

Abstract
Most cancer cells are aneuploid. The chromosomal instability hypothesis contends that aneuploidy is the catalyst for transformation, whereas the gene mutation hypothesis asserts that cancer is driven by mutations to proto-oncogenes and tumor-suppressor genes, with the aneuploidy a side effect of tumorigenesis. Because genotoxic stress induced by “culture shock” can obscure the transforming potential of exogenous genes, we cultured wild-type and p53-/- mouse embryo fibroblasts in a more physiological (serum-free) environment. Under these conditions, the cells were immortal and, more importantly, chromosomally stable. Expression of oncogenic H-RasV12 did not induce senescence, but sensitized these cells to p53-dependent apoptosis. In addition, H-RasV12 induced chromosomal instability, as well as accumulation and phosphorylation of p53. Significantly, whereas cells grown under standard conditions could be transformed by coexpression of H-RasV12 and E1A, the chromosomally stable cells were refractory to transformation, as measured by anchorage-independent growth and tumor formation in nude mice. These oncogenes required a third genetic alteration that abolished the p53 pathway to create a permissive environment that promotes rapid chromosomal instability and transformation. Oncogene-induced chromosomal instability and transformation was attenuated by antioxidants. These data indicate that chromosomal instability could be a catalyst for oncogenic transformation, and bring together aspects of the chromosomal instability hypothesis and the gene mutation hypothesis for tumorigenesis.
Keywords: Chromosomal instability, oncogenic Ras, p53, transformation
Most cancer biologists agree that genomic instability is a prerequisite for the onset of cancer. The majority of cancer cells are highly aneuploid, displaying dynamic karyotypic changes, including gain or loss of whole chromosomes, chromosomal rearrangements, and amplification or deletion of genetic material. There are two prevailing ideas that differ in the type of genomic alterations necessary for a normal cell to become an incipient cancer cell (Marx 2002). The chromosomal instability hypothesis contends that these gross genomic alterations are the root cause of cancer. The gene mutation hypothesis says that cancer is driven by mutations in growth-controlling oncogenes and tumor-suppressor genes, and the resulting aneuploidy is a side effect of tumorigenesis.
There are extensive data that support the chromosomal instability hypothesis for the genesis of cancer. Translocations and the loss or gain of whole chromosomes are seen in virtually every cancer of epithelial origin. Tumor staging reveals that >90% of premalignant colorectal adenomas display a high level of allelic loss, confirming that chromosomal instability does occur early during tumorigenesis (Shih et al. 2001). Furthermore, sequence analysis of colorectal tumor DNA showed only one mutation per megabase, which is a rate of mutation similar to that of normal cells (Wang et al. 2002). This indicates that colorectal cancers do not display instability at the nucleotide level.
An alternative theory asserts that the genomic instability initially occurs at the nucleotide level, resulting in insertion or deletion of a few nucleotides, or base substitutions (for review, see Hahn and Weinberg 2002). It is believed that mutation to the right combination of oncogenes and tumor-suppressor genes would lead to transformation of the cell. In support of this, the gene coding for the tumor suppressor p53 is lost or mutated in >50% of human tumors (Nigro et al. 1989; Hollstein et al. 1996), and ∼30% of human tumors carry RAS gene mutations (Bos 1989). In addition, numerous transgenic mouse models, including those that harbor activated Rasalleles (Johnson et al. 2001) or nullizygous p53 alleles (Donehower et al. 1992), are highly predisposed to a broad spectrum of tumors.
In vitro transformation models have shown that primary rodent fibroblasts can be transformed by the ectopic expression of oncogenic Ras and either c-Myc or adenovirus E1A (Land et al. 1983; Ruley 1983). This does not rule out other stochastic changes that may be associated with activated oncogene expression. Indeed, the fact that primary human cells have been refractory to transformation by a similar combination of two ectopic genes (O'Brien et al. 1986; Stevenson and Volsky 1986) implies that additional genetic or epigenetic events were likely required to yield transformants, and suggests that mouse cells may be more prone to these events.
The transformation of primary human cells has now been achieved by coexpressing a combination of three different genetic elements such as H-RasV12, SV40 early region, and hTERT (Hahn et al. 1999; Rich et al. 2001; MacKenzie et al. 2002) or H-RasV12, E1A, and Mdm2 (Seger et al. 2002). In fact, Weinberg and colleagues (Hahn et al. 1999; Zimonjic et al. 2001) were able to transform human embryonic kidney (HEK) cells with three distinct genetic elements without widespread genomic instability. This indicates that, at least in some cell types, oncogenic transformation can occur in the absence of gross chromosomal changes. Reanalysis of the transformed HEK cells by Duesberg and coworkers reported the cells to be aneuploid (Li et al. 2000), which was dismissed by Weinberg, who suggested that Duesberg's culture conditions were too harsh (for review, see Marx 2002).
This raises an interesting point about cell culture practices in general. Primary mammalian cells have a finite life span when propagated under standard cell culture conditions. Explanted mouse embryonic fibroblasts (MEFs) can divide for 15-20 population doublings before undergoing senescence. Immortalized clones emerge from this senescent state and become established cell lines that by and large are aneuploid. Primary human fibroblasts also have a finite proliferative capacity, but they are capable of many more doublings (50-100) than their murine counterparts. Human cells, however, rarely bypass senescence to become established and tend to be genomically stable (Hayflick 1965). The greater proliferative potential of human cells may be indicative of more efficient mechanisms for coping with the damage imposed by the culture environment (Sherr and DePinho 2000).
In sharp contrast with primary cells, tumor cell lines have the ability to proliferate indefinitely in culture. It has been suggested that aneuploidy and/or defective growth control mechanisms found in tumor cells might allow them to divide in conditions inadequate for long-term proliferation of normal primary cells (Ramirez et al. 2001). It follows, therefore, if the shock of the culture environment can be minimized, then primary cells may have an extended proliferative capacity. Indeed, human keratinocytes have an in vitro life span of 15-20 doublings in chemically defined media, whereas keratinocytes grown on feeder layers permitted up to 50 doublings (Ramirez et al. 2001). Similarly, MEFs grown in the absence of serum, but with a specific cocktail of supplements (including EGF, PDGF, insulin, transferrin, HDL, and fibronectin), do not undergo senescence. These cells proliferate indefinitely without any of the significant karyotypic alterations seen in established mouse cell lines (Loo et al. 1987).
We sought to take advantage of the latter observations in an attempt to assess the role of chromosomal instability in neoplastic transformation, as well as immortalization, senescence, and apoptosis. In sharp contrast to MEFs grown under standard cell culture conditions, we observed that both wild-type (WT) and p53-null MEFs grown in a defined-supplement, serum-free medium (SF-MEFs) were immortal with no signs of senescence or karyotypic change, irrespective of passage number. As expected, MEFs grown in standard conditions could be transformed by coexpression of oncogenic Ras and E1A. Significantly, the wild-type SF-MEFs could not be transformed by coexpression of these two oncogenes. Similarly, either E1A or oncogenic Ras was sufficient in transforming p53-null MEFs grown in standard conditions, but was not sufficient in transforming p53-null SF-MEFs. Although oncogenic Ras and E1A together could not transform the chromosomally stable wild-type SF-MEFs, this combination of oncogenes could transform p53-null SF-MEFs after inducing chromosomal instability. These results define a link between oncogenic lesions and chromosomal instability, and shows that they collaborate to facilitate transformation.
Results
MEFs grown in serum-free conditions are immortal
It is a well-accepted practice to grow mammalian cells in serum-containing medium. Yet the problems associated with serum's complexity and undefined nature have long been recognized. In fact, serum is believed to contribute to a phenomenon known as “culture shock,” in which mammalian cells are exposed to an unrelenting onslaught of mitogenic signals that may lead to DNA damage, checkpoint activation, and senescence (for review, see Sherr and DePinho 2000). To overcome the pitfalls associated with culture shock, we adapted cell culture conditions similar to those described previously (Barnes and Sato 1980; Loo et al. 1987), where serum was replaced with epidermal growth factor (EGF), insulin, high density lipoprotein (HDL), and transferrin (see Materials and Methods for more details).
Wild-type (WT) and p53-/- mouse embryonic fibroblasts were cultured in either serum-containing medium (MEFs) or in serum-free medium (SF-MEFs) with the supplements outlined above. Cells were passaged every 3 d following a 3T3 subculture schedule (Todaro and Green 1963). Early-passage MEFs of either genotype grown with serum or SF media exhibited the typical elongated, spindle-shaped, fibroblast morphology, forming monolayers that were contact-inhibited (Fig. 1B; data not shown). The wild-type MEFs had a doubling time of ∼30 h for the first 12 generations (Fig. 1A). As expected, the growth rate then slowed and eventually reached a nondividing state even at subconfluent density. The cells displayed a refractile cytoplasm with a flat, enlarged morphology, and stained positive for a senescence-associated β-galactosidase (SA-β-gal; Fig. 1B). Moreover, these cells expressed increasing levels of p53 and p21CIP1/WAF1 (Fig. 1C), typical of senescing fibroblasts (Alcorta et al. 1996; Serrano et al. 1997).
Figure 1.

Wild-type and p53-/- SF-MEFs are immortal. (A) Representative growth curves of wild-type (WT) or p53-/- mouse embryo cultures passaged under 3T3 conditions. Mouse embryo fibroblasts were grown in either serum-containing media (MEFs) or in serum-free media (SF-MEFs). Fibroblasts derived from 15-16-d embryos were cultured by passaging 106 cells per 100-mm dish every 3 d as described in Materials and Methods. Some plates of wild-type SF-MEFs were supplemented with 10% FBS at day 39. (B) MEFs enter senescence in serum-containing but not serum-free conditions. Wild-type MEFs and wild-type SF-MEFs at population doubling (PD) 3 and 15 were stained for SA-β-gal (pH 5.5) activity. Photographs are at the same magnification. (C) Increased expression of p53 and p21CIP1/WAF1 in serum-containing but not serum-free conditions. Immunoblot of cell lysates from wild-type MEFs and wild-type SF-MEFs for p53 and p21CIP1/WAF1 at the indicated PD. Equal loading of lysates was confirmed by immunoblotting for tubulin. The postcrisis (PC) wild-type MEFs represents a sample taken from cells at ∼15 PD after the cells had bypassed senescence and became spontaneously immortalized.
The p53-/- MEFs started with a doubling time of ∼18 h and decreased to as little as 12 h (Fig. 1A), presumably reflecting the acquisition of mutations advantageous to growth. In comparison, SF-MEFs of both wild-type and p53-/- background had a doubling time of ∼24 h that was consistent through all generations. The wild-type SF-MEFs have yet to adopt the senescent morphology, did not stain for SA-β-gal (Fig. 1B), or show increasing p53 or p21CIP1/WAF1 levels with replicative age (Fig. 1C). The senescence program is intact in SF-MEFs, because adding serum to wild-type SF-MEFs slowed their doubling time and eventually resulted in senescence (Fig. 1A). Conversely, adding the serum-free supplements to the cells growing in serum had no discernable effects that differed from growth in serum alone (data not shown). Taken together, these results indicate that the onset of senescence typical in primary mouse cultures after only 15-20 generations is likely caused by inappropriate growth conditions.
Serum-free MEFs are chromosomally stable
We next analyzed the karyotypic profiles of the mouse embryo cultures. Wild-type MEFs at population doubling (PD) 3 were diploid with the normal 40 chromosomal complement (Fig. 2A). Just prior to the onset of senescence at PD 15, the majority of wild-type MEFs were still in the diploid range; however, >30% had become aneuploid (Fig. 2B). On the other hand, postcrisis, wild-type MEFs that spontaneously immortalized showed drastic chromosomal changes, with >90% becoming aneuploid (Fig. 2C). In comparison, the immortalized wild-type SF-MEFs were chromosomally stable even at PD 60, showing a predominantly diploid karyotype (Fig. 2D). As for the p53-/- MEFs, >60% exhibited gross chromosomal defects and aneuploidy by PD 3, which increased in severity with replicative age (Fig. 2E,F). The genomic instability of p53-/- MEFs in culture is in stark contrast to the genomic stability in somatic cells within p53-null mice (Purdie et al. 1994). Similar to the in vivo situation, p53-/- SF-MEFs were chromosomally stable, with a near-diploid karyotype even at PD 60 (Fig. 2G). This indicates that culturing MEFs in the serum-free media more closely approximates normal physiological conditions. It also confirms that loss of p53 does not in itself cause chromosomal damage. When serum was added to the p53-/- SF-MEFs at PD 60, the karyotype quickly destabilized and became aneuploid (Fig. 2H). This demonstrates that growth in serum exposes the cells to genotoxic stress that, without the protective surveillance provided by p53, leads to chromosomal instability. Although the presence of p53 is able to mitigate the genome destabilizing effects, even wild-type MEFs eventually succumb to the damage imposed by the culture environment.
Figure 2.

Wild-type and p53-/- SF-MEFs have stable karyotypes. Embryo fibroblasts at the indicated PD were harvested for karyotype analysis. (A) Wild-type MEFs at PD 3. (B) Wild-type MEFs at PD 15. (C) Postcrisis wild-type MEFs at PD 30 (these cells had doubled ∼15 times from the first splitting of a near-confluent flask of spontaneously immortalized cells that escaped senescence). (D) Wild-type SF-MEFs at PD 60. (E) p53-/- MEFs at PD 3. (F) p53-/- MEFs at PD 30. (G) p53-/- SF-MEFs at PD 60. (H) p53-/- SF-MEFs at PD 60 were supplemented with 10% FBS for 48 h and then prepared for metaphase spreads as outlined in Materials and Methods. Approximately 50 metaphases from two representative cultures were counted in each case. The exception was the wild-type MEFs at PD 15, where a total of 54 metaphases were counted from two representative cultures. These cells were approaching senescence, which limited the number of available metaphases.
Chromosomally stable serum-free mouse embryonic fibroblasts are resistant to H-RasV12 induced senescence
Expression of activated oncogenes in normal cells has the paradoxical effect of eliciting an antiproliferative response. Oncogenic Ras induces premature cell senescence in primary fibroblasts (Serrano et al. 1997), whereas adenovirus E1A sensitizes the cells to apoptosis induced by DNA damage or serum starvation (Debbas and White 1993; Lowe and Ruley 1993). To see what effects these oncogenes have on the SF-MEFs, constitutively activated Ras (H-RasV12) or the 12S E1A gene was introduced into the different mouse embryo cultures by retroviral-mediated gene transfer. To minimize the risk of stochastic changes that may occur during a prolonged infection and selection period, we generated high-titre retroviruses from stable retrovirus-producer cell lines. This allowed us to transduce the target cell populations quickly and efficiently (see Materials and Methods for more details). In all experiments, parallel infections using retroviruses expressing EGFP showed that MEFs and SF-MEFs were equally infectable, with >90% of cells transduced within 24 h (Supplementary Fig. S1). In addition, the expression level of H-RasV12 and E1A was comparable between the different cell types irrespective of the culture conditions (Supplementary Fig. S2).
As expected, expression of H-RasV12 in early-passage wild-type MEFs slowed down proliferation that culminated in growth arrest (Fig. 3A) with the morphological hallmarks of senescence. This was confirmed by staining for SA-β-gal (Fig. 3B). In contrast, the p53-/- MEFs transduced with H-RasV12 grew to a very high density, with no sign of growth arrest (data not shown), verifying the p53-dependence of the senescent pathway (Lin et al. 1998). Surprisingly, when H-RasV12 was introduced into the wild-type SF-MEFs, we did not observe the expected growth arrest or SA-β-gal staining (Fig. 3A,B). Given that wild-type SF-MEFs are chromosomally stable, these data suggest that cellular senescence caused by oncogenic Ras may require cooperation with genomic instability associated with culture shock.
Figure 3.

Oncogenic Ras fails to induce senescence in wild-type SF-MEFs. Wild-type MEFs and wild-type SF-MEFs at PD 3 were transduced with H-RasV12 or empty viral vector. Day 0 represents the point when infected cell populations were split for the first time onto three plates (∼48 h after initial exposure to the retroviruses). (A) Growth curves for the wild-type MEFs and wild-type SF-MEFs transduced with H-RasV12 or empty vector. Each time point represents the average from the triplicate plates, and each curve was performed three times. Error bars represent the standard error of the mean from the three independent experiments. (B) Representative photographs of the indicated cells stained for SA-β-gal (pH 5.5) activity at day 4. Photographs are at the same magnification.
Oncogenic Ras evokes a p53-dependent apoptotic response in serum-free mouse embryo fibroblasts
In the above experiment, wild-type SF-MEFs transduced with H-RasV12 grew at a slightly slower rate than the same cells infected with the control virus (doubling time 30 h vs. 24 h; Fig. 3A). Most activated oncogenes (E1A, E2F-1, or Myc) provoke an antiproliferative response in the form of programmed cell death rather than senescence (Debbas and White 1993; Lowe et al. 1993; Hermeking and Eick 1994; Qin et al. 1994; Wu and Levine 1994). The slower growth rate of the wild-type SF-MEFs transduced with H-RasV12 may therefore be indicative of apoptosis because this pattern of growth is similar to that observed in primary fibroblasts expressing E1A (Serrano et al. 1997).
To test this hypothesis, the SF-MEFs transduced with either H-RasV12 or E1A were grown on glass coverslips, and irradiated with 5 Gy of ionizing radiation to induce p53-dependent apoptosis. Apoptosis was monitored by staining the cells with DAPI followed by fluorescence microscopy to visualize the morphological characteristics associated with apoptotic cells, including nuclear breakdown and heterochromatin aggregation (Hendzel et al. 1998). Irradiating the wild-type SF-MEFs expressing E1A led to ∼34% of cells with apoptotic morphology within 16 h (Fig. 4A,B). Similarly, H-RasV12-transduced wild-type SF-MEFs were also sensitive to IR-induced apoptosis. In contrast, the p53-/- SF-MEFs had a severely attenuated apoptotic response, demonstrating that H-RasV12-initiated apoptosis is dependent on p53. The vector control cells did not have any of the morphological characteristics of apoptotic cells following γ-irradiation, confirming that H-RasV12 or E1A was required for sensitizing these cells to apoptosis. Abrogating oncogene-induced apoptosis through loss of p53 enhances cell growth and survival (Lowe et al. 1994) and may augment the transforming potential of activated oncogenes during tumorigenesis (see below).
Figure 4.

Oncogenic Ras promotes p53-dependent apoptosis in SF-MEFs. Wild-type or p53-/- SF-MEFs were infected with retroviruses expressing empty viral vector, E1A, or H-RasV12. The indicated cells were simultaneously treated with 5 mM NAC. (A) The infected cells were either untreated (Control) or exposed to 5 Gy of ionizing radiation (IR). The cells were fixed 16 h after IR, stained with DAPI, and analyzed for the morphological characteristics associated with apoptosis. Apoptotic values were calculated as the number of apoptotic cells relative to the total number of cells. The data represent the average of at least three independent experiments ± S.E.M. (>100 cells counted per experiment). (B) Representative fluorescent images of DAPI-stained cells. Arrows identify cells with heterochromatin aggregation typical of apoptotic cells.
We found that there was a consistently elevated basal level of apoptosis (∼7%) in unirradiated wild-type SF-MEFs transduced with H-RasV12 that was not seen in the vector control cells or the p53-/- cells with H-RasV12 (Fig. 4A). This suggests that oncogenic Ras can elicit a p53-dependent, DNA-damage-like response, leading to apoptosis in SF-MEFs even in the absence of an external stress like IR. Consistent with this, p53 levels were higher in H-RasV12-transduced cells compared with the vector control cells (Fig. 5). In addition to its accumulation, we found that p53 was phosphorylated on Ser 18 (equivalent to Ser 15 in human p53). Phosphorylation of Ser 18 is often linked to a DNA damage response (Shieh et al. 1997; Siliciano et al. 1997), suggesting that expression of oncogenic Ras may cause DNA damage. Indeed, several reports have linked expression of H-RasV12 with the induction of DNA damage, chromosomal instability, and aneuploidy (Denko et al. 1994; Lee et al. 1999). The chromosomal instability caused by H-RasV12 could be rescued by treating the cells with N-acetyl-cysteine (NAC; Lee et al. 1999), a precursor of reduced glutathione and a scavenger of reactive oxygen species (ROS; Aruoma et al. 1989; Burgunder et al. 1989). Consistent with these reports, we confirmed that H-RasV12 induced ROS in the SF-MEFs as detected by staining with the redox-sensitive fluorochrome, dichlorofluorescein diacetate (DCFDA), and NAC was effective in blocking the Ras-induced ROS production (Supplementary Fig. S3). Adding reduced glutathione to the medium was similarly effective in blocking Ras-induced ROS (data not shown).
Figure 5.
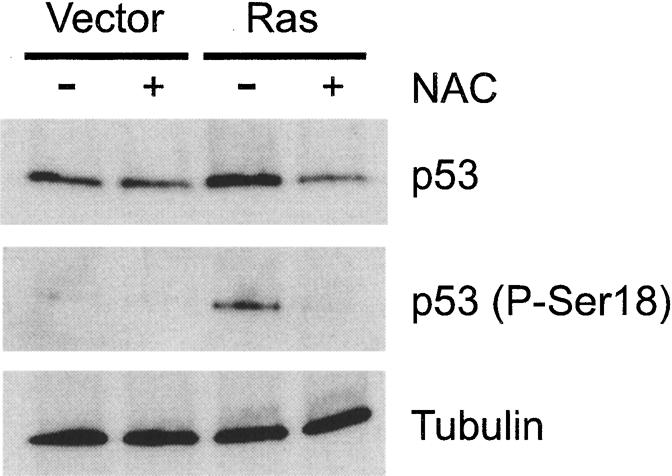
Expression of oncogenic Ras elicits a DNA-damage-like response leading to the induction and phosphorylation of p53. Wild-type SF-MEFs were infected with vector control or H-RasV12-expressing retrovirus. The indicated cells were simultaneously treated with 5 mM NAC. Cells were harvested ∼24 h after infection. Total cellular lysates were immunoblotted for total p53 and Ser 18-phosphorylated p53. Uniform loading of cell lysates was confirmed by immunoblotting for tubulin.
We found that NAC as well as reduced glutathione could block the basal level of apoptosis in the wild-type SF-MEFs induced by H-RasV12 (Fig. 4A; data not shown). In addition, the Ras-induced apoptosis, which manifests as a slower rate of growth shown in Figure 3A, could also be corrected by NAC (data not shown). Furthermore, NAC could block the accumulation and phosphorylation of p53 in the H-RasV12-transduced cells (Fig. 5). Collectively, these data indicate that activated Ras sensitizes SF-MEFs to p53-dependent apoptosis in unstressed and irradiated cells. Furthermore, activated Ras elicits a DNA damage-like response through the production of ROS, leading to p53 accumulation and phosphorylation. Evidence that oncogenic Ras induces DNA damage and chromosomal instability may represent a mechanism whereby activated oncogenes initiate tumor progression (see below).
Activated oncogenes H-RasV12 and E1A induce the rapid onset of chromosomal instability
The p53 response and the ensuing apoptosis suggest that expression of activated oncogenes induces DNA damage and chromosomal instability. To assess the effects of H-RasV12 and E1A, we analyzed the karyotype of the infected cultures. Both wild-type and p53-/- SF-MEFs infected with the vector control retroviruses maintained a normal diploid genome (Fig. 6A). Following H-RasV12 expression, the majority of wild-type SF-MEFs remained diploid. However, a significant proportion of p53-/- SF-MEFs became aneuploid in the presence of H-RasV12 (∼35% of cells had >45 chromosomes; Fig. 6B). Therefore, p53 can block the genome-destabilizing effects of activated Ras. The chromosomal-destabilizing effect of H-RasV12 was very rapid, as cells were harvested for karyotyping in no more than a few cell cycles after infection. NAC was able to compensate for the loss of p53 because pretreatment of the p53-/- SF-MEFs with the antioxidant prior to transduction with H-RasV12 prevented the chromosomal instability. Again, pretreating the cells with NAC did not affect infection efficiency because H-RasV12 was expressed equally well even in the presence of the antioxidant (Supplementary Fig. S2).
Figure 6.

Activated oncogenes H-RasV12 and E1A promote chromosomal instability. Wild-type or p53-/- MEFs or SF-MEFs were infected with (A) vector control, (B) H-RasV12, or (C) E1A and H-RasV12 retroviruses. At 24 h after infection, the cells were treated with nocodazole to induce metaphase arrest and then harvested for karyotype analysis. Between 30 and 40 metaphases from three independent infections were counted in each case. The indicated cultures were preincubated with 5 mM NAC before retroviral infection; these cells were maintained in NAC until the samples were harvested for metaphase spreads.
Coexpression of E1A and H-RasV12 had similar destabilizing effects on the genome. Early passage wild-type and p53-/- MEFs became highly aneuploid upon coexpressing E1A and oncogenic Ras (Fig. 6C). Therefore, the combined genome-destabilizing effects of activated oncogenes and serum stress on wild-type MEFs could not be ameliorated despite the presence of p53. Significantly, wild-type SF-MEFs transduced with E1A and H-RasV12 remained predominantly diploid. On the other hand, more than half of the p53-/- SF-MEFs expressing both oncogenes were aneuploid. This demonstrates that p53 is able to safeguard SF-MEFs from the genome-destabilizing effects induced by oncogenic Ras and E1A. Again, NAC was able to offset the loss of p53 because pretreatment of the p53-/- SF-MEFs with the antioxidant prevented the chromosomal instability induced by H-RasV12 and E1A. Taken together, these data show that the rapid onset of chromosomal instability induced by oncogenes is exacerbated by serum stress or the absence of p53.
Chromosomally stable serum-free mouse embryo fibroblasts are resistant to oncogenic transformation by E1A and H-RasV12
Classical studies showed that primary mouse cells grown in standard conditions could be transformed by two cooperating oncogenes (Land et al. 1983; Ruley 1983). Therefore, we sought to determine whether coexpression of E1A and oncogenic Ras could convert the SF-MEFs into tumorigenic cells. To that end, we serially introduced E1A followed by H-RasV12 into early-passage mouse embryo cultures. We then assayed for in vitro transformation as determined by the ability to grow in semisolid media, and to form tumors when injected subcutaneously into mice.
As expected, wild-type MEFs and p53-/- MEFs transduced with both E1A and H-RasV12 formed anchorage-independent colonies with a high degree of efficiency (Table 1; Fig. 7). The wild-type MEFs expressing either E1A or H-RasV12 alone did not form colonies in soft agarose. In comparison, p53-/- MEFs transduced with a single oncogene were able to form colonies in soft agarose. In marked contrast to MEFs grown in serum, the wild-type SF-MEFs transduced with both E1A and H-RasV12 did not form colonies in soft agarose even after 2 mo of observation. Similarly, p53-/- SF-MEFs transduced with a single oncogene did not form colonies.
Table 1.
Anchorage-independent growth of SF-MEFs
| Cells | Retroviral construct(s) | No. colonies/platea |
|---|---|---|
| WT MEFs | Vector | 0 |
| E1A | 0 | |
| Ras | 0 | |
| E1A + Ras | 12,258 ± 1746 | |
| p53−/− MEFs | Vector | 0 |
| E1A | 8234 ± 652 | |
| Ras | 11,350 ± 1206 | |
| E1A + Ras | 17,434 ± 3059 | |
| WT SF-MEFs | Vector | 0 |
| E1A | 0 | |
| Ras | 0 | |
| E1A + Ras | 0 | |
| p53−/− SF-MEFs | Vector | 0 |
| E1A | 0 | |
| Ras | 0 | |
| E1A + Ras | 5201 ± 642 | |
| p53−/− SF-MEFs + NACb | E1A + Ras | 48 ± 24 |
The number of colonies per 60-mm plate was determined by counting the number of colonies in 10 circles with a 5-mm diameter, then estimating the number of colonies over the entire surface area of the plate. The data represent the average from at least three independent experiments plus or minus the standard error of the mean.
Cells were preincubated with 5 mM NAC before retroviral infection and maintained in NAC for the duration of the assay.
Figure 7.

Loss of p53 promotes anchorage-independent growth of SF-MEFs transduced with E1A and H-RasV12. Wild-type or p53-/- MEFs or SF-MEFs infected with retroviruses expressing E1A and H-RasV12 were analyzed for anchorage-independent growth as described in Materials and Methods. The photographs are of wild-type MEFs (A), wild-type SF-MEFs (B), p53-/- MEFs (C), or p53-/- SF-MEFs (D). Colonies were counted (see Table 1) and photographs were taken after 14 d.
Unlike the chromosomally stable wild-type SF-MEFs, the p53-/- SF-MEFs were susceptible to transformation by two oncogenes. When both E1A and H-RasV12 were introduced into the p53-/- SF-MEFs, a significant number of colonies developed, but with only one-third the efficiency as the p53-/- MEFs with E1A and H-RasV12 (Table 1). In addition, the colonies were markedly smaller at the same end point (Fig. 7).
The same infected mouse embryo cultures were also tested for their ability to form tumors when injected subcutaneously into immunocompromised mice. The wild-type MEFs and p53-/- MEFs expressing E1A and H-RasV12 formed tumors in every instance (Table 2; Fig. 8). Notably, the wild-type MEFs had a longer latency period before tumors became perceptible. The wild-type SF-MEFs coexpressing E1A and H-RasV12 failed to form tumors before the 16-wk end point of the assay. In comparison, the p53-/- SF-MEFs expressing E1A and H-RasV12 were able to form tumors in eight of 12 cases. A cursory examination of the different mouse embryo cultures injected into syngeneic, immunocompetent mice produced similar results; however, the tumors had a longer latency period (data not shown). Taken together, these data indicate that the chromosomally stable wild-type SF-MEFs are not susceptible to oncogenic transformation by the combination of E1A and H-RasV12. However, the increase in chromosomal instability associated with the loss of p53 or serum stress facilitates oncogenic transformation.
Table 2.
Formation of subcutaneous tumors in nude mice by SF-MEFs
| Cells | No. tumors/no. injections | Onseta |
|---|---|---|
| p53−/− MEFs | 0/6 | NA |
| WT MEFs E1A/Ras | 6/6 | 10 d |
| p53−/− MEFs E1A/Ras | 6/6 | 6 d |
| WT SF-MEFs E1A/Ras | 0/12 | NA |
| p53−/− SF-MEFs E1A/Ras | 8/12 | 16 d |
| p53−/− SF-MEFs E1A/Ras + NACb | 1/18 | 58 d |
Average number of days from injection until the tumor mass was palpable (−2 mm diameter).
Cells were preincubated with 5 mM NAC before retroviral infection and maintained in NAC until the time of injection.
Figure 8.

Loss of p53 promotes the tumor-forming potential of SF-MEFs transduced with E1A and H-RasV12. Wild-type or p53-/- MEFs or SF-MEFs were transduced with E1A and H-RasV12. Cells (2 × 106) in 100 μL of PBS were injected subcutaneously into immunocompromised nude mice. The examples shown are mice injected with wild-type MEFs (A), wild-type SF-MEFs (B), p53-/- MEFs (C), or p53-/- SF-MEFs (D).
Because NAC could block oncogene-induced chromosomal instability, we sought to determine if it could have a similar effect on transformation. NAC treatment effectively blocked the anchorage-independent growth of the p53-/- SF-MEFs transduced with E1A and oncogenic Ras (Table 1; Fig. 7). On the other hand, the free-radical scavenger had no effect on the colony-forming ability of wild-type MEFs expressing E1A and H-RasV12, or the p53-/- MEFs expressing E1A and H-RasV12 (data not shown). This suggests that, like p53, the effect of NAC is insufficient to ameliorate the combined genome-destabilizing effects of activated oncogenes and serum. In addition, the efficiency of tumor formation by the p53-/- SF-MEFs expressing E1A and H-RasV12 was drastically reduced by pretreating the cells with NAC before the introduction of E1A and H-RasV12. We found that only a single tumor formed after a very long latency period (Table 2; Fig. 8). Because NAC was effective in blocking oncogene-induced chromosomal instability, it is likely that an additional stochastic event occurred in vivo for this single tumor to have formed. It is noteworthy that transformation of cells by coexpressing two or more genetic elements resulted in tumor cell populations that were polyclonal (Hahn et al. 1999; Seger et al. 2002). However, the pervasive nature in which oncogenic Ras and E1A induce chromosomal instability and karyotypic change demonstrates that tumor cells can undergo additional genetic alterations but still be polyclonal. A rare genetic event would not necessarily have to occur.
Discussion
Genomic stability prolongs cellular life span
It is generally assumed that the growth arrest of mouse embryonic fibroblasts after 15-20 population doublings represents replicative aging and senescence. However, we see here that MEFs cultured in a more suitable environment have greatly exceeded the previous limits. An immortalized culture has been subjectively defined as “one that doubles its maximal lifespan” (Ramirez et al. 2001). Using this definition, the SF-MEFs, which have exceeded 100 PD in our laboratory (Fig. 1A), could be considered immortal.
SF-MEFs exhibited a consistent, basal level of p53 and p21CIP1/WAF1, and an unaltered karyotype irrespective of replicative age (Figs. 1C, 2). In contrast, MEFs grown in serum show increasing levels of p53 and p21CIP1/WAF1 as well as chromosomal instability with replicative age. One hypothesis is that the culture environment may expose the cells to stress that induces DNA damage (Sherr and DePinho 2000). This is supported by the rapid onset of chromosomal instability when p53-/- SF-MEFs were switched to serum-containing media (Fig. 2H). In comparison, the same cells maintained continuously in serum-free culture preserve a diploid genome despite the absence of p53 (Fig. 2G). Therefore, maintaining a stable genome is favorable to extending the in vitro life span of mouse cells. Indeed, simply growing cells under physiological oxygen levels enables MEFs to proliferate for more than 60 PD with no sign of replicative senescence and fewer spontaneous DNA damage than when they are grown in higher oxygen conditions (Parrinello et al. 2003). This is further exemplified by hTERT expression in normal human somatic cells. Telomere attrition leads to genomic instability, typified by chromosomal end-to-end fusions resulting in nonreciprocal chromosomal translocations (Artandi et al. 2000). Cells with reconstituted telomerase activity maintain telomere structure (Masutomi et al. 2003), a diploid karyotype and have an extended replicative life span (Bodnar et al. 1998; Vaziri and Benchimol 1998) without any indication of being transformed (Jiang et al. 1999; Morales et al. 1999). Likewise, we found that wild-type SF-MEFs or p53-/- SF-MEFs are immortal but have stable diploid genomes (Fig. 2). Moreover, these cells were contact-inhibited, and were incapable of anchorage-independent growth.
Although we define the SF-MEFs as immortal, it may be possible for these cells to eventually reach their replicative capacity. An intriguing possibility is that telomere attrition eventually comes into effect. Mouse cells have telomeres about five times the length of human telomeres (60 kb vs. 12 kb). If we suppose that the rate of telomere attrition is comparable between mouse and human cells, then SF-MEFs may have five times the replicative potential of human cells. That could mean 250-500 doublings before the SF-MEFs reach a critically short telomere length. Indeed, telomere dysfunction becomes noticeable following extensive passaging of telomerase-deficient mouse cells or in early-passage cells from late-generation knockout mice (Blasco et al. 1997).
Influence of genomic instability on senescence and apoptosis
Replicative senescence of human cells is believed to be triggered by telomere attrition, whereas premature senescence induced by oncogenic Ras (Fig. 3) is provoked by hyperproliferative signals (Serrano et al. 1997). We found that SF-MEFs at similar passages did not undergo premature senescence when H-RasV12 was expressed (Fig. 3). Our results also suggest that mouse cells grown in serum undergo premature senescence caused by persistent mitogenic stimulation from serum factors. In this way, serum mimics the effects of oncogenic Ras, exposing the cell to hyperproliferative signals. However, oncogenic Ras appears to be more potent than serum, as demonstrated by the faster rate at which H-RasV12 caused senescence (cf. Figs. 1A, 3A).
Expression of activated oncogenes such as Ras or Myc can induce genomic instability in as little as one cell cycle (Denko et al. 1994; Felsher et al. 2000; Karlsson et al. 2003). These oncogenes induce genomic instability, at least in part, by increasing intracellular ROS (Lee et al. 1999; Vafa et al. 2002). This is the mechanism by which oncogenic Ras induces premature senescence, as the oxido-protective reagent NAC can prevent the genomic damage and rescue the cells from Ras-induced growth arrest (Lee et al. 1999). We found that H-RasV12 triggered observable chromosomal instability in just one or two cell cycles (Fig. 6). NAC blocked oncogene-induced chromosomal instability (Fig. 6), which alleviated the need for p53 accumulation and phosphorylation (Fig. 5). Similar to activated Ras, normal mitogens in serum could produce hyperproliferative signals. These hyperproliferative signals lead to increased oxidative metabolism and elevated ROS levels (Irani et al. 1997). It has been suggested that activated oncogenes produce higher or sustained levels of ROS compared to normal mitogens (Vafa et al. 2002). This may account for the greater potency of oncogenic Ras in inducing senescence and eliciting a p53-dependent DNA damage response in our studies.
In SF-MEFs, the genome-destabilizing effects of oncogenic Ras leads to apoptosis (Fig. 4), whereas serum reestablishes the senescence pathway that had been averted by growth in the absence of serum (Fig. 1). This may suggest that survival factors in serum nullify the apoptotic signals. The presence of survival factors may explain why oncogenic Ras-transduced MEFs undergo senescence, whereas oncogenic Ras-transduced SF-MEFs undergo apoptosis. A second possibility (not necessarily exclusive of the first) is that there may be senescence-promoting factors in serum, the effects of which are accentuated by oncogenic Ras. On the other hand, SF-MEFs would not be exposed to survival factors or senescence-promoting factors; therefore, apoptotic signals would go unabated.
Oncogenes collaborate with chromosomal instability during tumorigenesis
An extensive body of evidence has shown that several cooperating genetic elements can lead to the in vitro transformation of rodent and human cells (Land et al. 1983; Ruley 1983; Hahn et al. 1999; Rich et al. 2001; MacKenzie et al. 2002; Seger et al. 2002). However, it has been more difficult to define the contribution of gross genetic alterations that are characteristic of nearly every tumor cell. This is in part due to the genome-destabilizing effects inherent to in vitro cell culture (Sherr and DePinho 2000; Soucek and Evan 2002).
We found that primary mouse fibroblasts are more difficult to transform than what had previously been envisioned. We verified that MEFs grown in standard conditions were transformed by cooperation of two oncogenes (Tables 1, 2). In contrast, MEFs grown in an extracellular milieu that is favorable to maintaining long-term genomic stability were not transformed by the cooperative action of E1A and oncogenic Ras. These oncogenes required a third genetic alteration that abolished the p53 pathway. We showed that introduction of E1A and H-RasV12 into p53-/- SF-MEFs caused rapid chromosomal instability (Fig. 6). Induction of chromosomal instability by H-RasV12 was likely through the production of ROS, as chromosomal instability was blocked by NAC (Fig. 6). Expression of H-RasV12 led to the accumulation and phosphorylation of p53, analogous to a DNA-damage response (Fig. 5). Inactivation of the p53 pathway likely exacerbates oncogene-induced chromosomal damage by allowing the damage to go unchecked. Given that E1A and H-RasV12 induced apoptosis in wild-type SF-MEFs (Fig. 4), the instability is likely prevented in wild-type SF-MEFs by the action of p53. Any genomic instability would be met with a p53-dependent apoptotic response, thus eliminating any preneoplastic cells. This may explain, in part, why wild-type SF-MEFs were refractory to transformation by two cooperating oncogenes. Similarly, the transforming potential of E1A and H-RasV12 in p53-/- SF-MEFs was restricted by the antioxidant NAC with a concomitant attenuation of the chromosomal instability (Tables 1, 2). Therefore, in SF-MEFs, two activated oncogenes plus the loss of one tumor suppressor gene cooperate to induce chromosomal instability that leads to transformation.
In contrast with our findings, others have observed that SF-MEFs could be coerced into forming colonies upon the cointroduction of oncogenic Ras and Myc (Rawson et al. 1991). However, their experiments used cells derived from Balb/c mice, which have since been shown to be mutated for p16INK4a (Zhang et al. 1998). Furthermore, their transformed clones were derived from cells grown in the absence of EGF, thus prior selection for a transformed phenotype may have been imposed.
Chromosomal instability on its own was unable to cause cellular transformation. The genomically unstable p53-/- MEFs could not form tumors when injected into nude mice (Table 2; Fig. 6). Similarly, p53-/- SF-MEFs transduced with H-RasV12 were aneuploid but remained untransformed. These data show that in addition to promoting chromosomal instability, oncogenic Ras and E1A activate signal transduction pathways that are important for tumorigenesis. Therefore, this in vitro model demonstrates that deregulation of oncogenic and tumor suppressor pathways promotes and collaborates with chromosomal instability during tumorigenesis. Although it is difficult to formally exclude the possibility that additional gene mutations occurred during the experiments, the simplest interpretation of the data is that activated oncogenes induce ROS, leading to chromosomal instability and oncogenic transformation.
Our combination of E1A, oncogenic Ras, and nullizygous p53 alleles is similar to a report in which human fibroblasts were transformed with E1A, oncogenic Ras, and Mdm2 (Seger et al. 2002). Similar to cells described here, the transformed human fibroblasts too are highly aneuploid. In contrast, the combination of telomerase, SV40 early region, and oncogenic Ras was shown to transform human embryonic kidney cells without the need for chromosomal instability (Hahn et al. 1999; Zimonjic et al. 2001). However, it was found that the same combination of genetic elements introduced into either human mammary epithelial cells or BJ fibroblasts induced chromosomal translocations resulting in gene amplification or aneuploidy, respectively (Hahn et al. 1999; Elenbaas et al. 2001). This suggests that the involvement of chromosomal instability during transformation may depend on the cell type being analyzed.
Our results reinforce the notion that genomic instability, in the form of mutant genes and gain or loss of whole chromosomes, is a prerequisite for neoplastic transformation. Here, the mutant genes are represented by two activated oncogenes and the loss of one tumor-suppressor gene. The activated oncogenes induce chromosomal instability through production of ROS because the antioxidant NAC attenuates the karyotypic changes. Reactive oxygen can act directly on DNA to cause chromosomal breaks (Karanjawala et al. 2002), or indirectly through activation of topoisomerase (Li et al. 1999). Fusion of chromosomal breaks by nonhomologous end joining may generate dicentric chromosomes that can shear randomly between the centromeres during mitosis, resulting in uneven distribution of genetic material to the two daughter cells. These breakage-fusion cycles can give rise to aneuploidy, gene amplification, and gene deletion (Pihan and Doxsey 2003). In agreement with the gene mutation hypothesis, we find that cancer can be driven by activated oncogenes; this coupled with the loss of p53 creates a permissive environment that allows propagation of genomic lesions and cancer-prone cells. In support of the chromosomal instability hypothesis, the combination of H-RasV12, E1A, and loss of p53 rapidly destabilized the genome leading to aneuploidy. The cooperative effects of these gene mutations and aneuploidy facilitate the conversion of normal cells into cancer cells.
Materials and methods
Materials
All reagents were obtained from Sigma-Aldrich unless stated otherwise.
Preparation of MEFs and cell culture conditions
Pairs of p53+/- mice of C57BL/6 genetic background (JAX Research Systems) were mated. Timed pregnant females at 15-16 d of gestation were killed, followed by uterine dissection to isolate individual embryos. Each embryo was washed thoroughly with PBS (pH 7.2), followed by removal of the head and liver. The embryo body was suspended in 2 mL of 0.25% trypsin, and then forced through a 1-cc syringe with an 18-gauge needle. The tissue homogenate was incubated for 30 min at 37°C, triturated by drawing the suspension through a pipette, and then evenly divided into two 100-mm tissue culture dishes in Dulbecco's modified Eagle's medium (DMEM) with either 10% fetal bovine serum (FBS) or serum-free supplements (see below). Once these cells became confluent, we followed a 3T3 subculture schedule by counting the total number of cells using a hemocytometer, and plating 1 × 106 cells per 100-mm dish on a total of three dishes. Every 3 d, the three plates were trypsinized and the cells were pooled and counted to determine the number of population doublings, and then plated onto three new dishes as before.
The serum-free DMEM was supplemented with EGF (75 ng/mL), insulin (10 μg/mL; Roche), HDL (25 μg/mL; we used a lot that contained 80% HDL and 10% LDL), transferrin (30 μg/mL), sodium selenite (10 nM), penicillin, and streptomycin. For subculturing, the cells were trypsinized with trypsin (0.05%) containing EDTA (1 mM) and pluronic F68 (0.05%). The SF-MEFs were grown on dishes that were precoated with poly-L-lysine prior to plating. Cells from at least two embryos of each genotype were examined. Mice and mouse embryos were genotyped according to protocols described by JAX Research Systems without modification.
Retroviral-mediated gene transfer
The cDNAs for H-RasV12 (Guthrie cDNA Resource Center) or enhanced green fluorescent protein (EGFP) were subcloned into the retroviral vector pLNCX2 (Clontech). To create stable retroviral producer cell lines, the packaging cell line PT67 (Clontech) was transfected with the above constructs or empty vector using lipofectamine (Invitrogen Life Technologies). Following transfection and a 24-h recovery period, the cells were placed in medium containing G418 (350 μg/mL; Invitrogen Life Technologies). Selective medium was changed every 3-5 d for ∼2 wk. Drug-resistant clones were isolated using cloning cylinders. Stable integration of the H-RasV12 or EGFP cDNAs was confirmed by PCR (data not shown). The stable retroviral producer cell line ψ2-12s E1A (American Type Culture Collection) has been described (Cone et al. 1988). The retroviral producer cell lines were maintained in DMEM supplemented with the serum-free supplements. The virus-containing medium was filtered through a 0.45-μm filter, mixed 1:1 with the appropriate fresh medium supplemented with 4 μg/mL polybrene. Target fibroblasts (between PD 3 and PD 6) were plated at a density of 1 × 106 cells per 100-mm dish, 12-18 h before infection. For infection, the culture medium was removed and replaced with the virus-containing medium. The cells were incubated at 37°C; and the virus-containing medium was replaced with fresh virus every 4 h for 24 h. For double infections, the cells were infected with E1A-expressing retrovirus for the first 24 h, followed by a 12-h recovery, and then infected with the H-RasV12-expressing retrovirus over the next 24 h. The cells were used in the various assays within 48 h after being infected.
SA-β-gal activity assay
SA-β-gal activity was detected as described (Dimri et al. 1995) with slight modifications. Cells were washed with PBS (pH 7.2), fixed with 0.05% glutaraldehyde in PBS, and washed again in PBS (pH 7.2) supplemented with 1 mM MgCl2. Cells were stained with a solution containing 1 mg/mL X-gal, 0.12 mM K3Fe[CN]6, 0.12 mM K4Fe[CN]6, 1 mM MgCl2 in PBS (pH 5.5) overnight. Color photographs were taken using a Nikon CCD camera mounted on an Olympus phase-contrast microscope.
Immunoblotting
Immunoblotting was performed as described previously (Poon et al. 1995). Rat monoclonal antibody YL1/2 against tubulin was a gift from Tim Hunt (Cancer Research UK). Polyclonal antibodies against p53 (sc-6243), p21CIP1/WAF1 (sc-397), and E1A (sc-430) were obtained from Santa Cruz Biotechnology. Anti-Ser 15 (Ser 18 in mouse) phosphorylated p53 antibodies were obtained from Cell Signaling Technology. Monoclonal antibodies against Ras (clone 10) were from Upstate.
Karyotyping
Cells were treated with nocodazole (0.1 μg/mL) for 2 h to induce metaphase arrest. Following trypsinization, the cells were re-suspended in 5 mL of 75 mM KCl for 20 min at 37°C, then fixed with 1 mL of Carnoy's fixative (3:1 methanol:glacial acetic acid). The cells were collected by low-speed centrifugation (800 rpm) for 5 min, and then resuspended in an appropriate volume of fixative. The cell suspension was dropped onto glass slides, air dried, and then stained with 4′,6′-diamidino-2-phenylindole (DAPI). Metaphase spreads were imaged with a Nikon epifluorescence microscope equipped with a CCD camera (RS Photometrics).
ROS assay
Retroviral-infected cells grown on glass coverslips were assessed for ROS using the redox-sensitive fluorochrome 2′-7′-dichlorofluorescein diacetate (DCFDA; Molecular Probes) as described (Lee et al. 1999). Fresh stock of DCFDA (in DMSO) was added directly to the cell culture (final concentration 10 μM) and incubated for 30 min at 37°C. Samples were washed three times with PBS and mounted on glass slides. Cells were visualized using an epifluorescence microscope (Nikon), and digital images were collected using a CCD camera (RS Photometrics) at a designated exposure setting for all images. Captured images were quantified for fluorescence intensity using Image J software (National Institutes of Health). Relative mean fluorescence was obtained for ∼65 cells from multiple random fields.
Analysis of apoptotic morphology
Cells grown on glass coverslips in 60-mm dishes were exposed to a 137Cs γ-ray source (MDS Nordion) at a dose rate of 3 Gy/min for 1 min, 40 sec. Sixteen hours later, the cells were fixed in 3% formaldehyde for 5 min. Coverslips were washed with PBS, then mounted onto slides in 50% glycerol containing 10 μg/mL of DAPI. Nuclei were visualized using an epifluorescence microscope (Nikon), and digital images were collected using a CCD camera (RS Photometrics). Apoptotic cells were scored for changes in nuclear morphology. Apoptotic values were calculated as the number of apoptotic cells relative to the total number of cells in 15-20 random fields (>100 cells) and represent the average of three independent experiments ± the standard error of the mean.
Anchorage-independent growth
Approximately 105 cells were seeded in 3 mL of 0.35% lowmelting-point agarose in growth media (DMEM with either 10% FBS or serum-free supplements). The cell suspension was cast onto 60-mm plates with 3 mL of 0.5% agarose in growth media as an underlay. Fresh top agarose in growth media was added weekly. The assay was performed three times using cells from three separate infections. Photographs were taken 14 d after plating.
Tumorigenicity assay
The following experimental protocol was evaluated and approved by the Animal Care Committee at HKUST. Nude mice (Balb/cnu) 6-8 wk old were maintained in pathogen-free conditions. Cells (2 × 106) in a volume of 100 μL of PBS (pH 7.2) were injected subcutaneously into an area overlying the hind flank. Mice were regularly checked for tumor formation, and sacrificed when tumors reached 10-12 mm in diameter, or after 16 wk of monitoring.
Acknowledgments
We thank Drs. Tim Hunt, Faris Farassatti, and Melissa Jack for generous gifts of reagents. This work was supported in part by the Research Grants Council grants HKUST6129/02M and CA02/03.SC02 to R.Y.C.P. R.Y.C.P. is a Croucher Foundation senior fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1165204.
References
- Alcorta, D.A., Xiong, Y., Phelps, D., Hannon, G., Beach, D., and Barrett, J.C. 1996. Involvement of the cyclin-dependent kinase inhibitor p16INK4a in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. 93: 13742-13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi, S.E., Chang, S., Lee, S.L., Alson, S., Gottlieb, G.J., Chin, L., and DePinho, R.A. 2000. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406: 641-645. [DOI] [PubMed] [Google Scholar]
- Aruoma, O.I., Halliwell, B., Hoey, B.M., and Butler, J. 1989. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 6: 593-597. [DOI] [PubMed] [Google Scholar]
- Barnes, D. and Sato, G. 1980. Methods for growth of cultured cells in serum-free medium. Anal. Biochem. 102: 255-270. [DOI] [PubMed] [Google Scholar]
- Blasco, M.A., Lee, H.W., Hande, M.P., Samper, E., Lansdorp, P.M., DePinho, R.A., and Greider, C.W. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25-34. [DOI] [PubMed] [Google Scholar]
- Bodnar, A.G., Ouellette, M., Frolkis, M., Holt, S.E., Chiu, C.P., Morin, G.B., Harley, C.B., Shay, J.W., Lichtsteiner, S., and Wright, W.E. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349-352. [DOI] [PubMed] [Google Scholar]
- Bos, J.L. 1989. ras oncogenes in human cancer: A review. Cancer Res. 49: 4682-4689. [PubMed] [Google Scholar]
- Burgunder, J.M., Varriale, A., and Lauterburg, B.H. 1989. Effect of N-acetylcysteine on plasma cysteine and glutathione following paracetamol administration. Eur. J. Clin. Pharmacol. 36: 127-131. [DOI] [PubMed] [Google Scholar]
- Cone, R.D., Grodzicker, T., and Jaramillo, M. 1988. A retrovirus expressing the 12S adenoviral E1A gene product can immortalize epithelial cells from a broad range of rat tissues. Mol. Cell. Biol. 8: 1036-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbas, M. and White, E. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes & Dev. 7: 546-554. [DOI] [PubMed] [Google Scholar]
- Denko, N.C., Giaccia, A.J., Stringer, J.R., and Stambrook, P.J. 1994. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc. Natl. Acad. Sci. 91: 5124-5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri, G.P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E.E., Linskens, M., Rubelj, I., Pereira-Smith, O., et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. 92: 9363-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower, L.A., Harvey, M., Slagle, B.L., McArthur, M.J., Montgomery Jr., C.A., Butel, J.S., and Bradley, A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215-221. [DOI] [PubMed] [Google Scholar]
- Elenbaas, B., Spirio, L., Koerner, F., Fleming, M.D., Zimonjic, D.B., Donaher, J.L., Popescu, N.C., Hahn, W.C., and Weinberg, R.A. 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes & Dev. 15: 50-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher, D.W., Zetterberg, A., Zhu, J., Tlsty, T., and Bishop, J.M. 2000. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc. Natl. Acad. Sci. 97: 10544-10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, W.C. and Weinberg, R.A. 2002. Rules for making human tumor cells. N. Engl. J. Med. 347: 1593-1603. [DOI] [PubMed] [Google Scholar]
- Hahn, W.C., Counter, C.M., Lundberg, A.S., Beijersbergen, R.L., Brooks, M.W., and Weinberg, R.A. 1999. Creation of human tumour cells with defined genetic elements. Nature 400: 464-468. [DOI] [PubMed] [Google Scholar]
- Hayflick, L. 1965. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37: 614-636. [DOI] [PubMed] [Google Scholar]
- Hendzel, M.J., Nishioka, W.K., Raymond, Y., Allis, C.D., Bazett-Jones, D.P., and Th'ng, J.P. 1998. Chromatin condensation is not associated with apoptosis. J. Biol. Chem. 273: 24470-24478. [DOI] [PubMed] [Google Scholar]
- Hermeking, H. and Eick, D. 1994. Mediation of c-Myc-induced apoptosis by p53. Science 265: 2091-2093. [DOI] [PubMed] [Google Scholar]
- Hollstein, M., Shomer, B., Greenblatt, M., Soussi, T., Hovig, E., Montesano, R., and Harris, C.D. 1996. Somatic point mutations in the p53 gene of human tumors and cell lines: Updated compilation. Nucleic Acids Res. 24: 141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani, K., Xia, Y., Zweier, J.L., Sollott, S.J., Der, C.J., Fearon, E.R., Sundaresan, M., Finkel, T., and Goldschmidt-Clermont, P.J. 1997. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275: 1649-1652. [DOI] [PubMed] [Google Scholar]
- Jiang, X.R., Jimenez, G., Chang, E., Frolkis, M., Kusler, B., Sage, M., Beeche, M., Bodnar, A.G., Wahl, G.M., Tlsty, T.D., et al. 1999. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21: 111-114. [DOI] [PubMed] [Google Scholar]
- Johnson, L., Mercer, K., Greenbaum, D., Bronson, R.T., Crowley, D., Tuveson, D.A., and Jacks, T. 2001. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410: 1111-1116. [DOI] [PubMed] [Google Scholar]
- Karanjawala, Z.E., Murphy, N., Hinton, D.R., Hsieh, C.L., and Lieber, M.R. 2002. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr. Biol. 12: 397-402. [DOI] [PubMed] [Google Scholar]
- Karlsson, A., Deb-Basu, D., Cherry, A., Turner, S., Ford, J., and Felsher, D.W. 2003. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. 100: 9974-9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land, H., Parada, L.F., and Weinberg, R.A. 1983. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596-602. [DOI] [PubMed] [Google Scholar]
- Lee, A.C., Fenster, B.E., Ito, H., Takeda, K., Bae, N.S., Hirai, T., Yu, Z.X., Ferrans, V.J., Howard, B.H., and Finkel, T. 1999. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 274: 7936-7940. [DOI] [PubMed] [Google Scholar]
- Li, T.K., Chen. A.Y., Yu, C., Mao, Y., Wang, H., and Liu, L.F. 1999. Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes & Dev. 13: 1553-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., Sonik, A., Stindl, R., Rasnick, D., and Duesberg, P. 2000. Aneuploidy vs. gene mutation hypothesis of cancer: Recent study claims mutation but is found to support aneuploidy. Proc. Natl. Acad. Sci. 97: 3236-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A.W., Barradas, M., Stone, J.C., van Aelst, L., Serrano, M., and Lowe, S.W. 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & Dev. 12: 3008-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, D.T., Fuquay, J.I., Rawson, C.L., and Barnes, D.W. 1987. Extended culture of mouse embryo cells without senescence: Inhibition by serum. Science 236: 200-202. [DOI] [PubMed] [Google Scholar]
- Lowe, S.W. and Ruley, H.E. 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes & Dev. 7: 535-545. [DOI] [PubMed] [Google Scholar]
- Lowe, S.W., Ruley, H.E., Jacks, T., and Housman, D.E. 1993. p53-dependent apoptosis modulates the cytotoxicity of anti-cancer agents. Cell 74: 957-967. [DOI] [PubMed] [Google Scholar]
- Lowe, S.W., Jacks, T., Housman, D.E., and Ruley, H.E. 1994. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc. Natl. Acad. Sci. 91: 2026-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie, K.L., Franco, S., Naiyer, A.J., May, C., Sadelain, M., Rafii, S., and Moore, M.A. 2002. Multiple stages of malignant transformation of human endothelial cells modelled by co-expression of telomerase reverse transcriptase, SV40 T antigen and oncogenic N-ras. Oncogene 21: 4200-4211. [DOI] [PubMed] [Google Scholar]
- Marx, J. 2002. Debate surges over the origins of genomic defects in cancer. Science 297: 544-546. [DOI] [PubMed] [Google Scholar]
- Masutomi, K., Yu, E.Y., Khurts, S., Ben-Porath, I., Currier, J.L., Metz, G.B., Brooks, M.W., Kaneko, S., Murakami, S., DeCaprio, J.A., et al. 2003. Telomerase maintains telomere structure in normal human cells. Cell 114: 241-253. [DOI] [PubMed] [Google Scholar]
- Morales, C.P., Holt, S.E., Ouellette, M., Kaur, K.J., Yan, Y., Wilson, K.S., White, M.A., Wright, W.E., and Shay, J.W. 1999. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 21: 115-118. [DOI] [PubMed] [Google Scholar]
- Nigro, J.M., Baker, S.J., Preisinger, A.C., Jessup, J.M., Hostetter, R., Cleary, K., Bigner, S.H., Davidson, N., Baylin, S., Devilee, P., et al. 1989. Mutations in the p53 gene occur in diverse human tumour types. Nature 342: 705-708. [DOI] [PubMed] [Google Scholar]
- O'Brien, W., Stenman, G., and Sager, R. 1986. Suppression of tumor growth by senescence in virally transformed human fibroblasts. Proc. Natl. Acad. Sci. 83: 8659-8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello, S., Samper, E., Krtolica, A., Goldstein, J., Melov, S., and Campisi, J. 2003. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5: 741-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan, G. and Doxsey, S.J. 2003. Mutations and aneuploidy: Co-conspirators in cancer? Cancer Cell 4: 89-94. [DOI] [PubMed] [Google Scholar]
- Poon, R.Y.C., Toyoshima, H., and Hunter, T. 1995. Redistribution of the CDK inhibitor p27 between different cyclin · CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol. Biol. Cell 6: 1197-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdie, C.A., Harrison, D.J., Peter, A., Dobbie, L., White, S., Howie, S.E., Salter, D.M., Bird, C.C., Wyllie, A.H., Hooper, M.L., et al. 1994. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 9: 603-609. [PubMed] [Google Scholar]
- Qin, X.Q., Livingston, D.M., Kaelin Jr., W.G., and Adams, P.D. 1994. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. 91: 10918-10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, R.D., Morales, C.P., Herbert, B.S., Rohde, J.M., Passons, C., Shay, J.W., and Wright, W.E. 2001. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes & Dev. 15: 398-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson, C., Shirahata, S., Collodi, P., Natsuno, T., and Barnes, D. 1991. Oncogene transformation frequency of nonsenescent SFME cells is increased by c-myc. Oncogene 6: 487-489. [PubMed] [Google Scholar]
- Rich, J.N., Guo, C., McLendon, R.E., Bigner, D.D., Wang, X.F., and Counter, C.M. 2001. A genetically tractable model of human glioma formation. Cancer Res. 61: 3556-3560. [PubMed] [Google Scholar]
- Ruley, H.E. 1983. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 304: 602-606. [DOI] [PubMed] [Google Scholar]
- Seger, Y.R., Garcia-Cao, M., Piccinin, S., Cunsolo, C.L., Doglioni, C., Blasco, M.A., Hannon, G.J., and Maestro, R. 2002. Transformation of normal human cells in the absence of telomerase activation. Cancer Cell 2: 401-413. [DOI] [PubMed] [Google Scholar]
- Serrano, M., Lin, A.W., McCurrach, M.E., Beach, D., and Lowe, S.W. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593-602. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. and DePinho, R.A. 2000. Cellular senescence: Mitotic clock or culture shock? Cell 102: 407-410. [DOI] [PubMed] [Google Scholar]
- Shieh, S.Y., Ikeda, M., Taya, Y., and Prives, C. 1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325-334. [DOI] [PubMed] [Google Scholar]
- Shih, I.M., Zhou, W., Goodman, S.N., Lengauer, C., Kinzler, K.W., and Vogelstein, B. 2001. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 61: 818-822. [PubMed] [Google Scholar]
- Siliciano, J.D., Canman, C.E., Taya, Y., Sakaguchi, K., Appella, E., and Kastan, M.B. 1997. DNA damage induces phosphorylation of the amino terminus of p53. Genes & Dev. 11: 3471-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek, L. and Evan, G. 2002. Myc—Is this the oncogene from Hell? Cancer Cell 1: 406-408. [DOI] [PubMed] [Google Scholar]
- Stevenson, M. and Volsky, D.J. 1986. Activated v-myc and v-ras oncogenes do not transform normal human lymphocytes. Mol. Cell. Biol. 6: 3410-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro, G.J. and Green, H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17: 299-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa, O., Wade, M., Kern, S., Beeche, M., Pandita, T.K., Hampton, G.M., and Wahl, G.M. 2002. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 9: 1031-1044. [DOI] [PubMed] [Google Scholar]
- Vaziri, H. and Benchimol, S. 1998. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 8: 279-282. [DOI] [PubMed] [Google Scholar]
- Wang, T.L., Rago, C., Silliman, N., Ptak, J., Markowitz, S., Willson, J.K., Parmigiani, G., Kinzler, K.W., Vogelstein, B., and Velculescu, V.E. 2002. Prevalence of somatic alterations in the colorectal cancer cell genome. Proc. Natl. Acad. Sci. 99: 3076-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. and Levine, A.J. 1994. p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. 91: 3602-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., Ramsay, E.S., and Mock, B.A. 1998. Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16INK4a and p19ARF, is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc. Natl. Acad. Sci. 95: 2429-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimonjic, D., Brooks, M.W., Popescu, N., Weinberg, R.A., and Hahn, W.C. 2001. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 61: 8838-8844. [PubMed] [Google Scholar]