

Abstract
Oligo- and polysaccharides are infamous for being extremely flexible molecules, populating a series of well-defined rotational isomeric states under physiological conditions. Characterization of this heterogeneous conformational ensemble has been a major obstacle impeding high-resolution structure determination of carbohydrates and acting as a bottleneck in the effort to understand the relationship between the carbohydrate structure and function. This challenge has compelled the field to develop and apply theoretical and experimental methods that can explore conformational ensembles by both capturing and deconvoluting the structural and dynamic properties of carbohydrates. This review focuses on computational approaches that have been successfully used in combination with experiment to detail the three-dimensional structure of carbohydrates in a solution and in a complex with proteins. In addition, emerging experimental techniques for three-dimensional structural characterization of carbohydrate–protein complexes and future challenges in the field of structural glycobiology are discussed. The review is divided into five sections: (1) The complexity and plasticity of carbohydrates, (2) Predicting carbohydrate–protein interactions, (3) Calculating relative and absolute binding free energies for carbohydrate–protein complexes, (4) Emerging and evolving techniques for experimental characterization of carbohydrate–protein structures, and (5) Current challenges in structural glycoscience.
Keywords: Binding free energy, docking, molecular dynamics, nomenclature, oxidative footprinting
The complexity and plasticity of carbohydrates
Carbohydrates occupy a pivotal functional position in biological recognition processes (Sharon and Lis 1993; Varki 1993; Dwek 1996). The complex shape, functionality, and dynamic properties of oligo- and polysaccharides (hereafter denoted simply as “carbohydrates”) allow these molecules to function in intermolecular interactions as encoders of biological information. For instance, carbohydrate recognition is an integral part of normal biological development (Haltiwanger and Lowe 2004) and the immune defense against pathogens via the identification of exogenous carbohydrates (Brown and Gordon 2001; Cobb and Kasper 2005). Conversely, many bacterial and viral pathogens (such as Escherichia coli or Haemophilus influenza) initially adhere to host tissues by binding specifically to carbohydrates on the host’s cell surfaces (Karlsson 1986, 1989; Rostand and Esko 1997). Thus, there is an interest in developing therapeutic agents that can interfere with, modulate, or exploit carbohydrate-based host-pathogen interactions. Examples of therapeutic agents interfering with carbohydrate-specific interactions include the neuraminidase inhibitors zanamivir and oseltamivir used in the treatment of influenza infections (Dreitlein et al. 2001). Vaccines that employ bacterial polysaccharides, conjugated to carrier proteins, have been particularly effective, for example against H. influenza (Jennings 1992). Because abnormal glycosylation is also a marker for certain types of cancer (Hakomori 1989; Fukuda 1996) and other diseases, such as IgA nephropathy (Coppo and Amore 2004; Moura et al. 2004), inflammatory bowel disease (Campbell et al. 2001), and rheumatoid arthritis (Parekh et al. 1985; Malhotra et al. 1995), there is a growing interest in exploiting these variations in the development of therapeutics (Lo-Man et al. 2004; Buskas et al. 2005; Xu et al. 2005). In certain diseases, such as congenital disorders of glycosylation (Freeze 2001) or lysosomal storage diseases (Neufeld 1991), the origin of the observed glycosylation defects can be traced back to mutations in the glycan-processing pathway, suggesting a role for gene therapy and possibly glycosidase/transferase inhibition (Platt et al. 1994; Sly and Vogler 2002; Grabowski and Hopkin 2003).
Thus far, only rarely has the design of carbohydrate-based therapeutic agents made extensive use of 3D structural information, reflecting in part the difficulties of determining carbohydrate conformation, as well as a paucity of structural data for many carbohydrate—protein complexes. To help reverse this trend, computational approaches have emerged to complement experimental techniques in the analysis of structure–function relationships of carbohydrate–protein interactions.
A significant challenge in the characterization of the conformational properties of carbohydrates is that they are flexible, populating multiple (defined) conformational states under physiological conditions. This property necessitates a modification in the way we think about biological recognition processes. A rigid molecule can be fully characterized by a single conformational state, but not so for a flexible one. This raises an interesting question: How are flexible molecules recognized in nature? Does the receptor protein preferentially bind to the most frequently populated shape, or to the average shape, or to a relatively rare “bioactive” conformation, or does binding induce a unique conformation?
To help explore the concepts of the carbohydrate structure and recognition, let us compare carbohydrates to another flexible object, a snake. To the extent that a living snake is a flexible 3D object that is not random in its motional properties, it serves as a useful analogy for carbohydrate structure and recognition. The shape and motion (as well as color and sound) of a cobra are clearly distinct from those of a rattlesnake. Both are generally long, skinny, and wiggly, but each is recognizably different. Yet, the average shape of each snake would be remarkably similar; if each one were to wiggle to an equal extent to the right and left its average shape would be a straight line! So it goes with all flexible objects, including glycans; depending on the extent of the motion the average shape may be a very poor description of any instantaneous conformation. That is not to say that the average properties are not useful; most experimentally observable data are averages of a conformational ensemble. For example, NMR intensities are the average of contributions from all of the conformational states observed on the NMR timescale. This averaging means that NMR data must be used with care when deriving a 3D model for a flexible carbohydrate, as the data could point to a virtual conformation. Nevertheless, the NMR data are extremely important in characterizing the carbohydrate structure and dynamics, and for validating computational predictions (Sayers and Prestegard 2000; Kirschner and Woods 2001; Sayers and Prestegard 2002; Gonzalez-Outeirino et al. 2005, 2006).
If the average shape is not what is recognized, then a particular conformation must somehow be selected from the conformational ensemble. From a statistical perspective, recognition of the most frequently seen shape of the carbohydrate is more probable than recognition of a rarely populated conformation. Surveys of the Protein Data Bank (PDB) have found that in the majority of noncovalently associated carbohydrate–protein complexes (Petrescu et al. 1999; Imberty and Perez 2000) and glycoproteins (Petrescu et al. 1999), the glycosidic torsion angles were consistent with those displayed most commonly by glycans in a solution. Until recently, these types of statistical analyses were challenging to perform due to limited search software; however, a particularly convenient interface for this purpose (GlyTorsion) has been developed (Lütteke et al. 2005). Exceptions to the tendency to bind low energy conformations are found in carbohydrate-processing enzymes, in which ligand distortion in the active site may be integral to the enzyme’s function (Karaveg and Moremen 2005). Additionally, an induced fit in the carbohydrate has been observed in lectin binding (Casset et al. 1995; Imberty and Perez 2000). However, the induced fit may be the result of miss-matching a receptor with a ligand; that is, just because a protein is found to bind to a particular carbohydrate does not mean that it evolved to recognize that ligand. A miss-matched induced fit is potentially relevant to structures of plant lectins bound to mammalian glycans. These issues underlie challenges faced by computational predictions of carbohydrate epitopes.
Characterizing the extent of molecular flexibility is challenging for both experimental and theoretical methods. To include molecular flexibility accurately in a computational model requires a method that is able to generate states whose average properties are experimentally consistent. Current molecular dynamics (MD) simulations, when combined with force fields appropriate for carbohydrates, provide this capability (Vliegenhart and Woods 2006). A perpetual challenge to MD simulations of biomolecules is to achieve adequate sampling of molecular motions of the system under study. Fortunately, in the case of oligosaccharides, there are relatively few energetically accessible conformational states for each glycosidic linkage, and thus it is often possible to achieve good conformational sampling from an MD simulation of between 10 and 1000 ns, depending on the size and complexity of the oligosaccharide. Sampling efficiency may be enhanced by techniques such as replica exchange MD (Sugita and Okamoto 1999) and Monte Carlo (MC) simulations (Metropolis and Ulam 1949; Metropolis et al. 1953).
MD simulations can also provide insight into the dynamic properties of carbohydrate–protein complexes, given a reasonable initial structure for the complex. Crystallography continues to play a significant role in the characterization of carbohydrate–protein complexes; however, important new rungs have been added to the technological ladder. Most notably, advances in the a priori prediction of complexes by molecular docking have been made as well as NMR methods, such as saturation transfer difference (STD) NMR (Mayer and Meyer 2001; Meyer and Peters 2003). STD NMR experiments facilitate the identification of those regions of the carbohydrate that are proximal to the protein surface (the carbohydrate epitope). Thus, docking experiments may be validated by a qualitative comparison to STD NMR data (Haselhorst et al. 2004, 2007), or by quantitative computation of the NMR intensities in the complex (Wen et al. 2005). Integrating STD NMR data directly into docking algorithms is a potential next step. Emerging experiments included protein surface footprinting, based on either H/D exchange differences (King et al. 2002; Seyfried et al. 2007), or hydroxyl radical oxidation (Sharp et al. 2003, 2004; Hambly and Gross 2005; Takamoto and Chance 2006) that can identify carbohydrate interfaces on proteins, and as such may also provide a significant step up the technological ladder.
Here we summarize some recent reports that employ computational simulations to examine the 3D structures and dynamics of carbohydrates and carbohydrate–protein complexes. Our goal is to help promote awareness of the capabilities and limitations of current computational methods as applied to these systems and to illustrate how the integration of computation and experiment can advance efforts in the structure determination of carbohydrate–protein complexes (Figure 1).
Fig. 1.
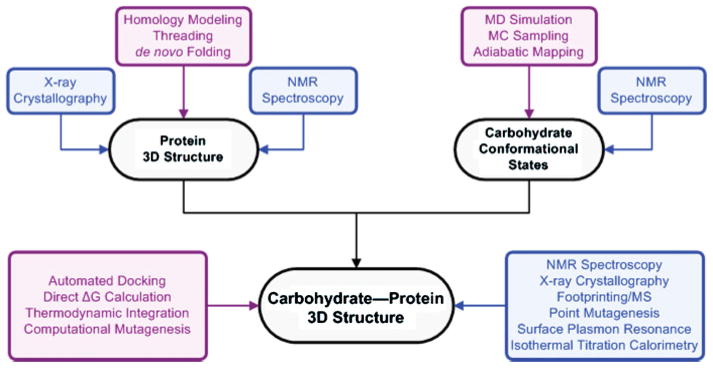
The roles of computational methods (purple) alongside experimental methods (blue) in structural glycobiology.
Predicting carbohydrate–protein interactions
Given the development of high-throughput affinity assays, employing immobilized microarrays of biologically relevant glycans, there is now a considerable amount of data on carbohydrate–protein interactions in vitro (Fukui et al. 2002; Blixt et al. 2004; Paulson et al. 2006). Microarray technologies are able to identify carbohydrate ligands for a protein receptor, providing an unprecedented level of information pertinent to carbohydrate recognition. But the picture is incomplete. While microarray data may identify a core carbohydrate recognition sequence, the precise manner in which the protein recognizes and binds to a ligand remains undefined. To complement experimental structural studies of carbohydrate–protein complexes, computational docking of carbohydrates to proteins can contribute to the understanding of carbohydrate recognition.
Drug design
Often host cell-surface carbohydrates are the targets for invading microbes and viruses (Karlsson 1986; Smith et al. 2004) and conversely may form targets for the design of novel therapeutic agents (von Itzstein et al. 1993). In just such a case, the GM1 binding site of the heat-labile enterotoxin produced by E. coli (a close structural analogue to the cholera toxin produced by Vibrio cholerae) was targeted for an inhibitor design (Minke, Diller, et al. 1999). Using crystal structures of the heat-labile enterotoxin with fragments of the carbohydrate head group of GM1, potential inhibitors were identified (Minke, Roach, et al. 1999). The program AutoDock (Goodsell and Olson 1990) was used to predict the structure of the toxin–inhibitor complexes. After the docking studies were complete, the structures of the inhibitor–toxin complexes were solved by X-ray crystallography (Merritt et al. 1997), providing a direct test of the docking protocol. In general, the experimentally obtained structures validated the models predicted via the docking procedure (Figure 2). In the case of one inhibitor, melibionic acid (galactose-α-(1-6)-gluconic acid), the majority of the highest ranked binding modes from the docking simulations placed the galactose residue in a similar position to that found in the crystal structure. For the gluconic acid residue there was no consensus among the top-ranked binding modes, as the ligand adopted several poses. It is noteworthy that this residue was also unresolved in the crystal structure (Merritt et al. 1997) (a common occurrence when dealing with flexible molecules like carbohydrates). Melibionic acid thus bound to the toxin primarily through interactions made by the galactose residue while the gluconic acid provided non-specific interactions that enhanced the affinity relative to free galactose.
Fig. 2.
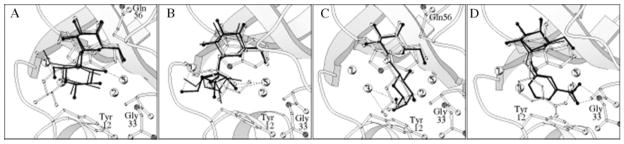
Inhibitors of the heat-labile enterotoxin from E. coli. Computationally docked galactose derivatives (white) were able to reproduce the experimentally observed binding modes (black) (reprinted from Minke, Diller, et al. 1999).
Identifying the protein contact residues
While the above example illustrates a successful use of docking in the characterization of the interaction between a protein and small carbohydrate derivatives, docking simulations have also been used to probe larger carbohydrate–protein interactions (Bitomsky and Wade 1999; Sachchidanand et al. 2002; Kadirvelraj et al. 2006). Heparin, a complex sulfated glycosaminoglycan, has several known protein-binding partners including antithrombin III and interleukin 8 (IL8). Lacking a structure of the heparin–IL8 complex, efforts have been undertaken to predict this carbohydrate–protein complex using a docking-based approach (Bitomsky and Wade 1999). The docking-based protocol included (1) using mono- and disaccharide heparin fragments to run a global docking search using several docking programs (GRID (Goodford 1985), AutoDock (Goodsell et al. 1996), and DOCK (Ewing and Kuntz 1997)); (2) calculating the probability that a particular protein residue would be found in the binding interface, based on the results from the global searches; and (3) docking a hexasaccharide fragment of heparin in the plausible binding regions. This method was first tested on three heparin-binding proteins with experimental structures, and the success of these test cases, along with available biological and spectroscopic data (Webb et al. 1993; Kuschert et al. 1998), supported the application of this protocol to the prediction of the heparin–IL8 complex. Using computational docking, it was possible to identify a shallow carbohydrate-binding site on the IL8 surface and construct 3D models of the binding mode of a complex and highly charged oligosaccharide.
Enzymatic pathway analysis
Computational docking can also assist in predicting carbohydrate–enzyme structures, and in defining enzymatic pathways. Recently the mechanism of action of a glycoside hydrolase in the N-glycan synthesis pathway, α-(1–2)-mannosidase I, was predicted using docking simulations (Mulakala et al. 2006), in line with previously predicted mechanisms, based on crystal structures of the enzyme with various substrates (Vallee et al. 2000; Karaveg et al. 2005). By computationally docking 16 α-D-manno-(1-2)-α-D-mannose conformers (generated by constraining relevant ring atoms and then minimizing the structure in vacuo) and evaluating binding energies and forces on the substrates (Figure 3) that were proposed to drive it toward the transition state (with AutoDock), a reasonable enzymatic pathway was deduced (Mulakala et al. 2006). The substrate of α-(1-2)-mannosidase I was predicted to populate conformations along the following pseudo-rotation pathway from its starting conformation (either 1C4 or 0S2) through to the transition state (3E): 1C4 → 3H2 → 0S2 → 3,0B → 3S1 → 3E (Mulakala et al. 2006).
Fig. 3.

Docking used to compute hydrogen-bonding forces on the substrate α-D-mannopyranosyl-(1-2)-α-D-mannopyranose (3H2) exerted by the active site of yeast α-(1-2)-mannosidase (measurements in pN) (reprinted from Mulakala et al. 2006).
Computational docking methods have evolved to help overcome difficulties in obtaining experimental 3D structures of multiple ligands to a common receptor. Docking strategies can utilize experimental or modeled protein structures to aid in the discovery new lead compounds for drug design. In the field of glycobiology docking simulations have several applications, in addition to drug design, as they provide computationally tractable methods to predict carbohydrate-binding sites on proteins, bound ligand conformations, and conformational pathways for carbohydrate-processing enzymes. Docking simulations are however far from infallible and benefit from experimental verification, as false positives and negatives are common. Typically, docking simulations do not generate a single model rather multiple plausible low energy poses. These results require ancillary information in order to select the most probable candidates. To facilitate selection, more rigorous post-docking approaches can be used to generate additional data for predicted carbohydrate–protein complexes; two such methods are highlighted in the following section.
Calculating relative and absolute binding free energies for carbohydrate–protein complexes
Characterization of the structural and thermodynamic properties of carbohydrate–protein interactions is desirable for many reasons, from understanding basic biological function to structure-based drug design. Two types of theoretical methods are commonly used to calculate the free energy of binding of a carbohydrate–protein complex: direct ΔG calculations and thermodynamic integration (TI) methods (Figure 4). Direct ΔG calculations use only the initial (free receptor and ligand) and final (complex) states of the cycle, which correspond to experimentally observable states. Information regarding the pathway is not required. Descriptions of the initial and final states are generated as structural ensembles collected from independent MD simulations, or from decomposition of a single MD simulation of the complex (reviewed in Swanson et al. 2004).
Fig. 4.
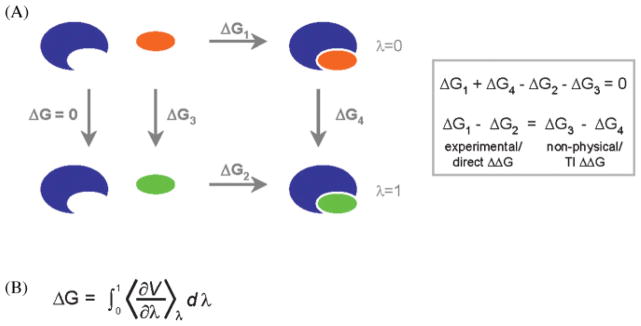
(A) The thermodynamic cycle for ligands (orange and green) binding to a protein receptor (blue). Direct ΔG calculations determine the free energy of binding of a ligand to a receptor (ΔG1), whereas TI methods calculate the free-energy difference between receptor–ligand complexes (ΔG4), where only the ligand is changed. (B) By considering the ΔG4 pathway as a series of nonphysical intermediate states (0 < λ < 1), the free energy difference between the real states λ = 0 and λ = 1 can be computed by taking the ensemble average of the first derivative of the potential energy (V) with respect to λ.
In TI calculations the relative ΔGbinding is computed for closely related systems by slowly transforming or perturbing the initial state to the final state (Figure 4). By employing an appropriate (although nonphysical) thermodynamic cycle and by proceeding in small discrete steps, the relative ΔGbinding may be computed (Zwanzig 1954).
Each method has advantages and disadvantages (as highlighted in Table I). TI calculations are best suited to a set of structurally similar ligands or a set of mutations within a binding site. As for example in the refining of a lead compound into a high affinity binder or in the design of higher affinity receptor proteins. To screen structurally dissimilar ligands, or a single ligand against multiple receptors, direct ΔG calculations are preferred. Both of these theoretical methods can provide a detailed structural explanation of the system under study; notably, they can provide an estimate of the contributions of specific residues and atoms to the free energy, as well as decomposition of energetic contributions from steric and electrostatic components, something that is difficult to achieve experimentally.
Table I.
A comparison of methods used to predict binding free energies
| Comparison | Automated docking | Direct ΔG calculations | Thermodynamic integration |
|---|---|---|---|
| Computational efficiency | High | Moderate | Low |
| Ligand set | Can be diverse | Can be diverse | Close structural analogues only |
| Binding free energy computed | Absolute ΔG | Absolute ΔG | Relative ΔG |
| Water model | Implicit | Implicit | Explicit |
| Accuracy strongly dependent on | Compounds used for calibration | Water model, force field, and sampling | Sampling and force field |
Direct ΔG calculations
Carbohydrate-binding proteins, known as lectins, exhibit a range of carbohydrate-binding specificities. Galectin-3, a mammalian lectin with relatively narrow binding specificities, has affinity for blood group A-oligosaccharides. At the other end of the spectrum are lectins such as galectin-1 that can bind a relatively diverse group of oligosaccharides (Consortium for Functional Glycomics (CFG), http://www.functionalglycomics.org). How is it that lectins are able to display such broad binding specificities for structurally distinct carbohydrate epitopes? To address this question, the ability of galectin-1 to bind a variety of oligosaccharides with varying specificity was explored using a combination of computational docking, MD simulations, and binding free energy calculations (Ford et al. 2003). Galectin-1 is known to bind to Gal-β-(1-4)-GlcNAc (LacNAc)-containing oligosaccharides and there are several crystal structures of it in complex with ligands including LacNAc (Liao et al. 1994). MD simulations, using the GLYCAM force field (Woods et al. 1995), of galectin-1 complexed (via docking simulations) with carbohydrate ligands containing the LacNAc core were run to determine the binding mechanism of LacNAc derivatives (Ford et al. 2003). From these simulations, it was concluded that various substitutions at the nonreducing end of the LacNAc core were tolerated with little or no disruption of key-binding interactions. Hydrogen bonding and aromatic stacking that occurred between LacNAc and galectin-1 were also found to be present for all of the modified LacNAc ligands. Direct ΔG calculations based on the results from the MD simulations provided good qualitative agreement with experimentally determined binding affinities for the ligands under study. In addition to known ligands, two simulations were run with negative controls (GlcNAc and N-acetylmaltosamine) and in both cases the ligands diffused away from the binding pocket early in the simulation. The inclusion of negative controls in MD simulations provides considerable confidence in the MD timescale and in the force field used for the simulation. MD simulations in combination with direct ΔG calculations predicted binding conformations that explained the weakly selective binding behavior of galectin-1 and qualitatively ranked the predicted free energies of binding in accordance with the experimentally determined binding affinities.
Similarly, a direct ΔG computational study using the GLYCAM force field (Woods et al. 1995) exploring the specificity of concanavalin A, a plant lectin specific for mannose-containing oligosaccharides (common to N-linked glycans), was successful in structurally accounting for the ligand preferences of the protein (Bryce et al. 2001). The results were further de-convoluted by decomposing binding energy into entropic and enthalpic contributions. Binding energy contributions were analyzed on a per-residue basis, identifying key interactions for the general recognition strategy and specificity of concanavalin A. The specific interactions of high-mannose fragments that led to the observed binding differences, could thus be ranked.
In addition to examining biological ligands, direct ΔG calculations have unraveled the energetics of binding of carbohydrate inhibitors for disease-related enzymes. Neuraminidase, a viral coat protein from influenza, is responsible for cleaving terminal neuraminic acid (Neu5Ac, sialic acid) residues on the host cell surface to promote viral fusion and release of viral progeny (Palese et al. 1974; Palese and Compans 1976; Liu et al. 1995). Due to its central role in influenza infection, viral neuraminidase has been selected as a drug target. Inhibitors based on the natural ligand, Neu5Ac, boosted the affinity from millimolar for the natural ligand (Potier et al. 1979; Jedrzejas et al. 1995) to nanomolar for some of the better inhibitors (von Itzstein et al. 1993; Finley et al. 1999; Babu et al. 2000; Chand et al. 2001). Direct ΔG calculations, using the AMBER force field, revealed that the energetic component that resulted in such a jump in potency was caused by hydrophobic contacts (Masukawa et al. 2003). The neuraminidase inhibitors tamiflu and relenza have increased van der Waals interactions over the natural ligand resulting in more direct contacts with the enzyme and fewer water-mediated interactions. Electrostatic contributions were less important in determining the binding affinity but were useful in correctly orienting the ligand within the active site.
The direct ΔG method is not only applicable to the study of small ligands, such as oligosaccharide fragments or small molecule carbohydrate derivatives, but also for large complex carbohydrates. Antibodies against capsular polysaccharides (CPS) from one strain of Streptococcus, such as Group B Streptococcus agalactiae type III (GBSIII), rarely cross-react with other strains, such as the CPS from Streptococcus pneumoniae type 14 (Pn14). GBSIII and Pn14 share a similar backbone sequence, but Pn14 lacks α-Neu5Ac in its side chains. This minor difference attenuates the antigenicity of Pn14-type CPS as compared to the sialated GBSIII type-CPS for antibodies raised against the sialated antigen (Jennings et al. 1981). To quantify differences in immunogenicity and antigenicity of CPS from a Gram-positive bacterium (Group B S. agalactiae), Kadirvelraj et al. (2006) used a combination of computational tools, including molecular modeling, docking, MD simulations, and direct ΔG calculations. While the conspicuous difference between GBSIII and Pn14 is the loss of an acidic monosaccharide, the primary contributions to differences in affinity were found to be entropic in origin, as determined via MD simulations of the immune complex and direct ΔG calculations with the GLYCAM force field (Kadirvelraj et al. 2006). Pn14 exhibited greater flexibility in solution, and thus paid a greater entropic penalty upon binding to the antibody, as compared to GBSIII. The absence of sialic acid in the side chains of Pn14 also decreased contributions to van der Waals stabilization energy and the electrostatic interaction energy. These factors combined to yield differences in affinity that accounted for differential antibody recognition of bacterial CPS and provided a structural framework for interpreting the observed immunological data.
Thermodynamic integration calculations
The application of TI calculations to the analysis of binding free energies of inhibitors to carbohydrate-binding enzyme includes α-D-glucose-based inhibitors of glycogen phosphorylase (Archontis et al. 2005). A potent inhibitor of glycogen phosphorylase, hydan (a spirohydantoin of glucopyranose), and two of its analogues (methyl-hydan and NH2-hydan) have been co-crystallized with the enzyme (Gregoriou et al. 1998; Oikonomakos et al. 2002; Watson et al. 2005). These structures provided a qualitative explanation for the differences in observed binding affinities (relative binding affinities: hydan > NH2-hydan > methyl-hydan ≫ α-D-glucose (Bichard et al. 1995; Watson et al. 2005)). The addition of data from TI simulations using the CHARMM22 glucose force field parameters (MacKerell et al. 1998) provided a quantitative description of the binding interaction energies (Archontis et al. 2005). The addition of functional groups (methyl and NH2 group) introduced a destabilizing van der Waals free energy component (the methyl group more so than the NH2) through unfavorable interactions of the ligands with an aspartate residue and a water molecule in the binding site. NH2-hydan improved with respect to electrostatic energies over hydan, but this was not sufficient to overcome the destabilizing van der Waals contacts. These results suggested alternative sites for hydan modifications that could yield more potent inhibitors.
Free energy calculations based on computational simulations of protein–carbohydrate complexes have also been employed to investigate antigenicitiy of carbohydrate–antibody interactions (Pathiaseril and Woods 2000). As a test of the ability of a method closely related to TI, free energy perturbation, to predict relative binding free energies for a series of haptens, the interaction energy of the trisaccharide epitope of the Salmonella serotype B O-antigen and a related monoclonal antibody fragment were analyzed. From calorimetric experiments, five structural analogues of the natural hapten were estimated to have similar binding affinities to the natural epitope, while one congener displayed very poor binding affinity (Bundle et al. 1994). Free energy perturbation simulations reproduced the experimentally determined relative free energies within an absolute error of 0.55 kcal/mol and revealed the likely protonation state of a histidine residue in the binding cleft. This computational study also demonstrated one of the challenges of free energy perturbation simulations, predicting relative free energies of ligands that differ only in their interactions with the solvent. This is also a challenge for the direct ΔG method, where accurately calculating the contributions of ligand–solvent and protein–solvent interactions tests the limits of the computational model.
Accurate representation of the effects of water as a solute is critical for the success of all of the computational methods discussed (MD, docking, and ΔG calculations). With an accurate force field and explicit water, MD simulations can generate conformational families that are consistent with experimentally observed structural data (Corzana et al. 2004; Gonzalez-Outeirino et al. 2006; Pereira et al. 2006). For docking studies, inclusion of conserved water molecules in the binding site can improve the success of the simulation (Minke, Diller, et al. 1999; Rarey et al. 1999; Österberg et al. 2002); however, the position of key waters is not always known. In the case of free energy simulations, the energetic contributions from direct interactions between the ligand and receptor can often be well modeled although the accurate treatment of contributions from ligand–solvent and protein–solvent interactions remains a challenge.
Emerging and evolving techniques for experimental characterization of carbohydrate–protein structures
Partially oriented NMR spectroscopy
Over the past decade, NMR structural constraints from anisotropies in spin interactions have greatly expanded the possibilities for biomolecular structure determination. By partially orienting a sample via medium- or field-induced alignment, observables that arise from anisotropic interactions such as residual dipolar couplings (RDCs), chemical shift anisotropy (CSA) offsets, and pseudocontact shifts in paramagnetic systems can be detected (reviewed in Prestegard 1998; Prestegard et al. 2004). In a recent application, a combination of NMR experiments, mass spectrometry (MS), and computational simulations, yielded a novel strategy to probe oligosaccharide conformation in a solution (Yu et al. 2007). In this experiment, a pentasaccharide fragment of chondroitin sulfate was isotopically enriched by replacing acetyl groups with 13C-labeled acetyl groups. The resulting 13C spectra provided structural constraints from the orientational dependence of RDCs and CSA offsets. To assign resonances, isotope ratios in the pentasaccharide, observed via the mass spectra, were correlated with enrichment levels seen in the NMR spectra. The 13C data alone were too sparse for structure determination, so additional structural constraints (NOEs, J-coupling constants, 1H–1H, and 13C–1H RDCs) were determined. In the final structure determination steps, a combination of computational tools was used: REDCAT (residual dipolar coupling analysis tool) to calculate alignment parameters (Valafar and Prestegard 2004) and XPLOR-NIH (using the GLYCAM force field; Kirschner et al. 2008) to optimize the structure via simulated annealing (Schwieters et al. 2003, 2006). The ultimate goal of this novel strategy is to examine oligosaccharides in complex with proteins, in which case the 13C data can be augmented with principal order parameters determined from the protein and conformational restraints for the oligosaccharide from computational simulations (Yu et al. 2007). This combination of NMR, MS, and computation highlights the creative cross-disciplinary approach necessary to tackle oligosaccharide–protein structure determination.
Hydroxyl radical protein footprinting with mass spectrometry
The concept of “footprinting” generally refers to techniques that probe macromolecular surface changes upon complex formation of two or more molecules, by modifying solvent-exposed surfaces. This technique has existed since the late 1970s when it was first applied to probe DNA–protein complexes (Galas and Schmitz 1978; Schmitz and Galas 1980). Since that time the concept has evolved to include footprinting of RNA–protein (Motoki et al. 1991; Wang and Padgett 1989) and protein–protein complexes (Sheshberadaran and Payne 1988). In glycoscience, footprinting is drawing attention for its potential to define carbohydrate-binding surfaces on proteins. Footprinting data could be used to complement computational predictions for systems that are not amenable to NMR spectroscopy or crystallography.
The highest resolution protein footprint technique currently available uses hydroxyl-radical oxidation of amino acid side chains, which can be detected and quantified by MS techniques to map the solvent accessible surfaces of proteins and protein complexes (reviewed in Takamoto and Chance 2006). Since many amino acid side chains react with hydroxyl radicals, this technique is capable of relatively high-resolution mapping. In contrast to H/D amide exchange MS, oxidative footprinting has two critical advantages, namely it modifies the amino acid side chains, rather than the backbone and it is not a chemically labile modification. Hydroxyl radicals can be generated using metal-dependent chemical generation from peroxide, by radiolysis of dilute peroxide solutions or by direct radiolysis of water. Hydroxyl radicals are ideal reagents to probe solvent accessible surfaces, as they are small and highly reactive. The relative reactivity of amino acid side chains has been characterized (Xu and Chance 2005) and all but the amino acids Gly, Ala, Asp, Asn, Ser, and Thr are sufficiently reactive to be useful probes. Since 14 types of amino acids are potentially reactive (Xu et al. 2003), this method boasts considerably higher structural resolution than residue-specific techniques (such as lysine modification), which provide sparse sampling of the protein surface. In addition, initial studies demonstrate that the extent of oxidation (reactivity) is strongly dependent on the solvent accessible surface area (SASA) of the amino acid (Charvátová, Foley, Bern, Sharp, Orlando, and Woods, in preparation) (Figure 5).
Fig. 5.
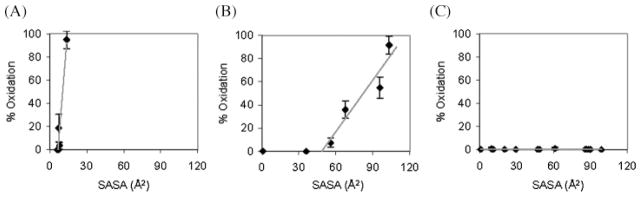
Correlation between reactivity and SASA (calculated from MD simulation) for amino acids from galectin-1: (A) phenylalanine (high reactivity), (B) proline (medium reactivity), and (C) asparagine (control/nonreactive).
After brief exposure to hydroxyl radicals the sample is subjected to protease digestion. To determine the modification sites and to quantify the extent of oxidation, MS/MS techniques are employed. Due to the potential for multiple oxidation states for some residues and oxidation combinations within a given proteolytic peptide, a far larger and more complex data set is obtained than typical for a proteomics analysis. Efficient and reliable identification of the locations and relative amounts of oxidation requires sophisticated emerging computational analysis.
Recently, we have been exploring hydroxyl radical footprinting techniques for the determination of binding surfaces of protein–carbohydrate complexes. In particular, we were interested in assessing the potential of this method to resolve carbohydrate-binding sites for future studies where the protein–carbohydrate complex may not be amenable to traditional structure determination methods (Figure 1), such as antibody–polysaccharide complexes. Initially, we used human galectin-1-lactose as a model system and pulse laser irradiation of a 1% hydrogen peroxide solution for the generation of hydroxyl radicals. By comparison with MD data based on the crystal structure of the galectin-1–lactose complex (Lopez-Lucendo et al. 2004), hydroxyl radical footprinting was able to identify those residues defining the binding site (Figure 6). Differences in relative oxidation levels correlated well with the difference in SASA calculated from MD simulations of galectin-1 with and without lactose (Figure 6D) (Charvátová and Woods, unpublished data).
Fig. 6.
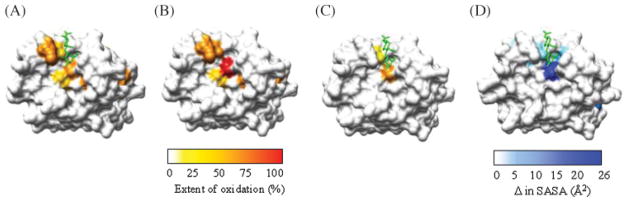
Extent of oxidation mapped onto the surface of the crystal structure of galectin-1 in the presence (A) and absence (B) of ligand. (C) The difference in the level of oxidation with and without ligand (with the ligand superimposed in green for reference) correlated with (D) the change in SASA of the protein from MD simulations with and without the ligand present.
Due to the large number of uncommon modifications that arose from the oxidation experiment, available proteomics (database-search) analysis software (Yates et al. 1995; Perkins et al. 1999) were poorly suited to assign the mass spectra of the modified peptides. A more efficient and sensitive identification of modified peptides was accomplished using the program ByOnic, which combines database searching with de novo analysis (Bern et al. 2007). Currently, the analysis of the MS data presents one of the most significant obstacles to hydroxyl radical footprinting technology, which must be overcome before footprinting makes the jump from “emerging” to established technology.
The ever-increasing pool of known carbohydrate-binding proteins and their preferred carbohydrate epitopes necessitates the development of efficient means to identify their interfaces; a challenge potentially well suited for hydroxyl radical footprinting and MS analysis.
Saturation transfer difference NMR
While footprinting may be used to identify the protein interface, STD NMR allows rapid identification of the regions of a carbohydrate ligand that are proximal to the protein surface in a complex (Mayer and Meyer 1999). STD NMR is now a mature technology; however, integrating STD NMR data with other methods, such as computational docking, is still evolving.
For the STD NMR experiment a single solution of the protein and carbohydrate ligand (with ~100-fold molar excess of ligand) is required, and two 1H-NMR spectra, at different saturation frequencies, are measured. The STD spectrum, or the ‘on-resonance’ spectrum, is acquired by irradiating at a small window of frequency where only the protein resonates (Figure 7). The protons from a ligand in exchange between the bound and free forms can also become differentially saturated when bound to the protein. The reference spectrum, or the ‘off-resonance’ spectrum, is acquired with irradiation far from the protein’s or ligand’s frequency range. The difference between the on- and off-resonance spectra, the difference STD NMR spectrum, identifies carbohydrate protons in close proximity to protons from the protein, revealing the carbohydrate epitope (Figure 7). A lack of observed intensities for carbohydrate protons does not necessarily indicate that they are distant from the protein surface, since carbohydrate interactions with regions of the protein surface with low-proton density, or interactions mediated by water do not facilitate magnetization transfer. Computational modeling used in conjunction with STD NMR can help explain such ambiguous cases, improving resolution of and confidence in the carbohydrate-binding epitope, or STD NMR can be used to validate computational predictions.
Fig. 7.

STD NMR characterized the carbohydrate-binding epitope of the siglec sialoadehesin at high resolution. (A) The STD NMR spectrum of sialyllactose in the presence of sialoadhesin and (B) the corresponding reference 1H NMR spectrum at 500 MHz. (C) The relative STD effects of sialyllactose bound to sialoadhesin. Percentages were calculated using the difference in individual signal intensities between the measured spectra and normalized to the largest STD effect (reprinted from Bhunia et al. 2004).
In addition to being a relatively straightforward experiment, STD NMR is advantageous because isotope labeling of the protein or ligand is not required (Meyer and Peters 2003). Some factors to consider when taking the spectra include the T1 relaxation times of the ligand protons and the effect of temperature on the measured spectra (Yan et al. 2003). Most critically, a fast off-rate of the ligand is required in order to observe saturation transfer to the ligand (Mayer and Meyer 1999).
STD NMR has been employed to characterize the carbohydrate-binding epitopes for a variety of classes of protein receptors, including glycosytransferases (Biet and Peters 2001; Macnaughtan et al. 2007), bacterial toxins (Haselhorst et al. 2004), viral and antiviral proteins (Sandstrom et al. 2004; Haselhorst et al. 2007), parasitic sialadases (Todeschini et al. 2002), lectins (Bhunia et al. 2004) (Figure 7), and antibodies (Maaheimo et al. 2000; Johnson and Pinto 2004). With data from STD experiments, validation of atomic resolution computational models of the protein–carbohydrate interaction is possible. In addition, using programs, such as CORCEMA (Moseley et al. 1995), nuclear Overhauser effect intensities can be computed for the complex allowing back-calculation of STD NMR intensities. Calculated STD NMR intensities have recently been employed in the validation of docking simulations (Bhunia et al. 2004; Haselhorst et al. 2004, 2007) and for refinement of docking poses (Jayalakshmi and Rama Krishna 2004).
Varieties of footprinting and saturation transfer NMR techniques have existed for decades, yet the application and modification of these methods to investigate problems in glycoscience has come about relatively recently. In a field where obstacles in structural determination abound, the potential synergy between these two methods is an appealing prospect for obtaining medium-to-high resolution structural information. Combining these methods with computational tools, such as protein homology modeling, MD simulations or docking, could provide experimentally guided/validated atomic resolution descriptions of protein–carbohydrate interactions.
Current challenges in structural glycoscience
Carbohydrate structure prediction
Given only the sequence of a protein, scientists have several tools for predicting its 3D structural properties, including homology or comparative modeling, threading and secondary structure prediction algorithms based on sequence similarity to other known proteins (reviewed in Marti-Renom et al. 2000), and more recently, de novo fold prediction algorithms (Baker 2000). Unfortunately, strategies based on sequence homology are unlikely candidates for application to carbohydrates. Two carbohydrate molecules may share a similar core structure, yet carbohydrate-residue modifications via addition, substitution, deletion, change in linkage configuration or chemical modification may yield molecules with vastly different structural properties. An example of this phenomenon is seen in amylose versus cellulose; both are linear polymers of glucose found in plants. The building blocks of amylose and cellulose are glucose-α-(1-4)-glucose and glucose-β-(1-4)-glucose, respectively. Because the building blocks are diasteriomers, the polymers have different structural and chemical properties leading to very different functions within plants and different susceptibilities to hydrolysis (digestion) in humans. Cellulose polymers are linear and rigid (Nishiyama et al. 2002, 2003), whereas amylose polymers are helical in shape and have more internal flexibility than cellulose (Gessler et al. 1999). For carbohydrates a single change, such as modifying an anomeric configuration or linkage position, is sufficient to create two biologically distinct molecules.
For proteins, mutations in the sequence can be classified in two general groups: conservative, such as Leu → Val, or nonconservative, such as Leu → Asp. However, carbohydrate mutations are not easily classified in such terms. While carbohydrate residues can be grouped into distinct families (hexoses, hexosamines, acidic, etc.), most carbohydrate residues share similar properties, particularly the existence of polar and nonpolar groups. Thus, such grouping is of limited usefulness since it is the great diversity in topological presentation of these groups that give carbohydrate residues their complexity. As discussed in the introduction, structural prediction is often further complicated by the existence of several low energy conformations under biological relevant conditions. Two computational methods, MC and MD simulations, have been extensively employed in the prediction of carbohydrate conformational families. Over the past decade, explicit solvent MD simulations have been successful in predicting conformational families, and often the relative populations of these families (Naidoo et al. 1997; Picard et al. 2000; Corzana et al. 2002, 2004; Eklund et al. 2005; Landersjo et al. 2005; Pereira et al. 2006). Evolution of new computational tools to predict the carbohydrate structure will benefit from the knowledge gained through experiments and computational simulations, and the challenge will be in finding ways to apply what we have learned to generate more efficient and accurate structure prediction tools.
Nomenclature
As part of the set of tools to probe the carbohydrate structure, nomenclature systems that effectively and precisely communicate carbohydrate structure—from monosaccharide composition to 3D shape—are essential. For many years glycoscientists have relied on the International Union of Pure and Applied Chemistry and the International Union of Biochemistry and Molecular Biology (IUPAC-IUBMB) standard (Figure 8) (McNaught 1997). Unfortunately, the IUPAC nomenclature system for carbohydrates is extremely complex, and unlike the IUPAC system for amino acids, peptides, and proteins, it is not suitable for many glycomics applications. In the short term, a glycomics nomenclature “wish-list” might include a standardized single-letter code for monosaccharides and a nomenclature system consistent with the PDB file format. Due to the format restrictions of PDB files (discussed below), a continuing objective is the development and implementation of a comprehensive nomenclature system appropriate for current and future glycomics applications. It is likely that future nomenclature systems will evolve to be machine readable, for reasons of accuracy and completeness, at the expense of human readability. Nevertheless, within the foreseeable future, the PDB format will continue to be the most widely supported and accepted file format for structural data.
Fig. 8.

A sample of carbohydrate nomenclature systems and schematic representations using a human blood group A antigen as an example. Due to the lengthy formats of GLYDE-II and GlycoCT, readers are directed to relevant websites to view sample XML codes.
For a standardized code for monosaccharides, we propose a one-letter code for monosaccharides and a two-letter code for their common derivatives (Table II). This system could be generalized for other applications or used to simplify existing nomenclature systems (Figure 8). This system is currently utilized in the GLYCAM carbohydrate and lipid force field as the foundation of its three-character PDB-style nomenclature system (http://www.glycam.com).
Table II.
Monosaccharide codes that form the core of the PDB-style GLYCAM nomenclature
| Symbol | Core monosaccharidesa | Symbol | Special cases and common derivativesb |
|---|---|---|---|
| Pentoses | 2-N-Acetylhexosamines | ||
| A | arabinose | GN | N-acetylglucosamine |
| R | ribose | LN | N-acetylgalactosamine |
| X | xylose | MN | N-acetylmannosamine |
| D | lyxose | ||
| Uronic Acids | |||
| Hexoses | GU | glucuronic acid | |
| G | glucose | LU | galacturonic acid |
| M | mannose | MU | manuronic acid |
| L | galactose | IU | iduronic acid |
| I | idose | ||
| Sialic Acids (9 carbon) | |||
| T | talose | ||
| 9N | N-acetylneuraminic acid | ||
| N | allose | ||
| 9G | N-glycolylneuraminic acid | ||
| E | altrose | ||
| 9O | 3-deoxy-D-manno-oct-2-ulosonic acid (KDO) | ||
| K | gulose | ||
| Hexuloses | Others | ||
| 8O | 3-deoxy-D-glycero-D-galacto-non-2-ulosonic acid (KDN) | ||
| C | fructose | ||
| P | psicose | ||
| S | sorbose | ||
| J | tagatose | ||
| 6-Deoxyhexoses | |||
| F | Fucose | ||
| Q | quinovose | ||
| H | rhamnose |
Case designates the isomeric configuration: D (upper case) and L (lower case).
The case of the first character designates the isomeric configuration: D (upper case) and L (lower case), with the exception of sialic acids and KDN. For the second character, case designates the anomeric configuration: α (upper case) or β (lower case).
Since, at present, all molecular visualization programs as well as many other types of biostructural software depend on the PDB format, a three-character code for carbohydrates is necessary. Creating a three-character system that captures the structural diversity of carbohydrate structures presents many challenges but would greatly facilitate structural searches and glycomics analysis. One of these challenges is the desire to explicitly state linkage information. When dealing with proteins, the linkage between amino acids is implicit in its sequence. Linkage information for carbohydrate molecules must be determined experimentally at each position (which is not a trivial task) and then explicitly defined in the nomenclature. To add another layer of complexity, carbohydrate residues can exist as D- or L-isomers, α- or β-anomers, and as pyranose or furanose ring forms. In animal systems, most monosaccharides exist as a single isomer (e.g. D-mannose), yet there are sufficient counter examples from outside the animal kingdom (Fichtinger-Schepman et al. 1979, 1981) to make the case for the inclusion of isomeric configurations in the nomenclature. This means that any standard code for carbohydrates residues would ideally include, at a minimum, five types of information: (1) residue type, (2) isomer, (3) anomer, (4) linkage, and (5) ring structure.
While it would be preferable for the PDB format restriction if all this information could fit into an uppercase three-letter residue identifier, it is not possible to specify all potential permutations of carbohydrate residues using only three noncase sensitive characters. As such, machine-readable nomenclature systems that dispense with the three-character PDB-constraint, such as the linearly formatted LINUCS (Bohne-Lang et al. 2001) and LinearCode (Banin et al. 2002), and the XML programming language codes Glyco-CT (Herget et al. 2007) and GLYDE-II (Sahoo et al. 2005), have been proposed for bioinformatics applications and described elsewhere (Figure 8). To accommodate the numerous biostructural applications that still make use of the PDB format, the GLYCAM PDB-style carbohydrate nomenclature system was created.
The development of the GLYCAM PDB-style nomenclature has evolved over the past 14 years (Woods et al. 1995; Tessier et al. 2007; Kirschner et al. 2008), originating from the necessity of having a descriptive and logical three-character naming system for carbohydrate residues for MD simulations with AMBER (Case et al. 2005). Here we propose a case sensitive three-character code that provides a descriptive, but concise nomenclature system for all monosaccharides and several common monosaccharide derivatives (Tables II–V) that can still meet the restriction of the PDB file format. A complete description of the nomenclature system can be found on the GLYCAM website (http://www.glycam.com). In general, the PDB-style GLYCAM code uses the first character to encode the linkage position(s) (Table III), the second to encode the residue (via the character, Table II) and the isomer (via case), and the third to encode anomeric configuration (via the character) and the ring size (via case). For specific monosaccharide derivatives, the third-placed character encodes the derivative class (via the character, Table II) and anomeric configuration (via case) at the expense of defining the ring size. Given only three characters, we have attempted to generate a comprehensive, relatively readable and flexible nomenclature system (Tables II–V). This naming convention, as well as the GLYCAM parameters, can be used with molecular mechanics programs that support AMBER input files (Case et al. 2005), including NAMD (Phillips et al. 2005) and NWChem (Kendall et al. 2000). Additionally, the GLYCAM nomenclature system can be implemented in other molecular mechanics programs such as GROMOS (Scott et al. 1999) and CHARMM (Brooks et al. 1983).
Table V.
Examples of PDB-style GLYCAM residue identifiers for common monosaccharide derivatives (N-acetylhexosamines and uronic acids) specifying linkage position (first position), monosaccharide residue (second and third position), D- or L-configuration (second position case), and anomeric configuration (third position case)
| Linkage |
N-Acetylhexosamines
|
Uronic acids
|
||||||
|---|---|---|---|---|---|---|---|---|
| D-Sugars
|
L-Sugars
|
D-Sugars
|
L-Sugars
|
|||||
| α-D-Glcp-Nac | β-D-Glcp-NAc | α-L-Glcp-Nac | β-L-Glcp-NAc | α-D-Glcp-A | β-D-Glcp-A | α-L-Glcp-A | β-L-Glcp-A | |
| 3- | 3GN | 3Gn | 3gN | 3gn | 3GU | 3Gu | 3gU | 3gu |
| 4- | 4GN | 4Gn | 4gN | 4gn | 4GU | 4Gu | 4gU | 4gu |
| 3,4- | WGN | WGn | WgN | Wgn | WGU | Wgu | WgU | Wgu |
Table III.
One-letter codes for specifying linkage positions in pyranoses and furanoses
| Symbol | Linkage | Symbol | Linkage |
|---|---|---|---|
| 0 | Terminal | v | 3,6- |
| 1 | 1- | U | 4,6- |
| 2 | 2- | M | 5,6- |
| 3 | 3- | T | 2,3,4- |
| 4 | 4- | L | 2,3,5- |
| 6 | 6- | s | 2,3,6- |
| Z | 2,3- | R | 2,4,6- |
| Y | 2,4- | K | 2,5,6- |
| O | 2,5- | Q | 3,4,6- |
| x | 2,6- | J | 3,5,6- |
| w | 3,4- | p | 2,3,4,6- |
| N | 3,5- | I | 2,3,5,6- |
While suitable for many monosaccharides, no three-character system is sufficiently robust to cover all possible monosaccharide derivatives. Due to this format-imposed limitation, the GLYCAM nomenclature system includes monosaccharides and common monosaccharide derivatives, omitting the more elaborate and rare derivatives. Another issue arises due to potential overlaps with existing PDB residue identifiers. For these reasons, we are working to implement an extended version of the GLYCAM code suited for the newer Research Collaboratory for Structural Bioinformatics standard format, macromolecular Crystallographic Information File (mmCIF). This newer format dispenses with the three-character limitation, which will permit the development of a highly descriptive and comprehensive, yet relatively user-friendly, nomenclature system.
Conclusion
The ability of proteins to recognize and distinguish carbohydrate molecules is at the heart of many critical biological processes, such as cell adhesion, adaptive and innate immunity, and cell signaling. Carbohydrates flex and bend, taking on different 3D shapes, yet somehow nature has evolved to recognize these complex and pliable molecules. Protein receptors are able to differentiate between (1) the varied 3D shapes any given carbohydrate may adopt and (2) closely related carbohydrates that differ only in such structural properties as configuration or linkage position. To develop a better understanding how nature employs the peculiar structural and dynamic properties of carbohydrates to its advantage, 3D characterization of carbohydrate–protein complexes is required. However, structure determination of flexible and dynamic molecules is a great challenge. Beginning with more rigid systems (often small carbohydrates in complex with proteins, or glycoproteins with deliberately trimmed glycans), crystallography and NMR spectroscopy added the first few rungs of the technical ladder that leads to more high throughput methods for carbohydrate structure determination. For the next rung of the ladder, MD simulations were employed, alongside NMR techniques, as a means to identify the 3D shapes, and relative populations, of more complex systems.
The latest major advance has been the creation and widespread use of glycan microarrays for receptor screening. With the emerging wealth of microarray data comes the urgent need for high throughput methods that provide structural insight into the mechanisms of carbohydrate recognition. By characterizing the interacting interfaces between ligands and protein surfaces, experimental and computational methods offer a new route to deriving the 3D structures of carbohydrate–protein complexes. Current computational techniques for carbohydrate–protein structural characterization include docking simulations and absolute and relative free energy calculations. On the experimental front, we have highlighted oxidative footprinting of the receptor surface, which is emerging as a method to complement epitope-mapping data from techniques, such as STD NMR. Also, NMR strategies that use partially oriented samples, in combination with computational simulations, are a promising avenue for 3D oligosaccharide–protein structure determination.
Table IV.
Example PDB-style GLYCAM residue identifiers for glucose. Specifying linkage position (first position), monosaccharide residue (second position: one-letter code), D- or L-configuration (second position case), anomeric configuration (third position), and pyranose of furanose (third position case)
| Linkage | Pyranoses
|
Furanoses
|
||||||
|---|---|---|---|---|---|---|---|---|
| D-Sugars
|
L-Sugars
|
D-Sugars
|
L-Sugars
|
|||||
| A-D-Glcp- | β-D-Glcp- | α-L-Glcp- | β-L-Glcp- | α-D-Glcf- | β-D-Glcf- | α-L-Glcf- | β-L-Glcf- | |
| 1- | 1GA | 1GB | 1gA | 1gB | 1Ga | 1Gb | 1ga | 1gb |
| 4- | 4GA | 4GB | 4gA | 4gB | 4Ga | 4Gb | 4ga | 4gb |
| 2,6- | XGA | XGB | XgA | XgB | Xga | XGb | Xga | Xgb |
| 2,3,6- | SGA | SGB | SgA | SgB | Sga | SGb | Sga | Sgb |
Acknowledgments
We would like to acknowledge Professors P. Hünenberger, A. Imberty, C. von der Lieth, and J. Naismith for their insightful comments regarding the development of the GLYCAM nomenclature system.
Abbreviations
- CSA
chemical shift anisotropy
- IL8
interleukin 8
- LacNAc
N-acetyllactosamine (βGal(1-4)GlcNAc)
- MC
Monte Carlo
- MD
molecular dynamics
- MS
mass spectrometry
- RDC
residual dipolar coupling
- SASA
solvent accessible surface area
- STD NMR
saturation transfer difference nuclear magnetic resonance
- TI
thermodynamic integration
Footnotes
Conflict of interest statement
There are no conflicts of interest.
References
- Archontis G, Watson KA, Xie Q, Andreou G, Chrysina ED, Zographos SE, Oikonomakos NG, Karplus M. Glycogen phosphorylase inhibitors: A free energy perturbation analysis of glucopyranose spirohydantoin analogues. Proteins. 2005;61:984–998. doi: 10.1002/prot.20641. [DOI] [PubMed] [Google Scholar]
- Babu YS, Chand P, Bantia S, Kotian P, Dehghani A, El-Kattan Y, Lin TH, Hutchison TL, Elliott AJ, et al. BCX-1812 (RWJ-270201): Discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J Med Chem. 2000;43:3482–3486. doi: 10.1021/jm0002679. [DOI] [PubMed] [Google Scholar]
- Baker D. A surprising simplicity to protein folding. Nature. 2000;405:39–42. doi: 10.1038/35011000. [DOI] [PubMed] [Google Scholar]
- Banin E, Neuberger Y, Altshuler Y, Halevi A, Inbar O, Nir D, Dukler A. A novel linear code nomenclature for complex carbohydrates. Trends Glycoscience Glycotechnol. 2002;14:127–137. [Google Scholar]
- Bern M, Cai Y, Goldberg D. Lookup peaks: A hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal Chem. 2007;79:1393–1400. doi: 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- Bhunia A, Jayalakshmi V, Benie AJ, Schuster O, Kelm S, Krishna NR, Peters T. Saturation transfer difference NMR and computational modeling of a sialoadhesin-sialyl lactose complex. Carbohydr Res. 2004;339:259–267. doi: 10.1016/j.carres.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Bichard CJF, Mitchell EP, Wormald MR, Watson KA, Johnson LN, Zographos SE, Koutra DD, Oikonomakos NG, Fleet GWJ. Potent inhibition of glycogen phosphorylase by a spirohydantoin of glucopyranose: First pyranose analogues of hydantocidin. Tetrahedron Lett. 1995;36:2145. [Google Scholar]
- Biet T, Peters T. Molecular recognition of UDP-Gal by beta-1,4-galactosyltransferase T1. Angew Chem, Int Ed. 2001;40:4189–4192. doi: 10.1002/1521-3773(20011119)40:22<4189::AID-ANIE4189>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Bitomsky W, Wade RC. Docking of glycosaminoglycans to heparin-binding proteins: Validation for aFGF, bFGF, and antithrombin and application to IL-8. J Am Chem Soc. 1999;121:3004–3013. [Google Scholar]
- Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci USA. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne-Lang A, Lang E, Forster T, von der Lieth C-W. LINUCS: Linear notation for unique description of carbohydrate sequences. Carbohydr Res. 2001;336:1. doi: 10.1016/s0008-6215(01)00230-0. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Brown GD, Gordon S. Immune recognition: A new receptor for b-glucans. Nature. 2001;413:36. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- Bryce RA, Hillier IH, Naismith JH. Carbohydrate-protein recognition: Molecular dynamics simulations and free energy analysis of oligosaccharide binding to concanavalin A. Biophys J. 2001;81:1373–1388. doi: 10.1016/S0006-3495(01)75793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundle DR, Eichler E, Gidney MA, Meldal M, Ragauskas A, Sigurskjold BW, Sinnott B, Watson DC, Yaguchi M, et al. Molecular recognition of a Salmonella trisaccharide epitope by monoclonal antibody Se155–4. Biochemistry. 1994;33:5172–5182. doi: 10.1021/bi00183a022. [DOI] [PubMed] [Google Scholar]
- Buskas T, Ingale S, Boons GJ. Towards a fully synthetic carbohydrate-based anticancer vaccine: Synthesis and immunological evaluation of a lipidated glycopeptide containing the tumor-associated Tn antigen. Angew Chem Int Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]
- Campbell BJ, Yu LG, Rhodes JM. Altered glycosylation in inflammatory bowel disease: A possible role in cancer development. Glycoconj J. 2001;18:851–858. doi: 10.1023/a:1022240107040. [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, et al. The AMBER biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casset F, Hamelryck T, Loris R, Brisson J-R, Tellier C, Dao-Thi M-H, Wyns L, Poortmans F, Pérez S, et al. NMR, molecular modeling, and crystallographic studies of lentil lectin-sucrose interaction. J Biol Chem. 1995;270:25619–25628. doi: 10.1074/jbc.270.43.25619. [DOI] [PubMed] [Google Scholar]
- Chand P, Kotian PL, Dehghani A, El-Kattan Y, Lin TH, Hutchison TL, Babu YS, Bantia S, Elliott AJ, et al. Systematic structure-based design and stereoselective synthesis of novel multisubstituted cyclopentane derivatives with potent antiinfluenza activity. J Med Chem. 2001;44:4379–4392. doi: 10.1021/jm010277p. [DOI] [PubMed] [Google Scholar]
- Cobb BA, Kasper DL. Coming of age: Carbohydrates and immunity. Eur J Immunol. 2005;35:352–356. doi: 10.1002/eji.200425889. [DOI] [PubMed] [Google Scholar]
- Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN) Kidney Int. 2004;65:1544–1547. doi: 10.1111/j.1523-1755.2004.05407.x. [DOI] [PubMed] [Google Scholar]
- Corzana F, Bettler E, Herve du Penhoat C, Tyrtysh TV, Bovin NV, Imberty A. Solution structure of two xenoantigens: aGal-LacNAc and aGal-Lewis X. Glycobiology. 2002;12:241–250. doi: 10.1093/glycob/12.4.241. [DOI] [PubMed] [Google Scholar]
- Corzana F, Motawia MS, Du Penhoat CH, Perez S, Tschampel SM, Woods RJ, Engelsen SB. A hydration study of (1→4) and (1→6) linked alpha-glucans by comparative 10 ns molecular dynamics simulations and 500-MHz NMR. J Comput Chem. 2004;25:573–586. doi: 10.1002/jcc.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreitlein WB, Maratos J, Brocavich J. Zanamivir and oseltamivir: Two new options for the treatment and prevention of influenza. Clin Ther. 2001;23:327–355. doi: 10.1016/s0149-2918(01)80042-4. [DOI] [PubMed] [Google Scholar]
- Dwek RA. Glycobiology: Toward understanding the function of sugars. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- Eklund R, Lycknert K, Soderman P, Widmalm G. A conformational dynamics study of α-L-Rhap-(1-2)[α-L-Rhap-(1-3)]-α-L-Rhap-OMe in solution by NMR experiments and molecular simulations. J Phys Chem B. 2005;109:19936–19945. doi: 10.1021/jp053198o. [DOI] [PubMed] [Google Scholar]
- Ewing TJ, AKuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- Fichtinger-Schepman AMJ, Kamerling JP, Versluis C, Vliegenthart JFG. Structural studies of the methylated, acidic polysaccharide associated with coccoliths of Emiliania huxleyi (lohmann) kamptner. Carbohydr Res. 1981;93:105. [Google Scholar]
- Fichtinger-Schepman AMJ, Kamerling JP, Vliegenthart JFG. Composition of a methylated, acidic polysaccharide associated with coccoliths of Emiliania huxleyi (lohmann) Carbohydr Res. 1979;69:181–189. [Google Scholar]
- Finley JB, Atigadda VR, Duarte F, Zhao JJ, Brouillette WJ, Air GM, Luo M. Novel aromatic inhibitors of influenza virus neuraminidase make selective interactions with conserved residues and water molecules in the active site. J Mol Biol. 1999;293:1107. doi: 10.1006/jmbi.1999.3180. [DOI] [PubMed] [Google Scholar]
- Ford MG, Weimar T, Kohli T, Woods RJ. Molecular dynamics simulations of galectin-1-oligosaccharide complexes reveal the molecular basis for ligand diversity. Proteins. 2003;53:229–240. doi: 10.1002/prot.10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH. Update and perspectives on congenital disorders of glycosylation. Glycobiology. 2001;11:129r–143r. [PubMed] [Google Scholar]
- Fukuda M. Possible roles of tumor-associated carbohydrate antigens. Cancer Res. 1996;56:2237–2244. [PubMed] [Google Scholar]
- Fukui S, Feizi T, Galustian C, Lawson AM, Chai W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat Biotech. 2002;20:1011. doi: 10.1038/nbt735. [DOI] [PubMed] [Google Scholar]
- Galas DJ, Schmitz A. DNAase footprinting – simple method for detection of protein-DNA binding specificity. Nucleic Acids Res. 1978;5:3157–3170. doi: 10.1093/nar/5.9.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessler K, Uson I, Takaha T, Krauss N, Smith SM, Okada S, Sheldrick GM, Saenger W. V-Amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucose residues (Cyclomaltohexaicosaose) Proc Natl Acad Sci USA. 1999;96:4246–4251. doi: 10.1073/pnas.96.8.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Outeirino J, Kadirvelraj R, Woods RJ. Structural elucidation of type III group B Streptococcus capsular polysaccharide using molecular dynamics simulations: The role of sialic acid. Carbohydr Res. 2005;340:1007–1018. doi: 10.1016/j.carres.2004.12.034. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Outeirino J, Kirschner KN, Thobhani S, Woods RJ. Reconciling solvent effects on rotamer populations in carbohydrates – a joint MD and NMR analysis. Can J Chem. 2006;84:569–579. doi: 10.1139/v06-036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodford PJ. A computational-procedure for determining energetically favorable binding-sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: Applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- Grabowski GA, Hopkin RJ. Enzyme therapy for lysosomal storage disease: Principles, practice, and prospects. Annu Rev Genom Hum Gen. 2003;4:403–436. doi: 10.1146/annurev.genom.4.070802.110415. [DOI] [PubMed] [Google Scholar]
- Gregoriou M, Noble MEM, Watson KA, Garman EF, Krulle TM, De-La-Fuente C, Fleet GWJ, Oikonomakos NG, Johnson LN. The structure of a glycogen phosphorylase glucopyranose spirohydantoin complex at 1.8 Å resolution and 100 K: The role of the water structure and its contribution to binding. Protein Sci. 1998;7:915–927. doi: 10.1002/pro.5560070409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakomori SI. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res. 1989;52:257–331. doi: 10.1016/s0065-230x(08)60215-8. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Haselhorst T, Blanchard H, Frank M, Kraschnefski MJ, Kiefel MJ, Szyczew AJ, Dyason JC, Fleming F, Holloway G, et al. STD NMR spectroscopy and molecular modeling investigation of the binding of N-acetylneuraminic acid derivatives to rhesus rotavirus VP8* core. Glycobiology. 2007;17:68–81. doi: 10.1093/glycob/cwl051. [DOI] [PubMed] [Google Scholar]
- Haselhorst T, Wilson JC, Thomson RJ, McAtamney S, Menting JG, Coppel RL, von Itzstein M. Saturation transfer difference (STD) H-NMR experiments and in silico docking experiments to probe the binding of N-acetylneuraminic acid and derivatives to Vibrio cholerae sialidase. Proteins. 2004;56:346–353. doi: 10.1002/prot.20143. [DOI] [PubMed] [Google Scholar]
- Herget S, Ranzinger R, von der Lieth CW. A sequence format and namespace for complex oligo- and polysaccharides. 2007 ( http://www.eurocarbdb.org/recommendations/encoding)
- Imberty A, Perez S. Structure, conformation, and dynamics of bioactive oligosaccharides: Theoretical approaches and experimental validations. Chem Rev. 2000;100:4567–4588. doi: 10.1021/cr990343j. [DOI] [PubMed] [Google Scholar]
- Jayalakshmi V, Rama Krishna N. CORCEMA refinement of the bound ligand conformation within the protein binding pocket in reversibly forming weak complexes using STD-NMR intensities. J Magn Reson. 2004;168:36. doi: 10.1016/j.jmr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Jedrzejas MJ, Singh S, Brouillette WJ, Air GM, Luo M. A strategy for theoretical binding constant, K-I, calculations for neuraminidase aromatic inhibitors designed on the basis of the active-site structure of influenza-virus neuraminidase. Proteins. 1995;23:264–277. doi: 10.1002/prot.340230215. [DOI] [PubMed] [Google Scholar]
- Jennings H. Further approaches for optimizing polysaccharide-protein conjugate vaccines for prevention of invasive bacterial disease. J Infect Dis. 1992;165(Suppl 1):S156–159. doi: 10.1093/infdis/165-supplement_1-s156. [DOI] [PubMed] [Google Scholar]
- Jennings HJ, Lugowski C, Kasper DL. Conformational aspects critical to the immunospecificity of the type III group B streptococcal polysaccharide. Biochemistry. 1981;20:4511–4518. doi: 10.1021/bi00519a001. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Pinto BM. Saturation-transfer difference NMR studies for the epitope mapping of a carbohydrate-mimetic peptide recognized by an anti-carbohydrate antibody. Bioorg Med Chem. 2004;12:295. doi: 10.1016/j.bmc.2003.09.041. [DOI] [PubMed] [Google Scholar]
- Kadirvelraj R, Gonzalez-Outeirino J, Foley BL, Beckham ML, Jennings HJ, Foote S, Ford MG, Woods RJ. Understanding the bacterial polysaccharide antigenicity of Streptococcus agalactiae versus Streptococcus pneumoniae. Proc Natl Acad Sci USA. 2006;103:8149–8154. doi: 10.1073/pnas.0602815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaveg K, Moremen KW. Energetics of substrate binding and catalysis by class 1 (Glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J Biol Chem. 2005;280:29837–29848. doi: 10.1074/jbc.M505130200. [DOI] [PubMed] [Google Scholar]
- Karaveg K, Siriwardena A, Tempel W, Liu Z-J, Glushka J, Wang B-C, Moremen KW. Mechanism of class 1 (Glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J Biol Chem. 2005;280:16197–16207. doi: 10.1074/jbc.M500119200. [DOI] [PubMed] [Google Scholar]
- Karlsson K-A. Animal glycolipids as attachment sites for microbes. Chem Phys Lipids. 1986;42:153. doi: 10.1016/0009-3084(86)90050-2. [DOI] [PubMed] [Google Scholar]
- Karlsson K-A. Animal glycosphingolipids as membrane attachment sites for bacteria. Annu Rev Biochem. 1989;58:309–350. doi: 10.1146/annurev.bi.58.070189.001521. [DOI] [PubMed] [Google Scholar]
- Kendall RA, Apra E, Bernholdt DE, Bylaska EJ, Dupuis M, Fann GI, Harrison RJ, Ju J, Nichols JA, et al. High performance computational chemistry: An overview of NWChem a distributed parallel application. Comput Phys Commun. 2000;128:260. [Google Scholar]
- King D, Lumpkin M, Bergmann C, Orlando R. Studying protein-carbohydrate interactions by amide hydrogen/deuterium exchange mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:1569–1574. doi: 10.1002/rcm.746. [DOI] [PubMed] [Google Scholar]
- Kirschner KN, Woods RJ. Solvent interactions determine carbohydrate conformation. Proc Natl Acad Sci USA. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner KN, Yongye A, Tschampel SM, González-Outeiriño J, Daniels C, Woods RJ. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschert GSV, Hoogewerf AJ, Proudfoot AEI, Chung Cw, Cooke RM, Hubbard RE, Wells TNC, Sanderson PN. Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry. 1998;37:11193–11201. doi: 10.1021/bi972867o. [DOI] [PubMed] [Google Scholar]
- Landersjo C, Jansson JLM, Maliniak A, Widmalm G. Conformational analysis of a tetrasaccharide based on NMR spectroscopy and molecular dynamics simulations. J Phys Chem B. 2005;109:17320–17326. doi: 10.1021/jp052206y. [DOI] [PubMed] [Google Scholar]
- Liao DI, Kapadia G, Ahmed H, Vasta GR, Herzberg O. Structure of S-lectin, a developmentally regulated vertebrate b-galactoside-binding protein. Proc Natl Acad Sci USA. 1994;91:1428–1432. doi: 10.1073/pnas.91.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Eichelberger MC, Compans RW, Air GM. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J Virol. 1995;69:1099–1106. doi: 10.1128/jvi.69.2.1099-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo-Man R, Vichier-Guerre S, Perraut R, Deriaud E, Huteau V, BenMohamed L, Diop OM, Livingston PO, Bay S, et al. A fully synthetic therapeutic vaccine candidate targeting carcinoma-associated Tn carbohydrate antigen induces tumor-specific antibodies in nonhuman primates. Cancer Res. 2004;64:4987–4994. doi: 10.1158/0008-5472.CAN-04-0252. [DOI] [PubMed] [Google Scholar]
- Lopez-Lucendo MF, Solis D, Andre S, Hirabayashi J, Kasai K-i, Kaltner H, Gabius H-J, Romero A. Growth-regulatory human galectin-1: Crystallographic characterisation of the structural changes induced by single-site mutations and their impact on the thermodynamics of ligand binding. J Mol Biol. 2004;343:957. doi: 10.1016/j.jmb.2004.08.078. [DOI] [PubMed] [Google Scholar]
- Lütteke T, Frank M, von der Lieth C-W. Carbohydrate structure suite (CSS): Analysis of carbohydrate 3D structures derived from the PDB. Nucl Acids Res. 2005;33:D242–D246. doi: 10.1093/nar/gki013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaheimo H, Kosma P, Brade L, Brade H, Peters T. Mapping the binding of synthetic disaccharides representing epitopes of chlamydial lipopolysaccharide to antibodies with NMR. Biochemistry. 2000;39:12778–12788. doi: 10.1021/bi000780o. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Macnaughtan MA, Kamar M, Alvarez-Manilla G, Venot A, Glushka J, Pierce JM, Prestegard JH. NMR structural characterization of substrates bound to N-acetylglucosaminyltransferase V. J Mol Biol. 2007;366:1266. doi: 10.1016/j.jmb.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Wormald MR, Rudd PM, Fishcher PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med. 1995;1:237–243. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Masukawa KM, Kollman PA, Kuntz ID. Investigation of neuraminidase-substrate recognition using molecular dynamics and free energy calculations. J Med Chem. 2003;46:5628–5637. doi: 10.1021/jm030060q. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem, Int Ed. 1999;38:1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- McNaught AD. Nomenclature of carbohydrates (recommendations 1996) Carbohydr Res. 1997;297:1. [PubMed] [Google Scholar]
- Merritt EA, Sarfaty S, Feil IK, Hol WG. Structural foundation for the design of receptor antagonists targeting Escherichia coli heat-labile enterotoxin. Structure. 1997;5:1485–1499. doi: 10.1016/s0969-2126(97)00298-0. [DOI] [PubMed] [Google Scholar]
- Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E. Equation of state calculations by fast computing machines. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- Metropolis N, Ulam S. The Monte Carlo method. J Am Stat Assoc. 1949;44:335–341. doi: 10.1080/01621459.1949.10483310. [DOI] [PubMed] [Google Scholar]
- Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem, Int Ed. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- Minke WE, Diller DJ, Hol WGJ, Verlinde CLMJ. The role of waters in docking strategies with incremental flexibility for carbohydrate derivatives: Heat-labile enterotoxin, a multivalent test case. J Med Chem. 1999;42:1778–1788. doi: 10.1021/jm980472c. [DOI] [PubMed] [Google Scholar]
- Minke WE, Roach C, Hol WGJ, Verlinde CLMJ. Structure-based exploration of the ganglioside GM1 binding sites of Escherichia coli heat-labile enterotoxin and cholera toxin for the discovery of receptor antagonists. Biochemistry. 1999;38:5684–5692. doi: 10.1021/bi982649a. [DOI] [PubMed] [Google Scholar]
- Moseley HNB, Curto EV, Krishna NR. Complete relaxation and conformational exchange matrix (CORCEMA) analysis of NOESY spectra of interacting systems – 2-dimensional transferred NOESY. J Magn Reson B. 1995;108:243–261. doi: 10.1006/jmrb.1995.1129. [DOI] [PubMed] [Google Scholar]
- Motoki I, Yosinari S, Watanabe K, Nishikawa K. Structural analyses on yeast tRNA(Tyr) and its complex with tyrosyl-tRNA synthetase by the use of hydroxyl radical ‘footprinting’. Nucleic Acids Symp Ser. 1991:173–174. [PubMed] [Google Scholar]
- Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, Vrtovsnik F, Haddad E, Chintalacharuvu KR, et al. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol. 2004;15:622–634. doi: 10.1097/01.asn.0000115401.07980.0c. [DOI] [PubMed] [Google Scholar]
- Mulakala C, Nerinckx W, Reilly PJ. Docking studies on glycoside hydrolase family 47 endoplasmic reticulum a-(1→2)-mannosidase I to elucidate the pathway to the substrate transition state. Carbohydr Res. 2006;341:2233. doi: 10.1016/j.carres.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Naidoo KJ, Denysyk D, Brady JW. Molecular dynamics simulations of the N-linked oligosaccharide of the lectin from Erythrina corallodendron. Protein Eng. 1997;10:1249–1261. doi: 10.1093/protein/10.11.1249. [DOI] [PubMed] [Google Scholar]
- Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, Langan P, Chanzy H. Crystal structure and hydrogen-bonding system in cellulose 1 beta from synchrotron x-ray and neutron fiber diffraction. J Am Chem Soc. 2002;124:9074–9082. doi: 10.1021/ja0257319. [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, Sugiyama J, Chanzy H, Langan P. Crystal structure and hydrogen bonding system in cellulose 1a, from synchrotron x-ray and neutron fiber diffraction. J Am Chem Soc. 2003;125:14300–14306. doi: 10.1021/ja037055w. [DOI] [PubMed] [Google Scholar]
- Oikonomakos NG, Skamnaki VT, Osz E, Szilagyi L, Somsak L, Docsa T, Toth B, Gergely P. Kinetic and crystallographic studies of glucopyranosylidene spirothiohydantoin binding to glycogen phosphorylase b. Bioorg Med Chem. 2002;10:261. doi: 10.1016/s0968-0896(01)00277-2. [DOI] [PubMed] [Google Scholar]
- Österberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- Palese P, Compans RW. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): Mechanism of action. J Gen Virol. 1976;33:159–163. doi: 10.1099/0022-1317-33-1-159. [DOI] [PubMed] [Google Scholar]
- Palese P, Tobita K, Ueda M, Compans RW. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, Rademacher TW, Mizuochi T, Taniguchi T, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature. 1985;316:452–457. doi: 10.1038/316452a0. [DOI] [PubMed] [Google Scholar]
- Pathiaseril A, Woods RJ. Relative energies of binding for antibody-carbohydrate-antigen complexes computed from free-energy simulations. J Am Chem Soc. 2000;122:331–338. doi: 10.1021/ja9914994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson JC, Blixt O, Collins BE. Sweet spots in functional glycomics. Nat Chem Biol. 2006;2:238. doi: 10.1038/nchembio785. [DOI] [PubMed] [Google Scholar]
- Pereira CS, Kony D, Baron R, Muller M, van Gunsteren WF, Hunenberger PH. Conformational and dynamical properties of disaccharides in water: A molecular dynamics study. Biophys J. 2006;90:4337–4344. doi: 10.1529/biophysj.106.081539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Petrescu AJ, Petrescu SM, Dwek RA, Wormald MR. A statistical analysis of N- and O-glycan linkage conformations from crystallographic data. Glycobiology. 1999;9:343–352. doi: 10.1093/glycob/9.4.343. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Gruza J, Derouet C, Renard C, Mazeau K, Koca J, Imberty A, du Penhoat CH. A conformational study of the xyloglucan oligomer, XXXG, by NMR spectroscopy and molecular modeling. Biopolymers. 2000;54:11–26. doi: 10.1002/(SICI)1097-0282(200007)54:1<11::AID-BIP20>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Platt FM, Neises GR, Dwek RA, Butters TD. N-Butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269:8362–8365. [PubMed] [Google Scholar]
- Potier M, Mameli L, Belisle M, Dallaire L, Melancon SB. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-α-D-N-acetylneuraminate) substrate. Anal Biochem. 1979;94:287–296. doi: 10.1016/0003-2697(79)90362-2. [DOI] [PubMed] [Google Scholar]
- Prestegard JH. New techniques in structural NMR – anisotropic interactions. Nat Struct Biol. 1998;5:517. doi: 10.1038/756. [DOI] [PubMed] [Google Scholar]
- Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- Rarey M, Kramer B, Lengauer T. The particle concept: Placing discrete water molecules during protein-ligand docking predictions. Proteins. 1999;34:17–28. [PubMed] [Google Scholar]
- Rostand KS, Esko JD. Microbial adherence to and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachchidanand, Lequin O, Staunton D, Mulloy B, Forster MJ, Yoshida K, Campbell ID. Mapping the heparin-binding site on the 13–14F3 fragment of fibronectin. J Biol Chem. 2002;277:50629–50635. doi: 10.1074/jbc.M208956200. [DOI] [PubMed] [Google Scholar]
- Sahoo SS, Thomas C, Sheth A, Henson C, York WS. GLYDE – an expressive XML standard for the representation of glycan structure. Carbohydr Res. 2005;340:2802. doi: 10.1016/j.carres.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Sandstrom C, Berteau O, Gemma E, Oscarson S, Kenne L, Gronenborn AM. Atomic mapping of the interactions between the antiviral agent cyanovirin-N and oligomannosides by saturation-transfer difference NMR. Biochemistry. 2004;43:13926–13931. doi: 10.1021/bi048676k. [DOI] [PubMed] [Google Scholar]
- Sayers EW, Prestegard JH. Solution conformations of a trimannoside from nuclear magnetic resonance and molecular dynamics simulations. Biophys J. 2000;79:3313–3329. doi: 10.1016/S0006-3495(00)76563-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Prestegard JH. Conformation of a trimannoside bound to mannose-binding protein by nuclear magnetic resonance and molecular dynamics simulations. Biophys J. 2002;82:2683–2699. doi: 10.1016/S0006-3495(02)75610-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Galas DJ. Sequence-specific interactions of the tight-binding I12-X86 lac repressor with non-operator DNA. Nucleic Acids Res. 1980;8:487–506. doi: 10.1093/nar/8.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48:47–62. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Kruger P, et al. The GROMOS biomolecular simulation program package. J Phys Chem. 1999;103:3596–3607. [Google Scholar]
- Seyfried NT, Atwood JA, Almond A, Day AJ, Orlando R, Woods RJ. Fourier transform mass spectrometry to monitor hyaluronan – protein interactions: Use of hydrogen/deuterium amide exchange. Rapid Commun Mass Spectrom. 2007;21:121–131. doi: 10.1002/rcm.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon N, Lis H. Carbohydrates in cell recognition. Sci Am. 1993;268:82–89. doi: 10.1038/scientificamerican0193-82. [DOI] [PubMed] [Google Scholar]
- Sharp JS, Becker JM, Hettich RL. Protein surface mapping by chemical oxidation: Structural analysis by mass spectrometry. Analyt Biochem. 2003;313:216–225. doi: 10.1016/s0003-2697(02)00612-7. [DOI] [PubMed] [Google Scholar]
- Sharp JS, Becker JM, Hettich RL. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal Biochem. 2004;76:672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- Sheshberadaran H, Payne LG. Protein antigen monoclonal-antibody contact sites investigated by limited proteolysis of monoclonal antibody-bound antigen – protein footprinting. Proc Natl Acad Sci USA. 1988;85:1–5. doi: 10.1073/pnas.85.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly WS, Vogler C. Brain-directed gene therapy for lysosomal storage disease: Going well beyond the blood-brain barrier. Proc Natl Acad Sci USA. 2002;99:5760–5762. doi: 10.1073/pnas.102175599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DC, Lord JM, Roberts LA, Johannes L. Glycosphingolipids as toxin receptors. Semin Cell Dev Biol. 2004;15:397–408. doi: 10.1016/j.semcdb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141. [Google Scholar]
- Swanson JMJ, Henchman RH, McCammon JA. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys J. 2004;86:67–74. doi: 10.1016/S0006-3495(04)74084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- Tessier MB, DeMarco ML, Yongye A, Woods RJ. Extension of the GLY-CAM06 biomolecular force field to lipids, lipid bilayers and glycolipids. Mol Sim. 2007 doi: 10.1080/08927020701710890. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todeschini AR, Girard MF, Wieruszeski J-M, Nunes MP, DosReis GA, Mendonca-Previato L, Previato JO. Trans-sialidase from Trypanosoma cruzi binds host T-lymphocytes in a lectin manner. J Biol Chem. 2002;277:45962–45968. doi: 10.1074/jbc.M203185200. [DOI] [PubMed] [Google Scholar]
- Valafar H, Prestegard JH. REDCAT: A residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Vallee F, Karaveg K, Herscovics A, Moremen KW, Howell PL. Structural basis for catalysis and inhibition of N-glycan processing class I alpha 1,2-mannosidases. J Biol Chem. 2000;275:41287–41298. doi: 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- Varki A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vliegenhart JFG, Woods RJ. NMR spectroscopy and computer modeling of carbohydrates. Recent advances. ACS Symp Ser 2006 [Google Scholar]
- von Itzstein M, Wu W-Y, Kok GB, Pegg MS, Dyason JC, Jin B, Phan TV, Smythe ML, White HF, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- Wang XD, Padgett RA. Hydroxyl radical footprinting of RNA –Application to pre-messenger RNA splicing complexes. Proc Natl Acad Sci USA. 1989;86:7795–7799. doi: 10.1073/pnas.86.20.7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson KA, Chrysina ED, Tsitsanou KE, Zographos SE, Archontis G, Fleet GWJ, Oikonomakos NG. Kinetic and crystallographic studies of glucopyranose spirohydantoin and glucopyranosylamine analogs inhibitors of glycogen phosphorylase. Proteins. 2005;61:966–983. doi: 10.1002/prot.20653. [DOI] [PubMed] [Google Scholar]
- Webb LMC, Ehrengruber MU, Clark-Lewis I, Baggiolini M, Rot A. Binding to heparan sulfate or heparin enhances neutrophil responses to interleukin 8. Proc Natl Acad Sci USA. 1993;90:7158–7162. doi: 10.1073/pnas.90.15.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Yuan Y, Kuntz DA, Rose DR, Pinto BM. A combined STD-NMR/molecular modeling protocol for predicting the binding modes of the glycosidase inhibitors kifunensine and salacinol to Golgi a-mannosidase II. Biochemistry. 2005;44:6729–6737. doi: 10.1021/bi0500426. [DOI] [PubMed] [Google Scholar]
- Woods RJ, Dwek RA, Edge CJ, Fraserreid B. Molecular mechanical and molecular dynamical simulations of glycoproteins and oligosaccharides: 1. GLYCAM-93 parameter development. J Phys Chem. 1995;99:3832–3846. [Google Scholar]
- Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- Xu G, Takamoto K, Chance MR. Radiolytic modification of basic amino acid residues in peptides: Probes for examining protein–protein interactions. Anal Chem. 2003;75:6995–7007. doi: 10.1021/ac035104h. [DOI] [PubMed] [Google Scholar]
- Xu Y, Sette A, Sidney J, Gendler SJ, Franco A. Tumor-associated carbohydrate antigens: A possible avenue for cancer prevention. Immunol Cell Biol. 2005;83:440–448. doi: 10.1111/j.1440-1711.2005.01347.x. [DOI] [PubMed] [Google Scholar]
- Yan J, Kline AD, Mo H, Shapiro MJ, Zartler ER. The effect of relaxation on the epitope mapping by saturation transfer difference NMR. J Magn Reson. 2003;163:270. doi: 10.1016/s1090-7807(03)00106-x. [DOI] [PubMed] [Google Scholar]
- Yates JR, Eng JK, Mccormack AL, Schieltz D. Method to correlate tandem mass-spectra of modified peptides to amino-acid-sequences in the protein database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- Yu F, Wolff JJ, Amster IJ, Prestegard JH. Conformational preferences of chondroitin sulfate oligomers using partially oriented NMR spectroscopy of 13C-labeled acetyl groups. J Am Chem Soc. 2007;129:13288–13297. doi: 10.1021/ja075272h. [DOI] [PubMed] [Google Scholar]
- Zwanzig RW. High-temperature equation of state by a perturbation method: 1. Nonpolar gases. J Chem Phys. 1954;22:1420–1426. [Google Scholar]