZ macrophages in LXRα-deficient mice. This is rather intriguing and implies that oxysterols, which are cholesterol metabolites that act as ligands for LXRα and LXRβ, may contribute to the development of a specific subset of macrophages. How this is accomplished remains to be characterized. However, note that LXRs are important regulators of phagocytosis (114), which involves lipid and cholesterol processing, and thus also likely oxysterols. Therefore, the ability to efficiently engage in its functions may actually synergize with the proper development of spleen MZ macrophages.
The studies presented above illustrate well how macrophages synergize with environmental cues to optimize their development. However, how these factors are transduced and integrated with PU.1 at the genomic level and whether there are PU.1-independent mechanism involved remains unknown. Similarly, the exact molecular mechanism(s) that these factors trigger to allow for effective epigenomic remodeling has yet to be characterized.
Insult-driven polarization leads to de novo enhancer establishment in macrophages
Although certainly required for efficient tissue functions under homeostatic conditions, perhaps the most defining role of macrophages is to counter microbial infections and promote repair following tissue injury. These latter two functions require substantial adaptation for macrophages to acquire competences that are typically not required to maintain healthy tissue functions. This entails important changes in their transcriptional output and this has been documented substantially by numerous studies throughout the years. Indeed, macrophage stimulation by pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), or various cytokines linked to diseases and loss of homeostasis results in a dramatic, stimulus-specific overhaul of the transcriptional output compared to the resting or quiescent states (115). The specificity of these responses, which arises by activating distinct signaling pathways and SDTFs, is required as different insults may require different combinations of macrophage effector mechanisms to be dealt with efficiently.
These observations raise an interesting parallel with the different subtypes of tissue macrophages. Indeed, given that the differential gene expression pattern that tissue macrophages exhibit among themselves is presumably derived to a large extent from different epigenomic configurations, the polarization of macrophages in contexts of insults is also likely to be associated with specific epigenomic arrangements.
We and others recently provided strong evidence validating this prediction. Stimulation of macrophages with various activating ligands leads to important epigenomic changes in macrophages. As mentioned earlier, TLR4 activation in mouse macrophages leads to a gain of de novo mono-methylation at H3K4 at 1,000 to 3,000 discrete regions in the genome compared to unstimulated macrophages (16, 116). These new enhancer-like regions have been also referred to as ‘latent-enhancers’. Similar epigenomic remodeling also occurred in response to the activation of other cytokine signaling pathways, including the interferon γ (IFN-γ) - STAT1 and the interleukin 4 (IL-4) – STAT6 axes (116). Importantly, many of these latent enhancers appear to persist in time and correlative evidence suggests that they may possess some functionality. Indeed, 72 h following an initial IL-4 stimulation, a second stimulation leads to quicker activation of the de novo gained enhancers, as assessed by H3K27Ac, compared to the first stimulation. Note, however, that a direct contribution of these enhancers to regulation of specific protein coding genes has yet to be established. Of note however, some of these unveiled latent-enhancers can also be activated at a later time by TFs not related to the first signaling pathway that implemented them.
Work by Kaikkonen et al. (16) and Ostuni et al. (116) provide important proof of concept possibly explaining how different macrophages, including tissue and insult-polarized, come to be distinct. Indeed, the frequency and/or magnitude of interactions of specific SDTFs with DNA appear to be a significant driver of epigenomic remodeling. Furthermore, that enhancers can be gained and maintained following macrophage activation is significant with respect to chronic inflammatory diseases associated with aging including cancer, metabolic deregulations, and neurodegenerative diseases. Conceptually speaking, many of those diseases are linked with the acquisition of different set-points of activating/inhibitory feed-forward/feed-back mechanisms (117). Given the predominant role that macrophages play in these diseases, we hypothesize that significant epigenomic remodeling may occur in these cells, which in turn may lead them on the one hand to promote disease activity and on the other hand de-sensitize their susceptibility to disengage from damaging activities.
Concluding remarks
The application of the epigenomic paradigm to macrophage studies has led to a major overhaul in our conceptualization of the mechanisms that regulate gene expression in these cells. In particular, the realization that most of the transcriptional activity of RNAP II in macrophages at any given time occurs at enhancers has been impactful. That enhancers constitute essential sites of integration of SDTFs with RNAP II complex activity, along with the identification of eRNAs, has allowed for an elegant way to introduce how the effects of natural genetic variation might translate into different phenotypes at the organism level.
Given that mutations in exon sequences typically negatively affect protein expression and/or functions, often leading to relatively strong phenotypes owing to qualitative, loss-of-functions effects among others, genetic variation at enhancers is, in contrast, more likely to have a quantitative effect, effecting the frequency and/or magnitude of transcription of associated protein-coding genes. In this scenario, any biochemical process a corresponding protein is part of should still run to completion, but with different dynamics and kinetics.
Many important questions remain unanswered. For example, in spite of the progress made in deciphering the DNA-binding behavior of PU.1 in mouse macrophages, current knowledge and mathematical models only predict at best 70 to 75% of the actual sites bound by PU.1. Thus, many additional, unidentified factors are likely to make important contributions to the binding behavior of PU.1 and that of other TFs for that matter. In particular, we note that given that human histones possess 130 PTM sites (118), much remains to be investigated regarding the contribution of chromatin states to the regulation of both transcription factor binding and gene transcription in general.
We predict that recent advancements in macrophage epigenomics will make significant contributions to medicine at both the diagnostic and the therapeutic levels. Epigenomics already has proven helpful in resolving paradoxical clinical observations in prostate cancer and in refining our understanding of the contribution of predisposing alleles to the etiology of atherosclerosis, among others (49, 119). With respect to therapeutics, given that expression of protein-coding genes can be inhibited by silencing targeted eRNAs, it is conceivable that this approach can be leveraged to inhibit genes whose expression promotes the etiology and/or maintenance of clinical diseases, in a gene and cell type specific manner. Therefore, work in epigenomics, by combining fundamental concepts of chemistry, genetics, and cell biology, has set the stage for promising advancements to human medicine.
Acknowledgments
We thank Kaitlyn Spees for assistance with the preparation of the manuscript. This work was supported by a Canadian Institutes of Health Research Fellowship to D.G., and NIH grants to C.K.G. The authors have no conflicts of interest to declare.
References
- 1.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkhurst CN, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohyama M, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature. 2009;457:318–321. doi: 10.1038/nature07472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 6.Hoeffel G, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209:1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epelman S, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 11.Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol. 2014;21:210–219. doi: 10.1038/nsmb.2784. [DOI] [PubMed] [Google Scholar]
- 13.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 16.Kaikkonen MU, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nady N, et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem. 2011;286:24300–2431. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelizzola M, Ecker JR. The DNA methylome. FEBS Lett. 2011;585:1994–2000. doi: 10.1016/j.febslet.2010.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghisletti S, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 23.Kumaki Y, et al. Analysis and synthesis of high-amplitude Cis-elements in the mammalian circadian clock. Proc Natl Acad Sci USA. 2008;105:14946–14951. doi: 10.1073/pnas.0802636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barish GD, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogdanovic O, et al. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 2012;22:2043–2053. doi: 10.1101/gr.134833.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, Sun L, Chen Z, Whitaker JW, Wang T, Wang W. Predicting enhancer transcription and activity from chromatin modifications. Nucl Acids Res. 2013;41:10032–10043. doi: 10.1093/nar/gkt826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem. 1999;274:1189–1192. doi: 10.1074/jbc.274.3.1189. [DOI] [PubMed] [Google Scholar]
- 30.Szerlong HJ, Prenni JE, Nyborg JK, Hansen JC. Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. J Biol Chem. 2010;285:31954–31964. doi: 10.1074/jbc.M110.148718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin Q, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 33.Wen YD, et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, et al. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. 2013;38:621–639. doi: 10.1016/j.tibs.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 39.Kruidenier L, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whyte WA, et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 42.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 44.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Santa F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hah N, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collis P, Antoniou M, Grosveld F. Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. The EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuan D, Kong S, Hu K. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci USA. 1992;89:11219–11223. doi: 10.1073/pnas.89.23.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashe HL, Monks J, Wijgerde M, Fraser P, Proudfoot NJ. Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev. 1997;11:2494–2509. doi: 10.1101/gad.11.19.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lam MT, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lam MT, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Step SE, et al. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARgamma-driven enhancers. Genes Dev. 2014;28:1018–1028. doi: 10.1101/gad.237628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamir I, et al. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin L, Lazar MA. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol. 2005;19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- 58.Melo CA, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 59.Li W, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsieh CL, et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci USA. 2014;111:7319–7324. doi: 10.1073/pnas.1324151111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mousavi K, et al. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. 2013;51:606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–1223. doi: 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magnani L, Eeckhoute J, Lupien M. Pioneer factors: directing transcriptional regulators within the chromatin environment. Trends Genet. 2011;27:465–474. doi: 10.1016/j.tig.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Serandour AA, et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011;21:555–565. doi: 10.1101/gr.111534.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng R, Blobel GA. GATA Transcription Factors and Cancer. Genes Cancer. 2010;1:1178–1188. doi: 10.1177/1947601911404223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKercher SR, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 68.DeKoter RP, Walsh JC, Singh H. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998;17:4456–4468. doi: 10.1093/emboj/17.15.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarrazin S, et al. MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell. 2009;138:300–313. doi: 10.1016/j.cell.2009.04.057. [DOI] [PubMed] [Google Scholar]
- 70.Herz HM, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26:2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33:4745–4754. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JE, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heinz S, et al. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barozzi I, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell. 2014;54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng R, et al. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci USA. 2008;105:6057–6062. doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jin F, Li Y, Ren B, Natarajan R. PU.1 and C/EBP(alpha) synergistically program distinct response to NF-kappaB activation through establishing monocyte specific enhancers. Proc Natl Acad Sci USA. 2011;108:5290–5295. doi: 10.1073/pnas.1017214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dahl R, et al. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol. 2003;4:1029–1036. doi: 10.1038/ni973. [DOI] [PubMed] [Google Scholar]
- 78.Zhang L, Friedman AD. SHP2 tyrosine phosphatase stimulates CEBPA gene expression to mediate cytokine-dependent granulopoiesis. Blood. 2011;118:2266–2274. doi: 10.1182/blood-2011-01-331157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jack GD, Zhang L, Friedman AD. M-CSF elevates c-Fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood. 2009;114:2172–2180. doi: 10.1182/blood-2008-11-191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Radomska HS, et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203:371–381. doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 83.Mirny LA. Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci USA. 2010;107:22534–22539. doi: 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller JA, Widom J. Collaborative competition mechanism for gene activation in vivo. Mol Cell Biol. 2003;23:1623–1632. doi: 10.1128/MCB.23.5.1623-1632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 86.Heinz S, Glass CK. Roles of lineage-determining transcription factors in establishing open chromatin: lessons from high-throughput studies. Curr Top Microbiol Immunol. 2012;356:1–15. doi: 10.1007/82_2011_142. [DOI] [PubMed] [Google Scholar]
- 87.He X, et al. Contribution of nucleosome binding preferences and co-occurring DNA sequences to transcription factor binding. BMC Genomics. 2013;14:428. doi: 10.1186/1471-2164-14-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Escalante CR, et al. Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol Cell. 2002;10:1097–1105. doi: 10.1016/s1097-2765(02)00703-7. [DOI] [PubMed] [Google Scholar]
- 89.Glasmacher E, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schlitzer A, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hambleton S, et al. IRF8 mutations and human dendritic-cell immunodeficiency. The N Engl J Med. 2011;365:127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kurotaki D, et al. Essential role of the IRF8-KLF4 transcription factor cascade in murine monocyte differentiation. Blood. 2013;121:1839–1849. doi: 10.1182/blood-2012-06-437863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.DeKoter RP, Singh H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science. 2000;288:1439–1441. doi: 10.1126/science.288.5470.1439. [DOI] [PubMed] [Google Scholar]
- 94.Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- 95.Wang H, et al. IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood. 2008;112:4028–4038. doi: 10.1182/blood-2008-01-129049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401:556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 97.Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science. 2013;341:670–673. doi: 10.1126/science.1240831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nerlov C, Querfurth E, Kulessa H, Graf T. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000;95:2543–2551. [PubMed] [Google Scholar]
- 99.Molawi K, Sieweke MH. Transcriptional control of macrophage identity, self-renewal, and function. Adv Immunol. 2013;120:269–300. doi: 10.1016/B978-0-12-417028-5.00010-7. [DOI] [PubMed] [Google Scholar]
- 100.Brass AL, Zhu AQ, Singh H. Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimers. EMBO J. 1999;18:977–991. doi: 10.1093/emboj/18.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kierdorf K, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16:273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 102.Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–152. doi: 10.1016/s0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 103.Elmore MR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Minten C, Terry R, Deffrasnes C, King NJ, Campbell IL. IFN regulatory factor 8 is a key constitutive determinant of the morphological and molecular properties of microglia in the CNS. PloS One. 2012;7:e49851. doi: 10.1371/journal.pone.0049851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Horiuchi M, et al. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J Neuroinflammation. 2012;9:227. doi: 10.1186/1742-2094-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Makwana M, et al. Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J Neurosci. 2007;27:11201–11213. doi: 10.1523/JNEUROSCI.2255-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Butovsky O, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abutbul S, et al. TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia. 2012;60:1160–1171. doi: 10.1002/glia.22343. [DOI] [PubMed] [Google Scholar]
- 109.Schilling T, Nitsch R, Heinemann U, Haas D, Eder C. Astrocyte-released cytokines induce ramification and outward K+ channel expression in microglia via distinct signalling pathways. Eur J Neurosci. 2001;14:463–473. doi: 10.1046/j.0953-816x.2001.01661.x. [DOI] [PubMed] [Google Scholar]
- 110.Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gautier EL, et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J Exp Med. 2014 doi: 10.1084/jem.20140570. pii: jem.20140570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosas M, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344:645–648. doi: 10.1126/science.1251414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.N AG, et al. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat Immunol. 2013;14:831–839. doi: 10.1038/ni.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.N AG, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xue J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 117.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 118.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harismendy O, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]