

Abstract
We compared nucleotide usage pattern conservation for related prokaryotes by examining the representation of DNA tetranucleotide combinations in 27 representative microbial genomes. For each of the organisms studied, tetranucleotide usage departures from expectations (TUD) were shared between related organisms using both Markov chain analysis and a zero-order Markov method. Individual strains, multiple chromosomes, plasmids, and bacteriophages share TUDs within a species. TUDs varied between coding and noncoding DNA. Grouping prokaryotes based on TUD profiles resulted in relationships with important differences from those based on 16S rRNA phylogenies, which may reflect unequal rates of evolution of nucleotide usage patterns following divergence of particular organisms from a common ancestor. By both symmetrical tree distance and likelihood analysis, phylogenetic trees based on TUD profiles demonstrate a level of congruence with 16S rRNA trees similar to that of both RpoA and RecA trees. Congruence of these trees indicates that there exists phylogenetic signal in TUD patterns, most prominent in coding region DNA. Because relationships demonstrated in TUD-based analyses utilize whole genomes, they should be considered complementary to phylogenies based on single genetic elements, such as 16S rRNA.
Biases in nucleotide composition and organization in prokaryotic genomes have long been recognized (Muto and Osawa 1987), with the representation of short oligonucleotide combinations as a focus of analysis (Henaut et al. 1996; Gelfand and Koonin 1997; Rocha et al. 1998). Dinucleotide frequencies within organisms represent genomic signatures, which may result from selective pressures as a result of dinucleotide stacking, DNA conformational tendencies, DNA replication and repair mechanisms, or selection by restriction endonucleases (Karlin et al. 1998), and codon usage also may influence nucleotide usage because it affects translational efficiency (Grantham et al. 1981; Grosjean and Freirs 1982; Sharp et al. 1993). However, constraints beyond dinucleotide frequencies and codon usage preferences can be identified only through analysis of longer oligonucleotide words (Pride and Blaser 2002). Methods available for determining the significance of oligonucleotide word frequencies include Markov chain analysis (Schbath et al. 1995; Cardon and Karlin 1994), which involves determining word frequencies by removing biases in their constituent oligonucleotides; however, the evolutionary significance of oligonucleotide word frequencies in prokaryotes has not been fully addressed.
Evolutionary inferences based on gene sequences, such as 16S rRNA (Woese and Fox 1977; Woese et al. 1990) are considered reliable indicators of prokaryotic ancestry; however, because evolutionary constraints are multidimensional (Koonin et al. 2000), analysis of a single gene is insufficient to fully understand the divergence between related life forms. The universally conserved 16S rRNA, with conservative rates of nucleotide substitution, is generally accepted as the standard for assessing microbial evolution; however, analysis of other gene loci often may not be phylogenetically congruent (Doolittle 1999). Such incongruencies often result from horizontal gene transfer, which obscures evidence of recent common ancestry (Holmes et al. 1999). With an increasing number of complete genomic sequences available, it now can be determined whether the relationships revealed from phylogenies based on 16S rRNA are reflected in the nucleotide usage patterns of individual organisms. Analysis of complete genomes can identify the extent to which nucleotide usage has evolved after divergence from recent common ancestors and can provide insight into selective pressures on usage not addressed by 16S rRNA sequences nor fully revealed in codon usage preference analyses.
Because analysis of tetranucleotide frequencies provides insights beyond those inferred from analysis of codon usage biases, we sought to develop an analytical method to examine their conservation across and between prokaryotic genomes. Our goals were to compare alternative models for determining tetranucleotide frequency divergences to understand the extent to which tetranucleotide usage is shared for multiple genomes and their plasmids and bacteriophages, and to determine whether tetranucleotide usage divergences exhibit phylogenetic signal compared with phylogenies based on 16S rRNA.
RESULTS
Representation of Tetranucleotide Combinations in Microbial Genomes
For the studied microbial genomes, we analyzed the tetranucleotide usage deviations from expectations (TUD) to determine whether the patterns of deviation are similar between closely related organisms. In a compromise between maximal information retrieval and minimal oligonucleotide length, tetranucleotides were selected for analysis because they offer both sufficient data points and provide data on nucleotide usage biases not inferred from codon usage analysis. We compared a zero-order Markov method that measures the deviation in usage of each tetranucleotide from that expected under a random mononucleotide distribution (Almagor 1983), and a Markov chain method (Cardon and Karlin 1994; Schbath et al. 1995) that measures the frequency divergence of tetranucleotides by removing the biases in their shorter oligonucleotide components. Although the TUD profile is unique for each microbial genome studied, closely related organisms are similar (Fig. 1). As expected, the TUD profiles for the two sequenced Helicobacter pylori strains are virtually superimposable (Fig. 1A). In other species (Neisseria meningitidis, Escherichia coli, Chlamydia pneumoniae, and Mycobacterium tuberculosis) for which two or more genomic sequences were analyzed, tetranucleotides with most extreme divergence and the extent of divergence were nearly identical for each member, indicating the existence of species-specific patterns (data not shown). Although H. pylori and Campylobacter jejuni differ in G + C content by 8.6% (Table 1), their TUD profiles are similar (Fig. 1A), including many of the most highly over- and underrepresented tetranucleotides, consistent with their close evolutionary relationship (Parkhill et al. 2000). As G + C content deviates from 50%, nucleotide usage is predicted to become less random (Muto and Osawa 1987; Sueoka 1988), however, even amongst organisms with G + C content near 50% (e.g., E. coli) their patterns of tetranucleotide usage are substantially deviated from expected (Fig. 1A). Of the organisms studied, the number of tetranucleotides with F(W) > |21.5| is highest forMethanococcus janaschii (34 tetranucleotides), followed by H. pylori (21), N. meningitidis (19), C. jejuni(12), and Deinococcus radiodurans (12). These organisms had the broadest range in tetranucleotide usage deviation using the zero-order Markov method. Similarity of profiles in related species is most clearly demonstrated by E. coli and Salmonella typhi; whereas M. tuberculosis and Mycobacterium leprae differ in profile to a greater degree (Fig. 1A). For both D. radiodurans and Vibrio cholerae, each of their two chromosomes had similar TUD profiles (data not shown).
Figure 1.

Frequency distribution of DNA tetranucleotide usage profiles of selected prokaryotes. The observed/expected tetranucleotide frequency divergence (F(W)) was determined for the 256 tetranucleotide combinations for each genome, using both Markov chain and zero-order Markov analysis as described in Methods section. The F(W) values were sorted within 0.25 intervals and the ordinate represents the number of tetranucleotide combinations within each interval. (A) Zero-order Markov analysis. (B) Markov chain analysis.
Table 1.
Bacterial Chromosomal and Plasmid Genomes Examined in This Study.
| Organism or Plasmid (Strain or Serogroup Designation) | Genome Size (bp) | Characteristic | GC Content (%) |
| Aeropyrum pernix(K1) | 1,669,695 | Archaea | 56.3 |
| Aquifex aeolicus (VF5) | 1,551,335 | Thermophilic bacteria | 43.5 |
| Bacillus halodurans(C-125) | 4,202,352 | Gram positive bacilli | 43.7 |
| Bacillus subtilis (168) | 4,214,814 | Gram positive bacilli | 43.5 |
| Campylobacter jejuni (NCTC11168) | 1,641,481 | Gram negative spiral bacilli | 30.6 |
| Chlamydia pneumoniae (CWL029) | 1,230,230 | Obligate eubacterial parasite | 40.6 |
| Chlamydia trachomatis (serovar D) | 1,042,519 | Obligate eubacterial parasite | 41.3 |
| Deinococcus radiodurans (R1) | 2,648,638a | Gram positive cocci | 67.0 |
| Chromosome 2 | 412,348 | 66.7 | |
| MP1 | 177,466 | Megaplasmid | 63.2 |
| CP1 | 45,704 | Plasmid | 56.2 |
| Escherichia coli (K12-MG1655) | 4,639,221 | Gram negative bacilli | 50.8 |
| Escherichia coli (O157:H7-EDL933) | 5,529,376 | Gram negative bacilli | 50.4 |
| pO157 | 92,084 | Plasmid | 47.6 |
| Haemophilus influenzae (KW20) | 1,830,138 | Gram negative bacilli | 38.2 |
| Helicobacter pylori (J99) | 1,643,831 | Gram negative spiral bacilli | 39.2 |
| Helicobacter pylori(26695) | 1,667,867 | Gram negative spiral bacilli | 38.9 |
| Lactococcus lactis(IL1403) | 2,365,589 | Gram positive cocci | 35.3 |
| Methanobacterium thermoautotrophicum | 1,751,377 | Archaea | 49.5 |
| (delta H) | |||
| Methanococcus janaschii (DSM2661) | 1,664,970 | Archaea | 31.3 |
| Mycobacterium leprae(TN) | 3,268,203 | Acid fast bacteria | 57.8 |
| Mycobacterium tuberculosis (H37RV) | 4,411,529 | Acid fast bacteria | 65.6 |
| Mycoplasma genitalium(G-37) | 580,074 | Obligate eubacterial parasite | 31.6 |
| Mycoplasma pneumoniae (M129) | 816,394 | Obligate eubacterial parasite | 49.9 |
| Neisseria meningitidis (serogroup A) | 2,184,406 | Gram negative cocci | 51.8 |
| Neisseria meningitidis (serogroup B) | 2,272,325 | Gram negative cocci | 51.5 |
| Pyrococcus abyssi (GE5) | 1,765,118 | Archaea | 44.9 |
| Pyrococcus horikoshii (OT3) | 1,738,505 | Archaea | 42.0 |
| Rickettsia prowazekii (Madrid E) | 1,111,523 | Spirochaete | 28.9 |
| Salmonella typhi | 4,809,037 | Gram negative bacilli | 52.1 |
| Synechocystis sp. (PCC6803) | 3,573,470 | Cyanobacteria | 47.7 |
| Thermotoga maritima (MSB8) | 1,860,725 | Thermophilic bacteria | 46.3 |
| Vibrio cholerae (El Tor N16961) | 2,961,149a | Gram negative curved bacilli | 47.7 |
| Chromosome 2 | 1,072,315 | 46.9 | |
| Yersinia pestis (CO92) | 4,653,728 | Gram negative bacilli | 47.6 |
| pCD1 | 70,559 | Plasmid | 44.8 |
The larger chromosome was designated as chromosome 1.
The zero-order Markov method yields a wider profile base with greater interspecies distinction than does the Markov chain method (Fig. 1). Although E. coli and M. tuberculosis have similar profiles in Markov chain analysis (Fig. 1B), they have unique profiles by zero-order Markov analysis (Fig. 1A). Thus, because the zero-order Markov method only removes the biases resulting from the frequencies of mononucleotides, the TUD calculated this way will incorporate the frequency biases of all the component oligonucleotides yielding distinct species-specific profiles.
Interchromosomal Tetranucleotide Comparisons
Pairwise genomic comparisons of TUD profiles within and between species illustrates that related organisms share common patterns (Fig. 2). Previous studies indicate that TUD patterns are highly conserved across prokaryotic genomes, with the exception of horizontally acquired genetic elements (Pride and Blaser 2002). Many of these elements, such as the cag island in H. pylori and the integron island in V. cholerae, have more similar TUD patterns to their host genomes than to other closely related organisms despite their horizontal acquisition (Table 2), and therefore were not excluded from the analysis. The two H. pylori strains have nearly identical profiles of tetranucleotide divergences (R2 > 0.99; Fig. 2A, B). These relationships are not based on G + C content, as randomly generated sequences designed with H. pylori G + C content show no correlation to either strain (R2 < 0.01) in TUD profiles. As expected by their evolutionary proximity (Parkhill et al. 2000), H. pylori and C. jejuni (Fig. 2C, D) have considerably more similarity in their TUD profiles than do H. pylori and H. influenzae(Fig. 2E, F), which have nearly identical G + C compositions (Table 1). The zero-order Markov method yields higher correlation in TUD profiles between H. pylori and C. jejuni or H. influenzae than does the Markov chain method, indicating that oligonucleotide (<4 nt) components contribute substantially to the similarity between species. Distantly related H. pylori and M. tuberculosis show no correlation (R2 < 0.03) in TUD patterns (data not shown; Appendix Table 1). Two Pyrococcus species show strong similarities to one another, whereas Bacillus subtilis and Bacillus halodurans are less similar (data not shown; Appendix Table 1). E. coli strains K12 and O157:H7 have nearly identical TUD (R2 > 0.99), despite the presence of 1387 additional open reading frames (ORFs) in O157:H7, a difference believed the result of horizontal gene transfer (Perna et al. 2001). For D. radiodurans that possesses two chromosomes, the TUD of each is nearly identical; a similar phenomenon was found for the two-chromosome V. cholerae as well (data not shown; Appendix Table 1).
Figure 2.

Linear regression analysis of DNA tetranucleotide usage profiles among selected genomes. F(W) was determined for each of the 256 tetranucleotide combinations for each genome as described in Methods section, and the profiles compared by linear regression analysis. (A, C, E) Zero-order Markov analysis (ZOM). (B, D, F) Markov chain analysis (MCM).
Table 2.
Comparison of Tetranucleotide Usage Deviation in Species-Specific Bacteriophages, Plasmids, and Horizontally Acquired Genetic Elements and Their Host Strains and Controls.
| Plasmid, Bacteriophage, or Island (bp) | Comparison Genome or Plasmid | R2a Zero-Order Markov | R2a Markov Chain |
| Deinococcus radiodurans megaplasmid | Chromosome 1 | 0.893 | 0.811 |
| (177,466) | Chromosome 2 | 0.915 | 0.805 |
| Bacillus subtilis | 0.721 | 0.053 | |
| D. radiodurans plasmid | Chromosome 1 | 0.573 | 0.348 |
| (45,704) | Chromosome 2 | 0.588 | 0.372 |
| Megaplasmid | 0.708 | 0.420 | |
| B. subtilis | 0.312 | 0.110 | |
| Escherichia coli O157:H7 plasmid pO157 | E. coli O157:H7 | 0.634 | 0.645 |
| (92,084) | E. coli K12 | 0.574 | 0.590 |
| Salmonella typhi | 0.533 | 0.615 | |
| Haemophilus influenzae | 0.185 | 0.121 | |
| Yersinia pestis plasmid pCD1 | Y. pestis | 0.800 | 0.684 |
| (70,559) | E. coli K12 | 0.669 | 0.503 |
| H. influenzae | 0.448 | 0.252 | |
| Enterobacterial-specific phage HKO22 | E. coli K12 | 0.805 | 0.592 |
| (40,751) | S. typhimurium | 0.717 | 0.606 |
| Mycobacterium tuberculosis | 0.269 | 0.108 | |
| Enterobacterial-specific phage P22 | E. coli K12 | 0.697 | 0.547 |
| (41,724) | S. typhimurium | 0.608 | 0.551 |
| M. tuberculosis | 0.195 | 0.127 | |
| Pseudomonas-specific phage D3 | Pseudomonas aeruginosa | 0.757 | 0.410 |
| (56,425) | E. coli K12 | 0.351 | 0.305 |
| Pseudomonas-specific phage ΦCTX | P. aeruginosa | 0.767 | 0.681 |
| (35,559) | E. coli K12 | 0.356 | 0.141 |
| Methanobacterium-specific phage ΘM2 | Methanobacterium thermoautotrophicum | 0.709 | 0.210 |
| (26,111) | Methanococcus janaschii | 0.344 | 0.090 |
| Pyrococcus abyssi | 0.275 | 0.122 | |
| Mycobacterium-specific phage D29 | M. tuberculosis | 0.450 | 0.266 |
| (49,136) | Mycobacterium leprae | 0.290 | 0.184 |
| Mycobacterium-specific phage I5 | M. tuberculosis | 0.428 | 0.342 |
| (52,297) | M. leprae | 0.270 | 0.218 |
| Helicobacter pylori J99 cag Island | H. pylori | 0.726 | 0.618 |
| (36,608) | C. jejuni | 0.632 | 0.144 |
| B. subtilis | 0.214 | 0.087 | |
| Vibrio cholerae integron island | Vibrio cholerae chromosome 1 | 0.394 | 0.047 |
| (125,300) | V. cholerae chromosome 2 | 0.499 | 0.205 |
| E. coliK12 | 0.220 | 0.005 | |
| B. subtilis | 0.208 | 0.010 | |
| Campylobacter jejuni extracellular | C. jejuni | 0.470 | 0.194 |
| polysaccaride biosynthesis cluster | H. pylori | 0.299 | 0.029 |
| (28,313) | B. subtilis | 0.114 | 0.048 |
Determined by linear regression comparisons of F(W) for all tetranucleotides.
Analysis of Plasmids, Species-Specific Phages, and Horizontally Acquired Genetic Elements
To determine whether organism-specific TUD patterns extend to horizontally acquired genetic elements, D. radiodurans was studied; its megaplasmid (177 kb) has similar patterns to the two chromosomes, but for its large (45 kb) plasmid, relationships are less close (Table 2). TUD profiles of pO157 found in E. coli O157:H7 are most similar with its host strain, less similar to E. coli strain K12 and to S. typhi, and dissimilar to the more distant H. influenzae. Similarly, Yersinia pestis plasmid pCD1 has TUD patterns highly similar to its host's chromosome, with less related bacteria progressively less similar. In general, smaller plasmids (<25 kb) share less similarity in TUD patterns to their host's genome than do larger plasmids (data not shown), consistent with their greater host range. Species-specific bacteriophages showed similar TUD patterns with their hosts (Table 2), which may hinder their ability to infect distantly related species. Whereas two Enterobacteriaceae-specific phages studied show parallel similarities to Enterobacteriaceae TUD patterns, larger differences are seen for two Mycobacterium-specific phages. Both the H. pylori cag island (Tomb et al. 1997) and the V. cholerae integron island (Heidelberg et al. 2000) have TUD patterns more similar to their host genomes than to other organisms studied (Table 2).
Intragenomic Comparisons of Tetranucleotide Usage
Although patterns of dinucleotide divergences in coding and noncoding DNA are essentially identical (Burge et al. 1992), our analysis of tetranucleotide usage deviations indicate that there are substantial differences in some prokaryotes (Table 3; Fig. 3). For H. pylori, although coding and noncoding DNA TUD profiles are strongly correlated (Fig. 3A, B), the most overrepresented tetranucleotides in coding and noncoding DNA differ (Table 3). Homopolymers CCCC and GGGG show substantial differences in representation between coding and noncoding DNA. That the most underrepresented tetranucleotides (GTAC, ACGT, and TCGA) in H. pylori are shared for both coding and noncoding DNA, indicates that factors beyond codon usage biases, such as restriction-endonuclease cognate sequence avoidance (Pride and Blaser 2002), influence their distribution (Table 3). For C. jejuni, the differences in TUD profiles in coding and noncoding DNA are greater than that for H. pylori (Table 3; Fig. 3C, D). B. subtilis has TUD profile differences in coding and noncoding DNA intermediate to that for H. pylori and C. jejuni (Table 3; Fig. 3E, F). Therefore, analysis of TUD profiles reveals greater differences between coding and noncoding DNA than would be predicted by analysis of dinucleotides.
Table 3.
Extremes of Tetranucleotide Usage Deviation in Coding and Noncoding DNA of Three Prokaryotic Genomes
| Zero Order Markov | Markov Chain | |||||||
| Coding | Noncoding | Coding | Noncoding | |||||
| Helicobacter pylori 26695 | (39.6%)a | (33.8%) | ||||||
| Overrepresented | AGCG | 3.178 | CCCC | 4.804 | CCGG | 1.475 | GAGT | 1.408 |
| CGCT | 3.066 | GGGG | 4.171 | AGCG | 1.456 | ACGA | 1.396 | |
| AAAA | 2.895 | CGCT | 2.969 | GTAT | 1.451 | GCGT | 1.379 | |
| TTTT | 2.830 | AGCG | 2.862 | ACCG | 1.443 | ACGC | 1.378 | |
| GGGG | 2.755 | GCGC | 2.667 | CGCT | 1.433 | ATAC | 1.355 | |
| Underrepresented | GTAC | 0.018 | GTAC | 0.053 | ACGT | 0.088 | ACGT | 0.111 |
| ACGT | 0.048 | ACGT | 0.055 | GTAC | 0.090 | GTAC | 0.204 | |
| TCGA | 0.049 | TCGA | 0.088 | TCGA | 0.111 | TCGA | 0.245 | |
| AGTA | 0.269 | ACTG | 0.316 | GGCC | 0.422 | GGCC | 0.403 | |
| TACT | 0.277 | TACG | 0.319 | CGCG | 0.427 | CGCG | 0.407 | |
| Campylobacter jejuni | (30.8%) | (25.5%) | ||||||
| Overrepresented | GCTT | 2.793 | GGGG | 5.123 | CAGG | 1.655 | GTCG | 1.993 |
| AAGC | 2.782 | GGGC | 3.656 | CCTG | 1.639 | ACGC | 1.578 | |
| AGCT | 2.343 | GCGG | 3.510 | GTCC | 1.483 | GACG | 1.533 | |
| TTGC | 2.275 | GCCC | 3.488 | CGCC | 1.473 | CAAG | 1.513 | |
| GCAA | 2.260 | CCCG | 3.443 | GGAC | 1.473 | CCCG | 1.496 | |
| Underrepresented | ACGT | 0.085 | ACGT | 0.125 | ACGT | 0.283 | ACGT | 0.344 |
| CCGG | 0.184 | GTAC | 0.294 | CCGG | 0.382 | GATC | 0.568 | |
| GACG | 0.288 | CATG | 0.308 | GGCC | 0.429 | GGCC | 0.622 | |
| CGAC | 0.288 | CTGT | 0.366 | GCGC | 0.457 | CTGT | 0.629 | |
| GTAC | 0.295 | CGTT | 0.386 | GGTC | 0.573 | GCGC | 0.634 | |
| Bacillus subtilis | (44.3%) | (38.6%) | ||||||
| Overrepresented | CAGC | 2.290 | TTTT | 2.445 | CGCC | 1.247 | GGAG | 1.376 |
| GCTG | 2.278 | AAAA | 2.371 | GGTG | 1.243 | CTCC | 1.372 | |
| AAAA | 2.265 | CCCC | 1.973 | GGCG | 1.235 | CCGC | 1.310 | |
| TTTT | 2.262 | CTCC | 1.957 | CACC | 1.227 | GGTG | 1.282 | |
| CGGC | 2.060 | GGAG | 1.937 | CGAT | 1.222 | CGGC | 1.282 | |
| Underrepresented | CTAG | 0.170 | CTAG | 0.401 | TAAG | 0.708 | GGCC | 0.694 |
| ACTA | 0.228 | CTAC | 0.422 | CTTA | 0.708 | CTCG | 0.729 | |
| TAGT | 0.234 | TAGT | 0.460 | GGCC | 0.743 | CTCA | 0.733 | |
| TAGG | 0.307 | ACTA | 0.467 | ATCT | 0.799 | TGAG | 0.743 | |
| CCTA | 0.314 | GTAG | 0.475 | CGCG | 0.802 | CGCG | 0.748 | |
G + C composition.
Figure 3.

Frequency distribution (A, C, E) and linear regression (B, D, F) of DNA tetranucleotide usage deviation profiles of selected prokaryotes. For each genome, the observed/expected tetranucleotide usage deviation (F(W)) was determined for the 256 combinations using zero-order Markov (ZOM) analysis as described in Methods section. The F(W) values were sorted within 0.25 intervals and the ordinate represents the number of tetranucleotide combinations within each interval.
Clustering of Organisms Based on Tetranucleotide Usage
Because TUD profiles appeared most similar between related organisms (Figs. 1, 2), we next sought to determine whether groupings based on such profiles resemble phylogenetic groupings based on 16S rRNA for 27 representative organisms. In the phylogram based on 16S rRNA, most Gram-negative organisms cluster together, with the archaea distant from the eubacteria, the thermophilic bacteria most proximate to the archaea, and the Chlamydia species and the Gram-positive organisms most proximate to the thermophilic bacteria (Fig. 4A). Because the zero-order Markov method yields distinct species-specific TUD profiles, we grouped organisms based on these profiles. The TUD profile-based phylogeny (Fig. 4B), shows different relationships from those based on 16S rRNA, including that: (1) Campylobacter, Helicobacter, and Rickettsia are more distant from the other Gram-negative organisms; (2) the relative distance between the archaea and the bacteria is decreased; (3) the Pyrococcus species are more distantly related to one another; (4) B. halodurans and B. subtilis are more distantly related to each other; (5) the relative distance between M. tuberculosis and M. lepraeis increased; (6) the relative distances between the Mycoplasma species are increased; and (7) the relative distances between the two N. meningitidis strains are increased. Groupings based on penta-, and hexanucleotide usage deviations are essentially identical to those based on tetranucleotides (data not shown). Thus, although the phylogenies produced have broad similarities, important differences are uncovered.
Figure 4.

Phylograms of 27 selected organisms for which genomic sequences are available. (A) 16S rRNA sequences were subjected to neighbor-joining analysis using HKY85 distance matrices. (B) The same organisms were grouped by using distance matrices based on the sums of the zero-order Markov F(W) differences from the other organisms for the 256 tetranucleotide combinations, and phylogenies created by neighbor-joining analysis. Bootstrap values based on 100 replicates are represented at each node, and branch length index is indicated in each panel. Gram-negative branches are indicated in green, Gram-positive in red, archaea and thermophilic bacteria in blue, and all other branches in black.
Analysis of Congruence Among 16S and Tetranucleotide Trees
The similarities between phylogenies created based on 16S rRNA and those created based on TUD profiles indicate that the latter contain phylogenetic signal. To determine the extent of the phylogenetic signal in TUD-based trees in comparison to 16S rRNA trees, topological differences between each were analyzed by symmetrical tree distances, which measure the number of clusters present exclusively in either tree (Penny and Hendy 1985). Of 100 trees based on 16S rRNA sequences, an average of nine clusters differ between each tree (green), while an average of 19 clusters differ between each TUD (red) tree (Fig. 5A). Comparisons of 16S rRNA vs. TUD trees show that an average of 27 clusters differ (blue), while neither 16S rRNA nor TUD trees has clusters in common with 100 random trees (Fig. 5A, black). Trees based on 16S rRNA and RpoA differ by an average of 23 clusters (Fig. 5B, blue), which indicates that the conservation of clusters for RpoA is similar to that for TUD. TUD trees based on coding DNA are similar to those for whole genomes, and have more clusters in common with 16S rRNA trees than those based on noncoding DNA (Fig. 5C, D), indicating that in prokaryotes, most of the phylogenetic signal exists in the coding regions. Importantly, 16S rRNA and TUD trees based on the Markov chain method differ by an average of 37 clusters (data not shown), demonstrating that phylogenetic signal is more conserved using the zero-order Markov method.
Figure 5.

Tree distance analysis of phylogenies of 27 prokaryotes. One hundred phylogenies were created using bootstrapping techniques for these organisms based on 16S rRNA or RpoA sequences, or tetranucleotide usage deviation (TUD). Tree distances were determined using symmetrical parameters (Penny and Hendy 1985) using Paup 4.0b8 (Swofford 1998). (A–D) The distances between each set of phylogenetic trees; black columns represent all comparisons with random trees. Tree comparisons represented are: (A) 16S rRNA and tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, tetranucleotide; blue, 16S vs. tetranucleotide); (B) 16S rRNA and RpoA trees (green, 16S rRNA; red, RpoA; blue, 16S vs. RpoA); (C) 16S rRNA and coding DNA tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, coding DNA tetranucleotide; blue, 16S vs. coding DNA tetranucleotide); (D) 16S rRNA and noncoding DNA tetranucleotide trees based on zero-order Markov criteria (green, 16S rRNA; red, noncoding DNA tetranucleotide; blue, 16S vs. noncoding DNA tetranucleotide).
Formal analysis of congruence between trees based on 16S rRNA and TUD was performed using likelihood analysis (Feil et al. 2001), a statistical test for comparison of tree topologies. The results are generally similar to those of the symmetrical tree distance analysis, with trees based on RpoA, RecA, GroE, and TUD revealing a high degree of similarity to 16S rRNA in topology (Fig. 6). In all cases, the differences in likelihoods (Δ-ln L) fall well outside those of 200 random trees (the 99th percentile of the random distribution), indicating a high degree of congruence among the trees. Trees for prokaryotic coding DNA TUD demonstrate more congruence with 16S rRNA trees than those of GroE, whole-genome TUD, and noncoding DNA TUD, and demonstrate a level of similarity to 16S rRNA trees parallel to that of RpoA and RecA trees. That coding DNA TUD trees are more congruent with 16S rRNA trees than noncoding DNA and whole-genome TUD trees confirms that the phylogenetic signal exists largely in the coding DNA. TUD phylogenies based on Markov chain analysis (Fig. 6G) and phylogenies based on whole-genome dinucleotide usage patterns (Fig. 6H), while demonstrating topological similarities to 16S rRNA, are far less congruent with 16S rRNA than the other trees analyzed.
Figure 6.

Likelihood analysis of phylogenetic congruence in prokaryotes. The phylogeny based on 16S rRNA is compared with phylogenies based on RpoA, GroE, RecA, whole-genome dinucleotide usage deviation, whole-genome tetranucleotide usage deviation (TUD), coding DNA TUD, or noncoding DNA TUD. The letters represent the locations of the distances in log likelihood (Δ-ln L) between the 16S rRNA phylogeny and: RpoA (A), GroE (B), RecA (C), whole-genome TUD based on zero-order Markov criteria (D), whole-genome TUD based on Markov chain analysis (E), coding DNA TUD based on zero-order Markov criteria (F), noncoding DNA TUD based on zero-order Markov criteria (G), and whole-genome dinucleotides based on zero-order Markov criteria (H). The 99th percentile of the likelihood differences between the 16S rRNA tree and the topologies from 200 random trees is indicated by the dotted line.
DISCUSSION
We analyzed prokaryotic genome TUD to determine whether common patterns are shared by related organisms. The Markov chain model, involving determining the expected frequency of a word by removing biases in its oligonucleotide components to find statistically meaningful deviations in word frequencies (Rocha et al. 1998), is the most common method for analysis of oligonucleotide word frequencies. However, by removing oligonucleotide component biases, cross-species comparisons become increasingly difficult, as these biases apparently contribute to the development of organism-specific nucleotide usage patterns. An alternative method, using zero-order Markov criteria (Almagor 1983), is based on comparing tetranucleotide frequencies across genomes by correcting for unequal base frequencies. Although there is no statistically meaningful way to compare differences observed using zero-order Markov and Markov chain criteria, the TUD developed by zero-order Markov analysis shows stronger relationships between like genomes (Figs. 1, 2).
Our data demonstrate that TUD patterns are well-conserved for both intra- and interspecies comparisons, and that similarity in these patterns is not based on G + C content. That the different chromosomes of D. radiodurans and V. cholerae demonstrate substantial TUD conservation, and that different H. pylori, E. coli, N. meningitidis, and C. pneumoniae strains share essentially identical TUD patterns, indicates their species specificity. That the closely related C. jejuni and H. pylori differing in G + C content by 8.6% demonstrate significant correlation in TUD patterns, while less closely related H. pylori and H. influenzae, which differ in G + C content only by 1%, have lower correlation, suggests that nucleotide usage patterns are relatively conserved despite evolution of G + C composition. The conservation in TUD patterns also extends to horizontally acquired genetic elements, plasmids, and bacteriophages with substantial correlation to their host organisms (Table 2). These findings further substantiate that there are organism-specific TUD patterns transmitted to horizontally acquired genetic elements, likely through the process of amelioration (Lawrence and Ochman 1997; Pride and Blaser 2002).
Phylogenetic reproduction based on prokaryotic nucleotide frequency divergences is not a novel concept, and is generally not believed to be as robust as standard phylogenetic methods based on 16S rRNA (Cardon and Karlin 1994; Leung et al. 1996). Our TUD-based analysis produces phylogenies similar to those based on 16S rRNA sequences, with several important differences. One explanation for these differences is that the 16S rRNA and TUD-based phylogenies result from unequal evolutionary rates after divergence of the studied organisms from common ancestors. For example, in contrast to 16S rRNA analysis, Gram-negative organisms E. coli, S. typhi, Y. pestis, H. influenzae, and N. meningitidis do not share a recent common ancestor with H. pylori and C. jejuni on TUD-based phylogenies. One hypothesis to explain the greater degree of difference between the Enterobacteriaceae and the Campylobacter/Helicobacter group is that the nucleotide usage patterns of H. pylori and C. jejuni are evolving more rapidly than their 16S rRNA sequences. In support of this hypothesis is that both H. pylori and C. jejuni demonstrate the greatest range in TUD of the organisms studied (Fig. 1A, B), and have substantial extremes of both tetranucleotide under- and overrepresentation. These extremes could result from lack of functional mismatch repair systems (Bhagwat and McClelland 1992) in both organisms (Tomb et al. 1997; Parkhill et al. 2000), or restriction-modification (R-M) induced pressures. R-M systems are believed to exert considerable selective pressures on nucleotide usage, as if restriction is intact but methylation incomplete, organisms avoiding the cognate sequences have a fitness advantage (Gelfand and Koonin 1997). Both H. pylori (Kong et al. 2000) and C. jejuni contain substantial numbers of R-M systems. The substantial underrepresentation of tetranucleotides ACGT, GTAC, and TCGA (Table 3), each the recognition sequence for known H. pylori R-M systems (Xu et al. 2000; V. Butkus, unpubl.), further suggests a role for these systems in shaping TUD patterns (Pride and Blaser 2002). That these tetranucleotides are underrepresented to similar extents in both coding and noncoding DNA (Table 3), supports this hypothesis, as R-M systems exert genome-wide pressures on nucleotide usage patterns, further demonstrating that the underrepresentation cannot be attributed to codon usage biases. Alternatively, natural competence and its control also could affect nucleotide usage patterns, as naturally competent organisms (e.g., M. janaschii, H. pylori, N. meningitidis, C. jejuni, and D. radiodurans) containing the largest numbers of R-M systems (Kong et al. 2000; Lin et al. 2001) possess the highest proportion of highly divergent tetranucleotides.
By analysis of congruence between phylogenetic trees (Feil et al. 2001) based on TUD profiles and on 16S rRNA, we demonstrate that there is phylogenetic signal in the whole-genome TUD patterns of prokaryotes, and that the signal is most prominent in coding DNA (Fig. 6). Phylogenetic trees for RpoA, RecA, and coding DNA TUD exhibit essentially identical levels of congruence with 16S rRNA phylogenies, and slightly higher levels of congruence than GroE and whole-genome TUDs. The lack of complete congruence among phylogenies based on housekeeping genes (such as RpoA, RecA, GroE) and 16S rRNA is usually attributed to frequent recombinational events (Holmes et al. 1999; Eisen 2000b), obscuring evidence of phylogenetic signal. Because nucleotide usage patterns in coding DNA are responsible for most of the phylogenetic signal, it is possible that recombination on a whole-genome level is reflected in the frequency of RecA and RpoA recombinational events, and that phylogenetic incongruencies between 16S rRNA and TUD trees may reflect differential levels of horizontal transfer events in certain prokaryotes. Trees for noncoding DNA TUDs and whole-genome TUDs based on Markov chain analysis are significantly correlated with 16S rRNA trees, but show much less congruence than trees based on housekeeping genes or zero-order Markov coding DNA TUDs, which indicates that little phylogenetic signal is conserved in noncoding or Markov chain TUD patterns (Fig. 6). That trees based on TUD also are substantially more congruent with 16S rRNA trees than those based on dinucleotide or codon usage frequencies (Fig. 6, and data not shown; Appendix Fig. 1), suggests that through analysis of longer oligonucleotide words, biases will be uncovered that contribute to phylogenetic signal. That TUD patterns have greater phylogenetic signal than codon frequencies supports the hypothesis that nucleotide organizational biases beyond those of codon usage are the basis for these results. Although previous studies indicate that there is considerable distance between the Mycoplasma species based on dinucleotide usage patterns (Karlin et al. 1997), in the TUD trees the Mycoplasma species cluster together, but with greater divergence than those based on 16S rRNA.
Although phylogenetic analysis of 16S rRNA provides the most widely accepted methodology for grouping organisms (Woese et al. 1990; Olsen et al. 1994; Pace 1997; Doolittle 1999), analysis of TUD patterns in microbial genomes provides a tool for examination of related organisms after their evolutionary divergence. We hypothesize that the differences indicate that organisms evolve nucleotide usage patterns more rapidly than 16S rRNA after diverging from their recent common ancestors, as is likely the explanation for the Mycoplasma clustering and for the Bacillus species. Thus, TUD analysis allows alternative insights into the selective forces governing microbial evolution, especially as a result of elements that might affect genomic structure, such as natural competence, lack of functional mismatch repair systems, and R-M systems. The benefits of the method are that it is easily reproducible, requires no foreknowledge of coding and noncoding sequences, requires no nucleotide or amino-acid alignments, and contains phylogenetic signal rivaling that of housekeeping genes. The drawbacks of the method include that it likely is subject to convergent evolution, in which external forces induce changes in genomic nucleotide usage patterns, giving unrelated organisms the appearance of recent common ancestry. This phenomenon of homoplasy also substantially influences phylogeny based on single genes, and is thus not unique to TUD analysis (Maynard Smith and Smith 1998). Another similar drawback is that the method may be subject to global influences (e.g., restriction endonucleases) that affect genomic structure, increasing the apparent distance between related organisms. These global forces should not be ignored, but may not be uniform for all organisms, probably affecting ancestral reproduction. The method also is influenced by horizontal transfer events. In organisms in which the proportion of horizontal transfer is large, such as Thermotoga maritima (Nelson et al. 1999), its phylogenetic position on TUD trees may be affected. This is offset at least partially by the phenomenon of amelioration (Table 2), thus dampening the effect of horizontal transfer events (Pride and Blaser 2002). For phylogenetic studies, use of TUD and other such whole genomic analyses (Sankoff et al. 1992; Fitz-Gibbon and House 1999; Snel et al. 1999; Eisen 2000a) should be considered complementary to analyses based on single gene products, such as 16S rRNA.
METHODS
Microbial Genomes, Phages, and Plasmids
Complete genome sequences of the bacteria, archaea, bacteriophages, and plasmids (all > 25 kb) studied were obtained from GenBank (ftp://ncbi.nlm.nih.gov/genbank/genomes/bacteria/, http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/phg.html, and http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/eub_p.html, respectively) (Tables 1 and 3). Coding regions of prokaryotic genomes were identified based on GenBank annotation using Swaap PH 1.0 (Pride, D.T. 2001. Swaap PH 1.0: A tool for analyzing nucleotide usage patterns in coding and noncoding portions of microbial genomes. Distributed by the author, Department of Microbiology and Immunology, Vanderbilt University, Nashville, Tennessee, available at http://www.bacteriamuseum.org/SWAAP/SwaapPage.htm), and noncoding regions were classified as all other DNA sequences.
Analysis of Representation of Nucleotide Combinations
To determine the tetranucleotide usage departures from expectations among prokaryotic genomes, two different Markov methods were used. The zero-order Markov method (Almagor 1983) is designed to determine the expected number of tetranucleotides by removing biases in mononucleotide frequencies. The expected number of tetranucleotides is determined by the equation: E(W) = [(Aa * Cc * Gg * Tt) * N], where A, C, G, and T represent the frequency of nucleotides A, C, G, and T within the window being evaluated, respectively, a, c, g, and t represent the number of nucleotides A, C, G, and T in each tetranucleotide, respectively, and N represents the length of the window being evaluated. The frequency of divergence of the word F(W) is expressed as the ratio of the observed O(W) to the expected E(W). Markov chain analysis (Cardon and Karlin 1994; Schbath et al. 1995) determines the expected frequency of oligonucleotide words by removing biases in their oligonucleotide components. Briefly, as described (Rocha et al. 1998), W = (w1w2…wm) denotes the word formed by the concatenation of m nucleotides, and N(W) is its observed count in a sequence of length n. The expected count E(W) of W is:
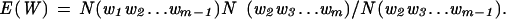 |
For each genome analyzed, comparisons of F(W) for each tetranucleotide combination, and for the reverse-complement of each combination by linear regression analysis yielded R2 values = 0.99; therefore, analyses concentrated only on the documented clockwise strand F(W) values. The profile of TUD for all tetranucleotides was determined for each organism studied (Table 1) using Swaap 1.0.0 (Pride, D.T. 2001. Swaap 1.0.0: A tool for analyzing substitutions and similarity in multiple alignments. Distributed by the author, Department of Microbiology and Immunology, Vanderbilt University in Nashville, Tennessee, available at http://www.bacteriamuseum.org/SWAAP/SwaapPage.htm), and their relative intra- and intergenomic abundance compared by linear regression analysis using Microsoft Excel 2000 (Microsoft Corp., Inc.).
Cluster Analysis of Prokaryotes
Distances based on tetranucleotide frequency divergences were determined: Dt = ¼N * |F1(W) − F2(W)|, where N equals the length of the nucleotide word, F1(W)and F2(W) represent F(W) for each of the 256 tetranucleotides for organisms 1 and 2 (analogous to computations derived by Cardon and Karlin [1994]). Bootstrapping was performed by sampling with replacement of each of the 256 tetranucleotide frequencies using Swaap PH 1.0 (Pride, D.T. 2001. Swaap PH 1.0: A tool for analyzing nucleotide usage patterns in coding and noncoding portions of microbial genomes. Distributed by the author, Department of Microbiology and Immunology, Vanderbilt University in Nashville, Tennessee, available at http://www.bacteriamuseum.org/SWAAP/SwaapPage.htm), and phylograms were created based on distance matrices using Phylip 3.5 (Felsenstein 1989), and displayed using Treeview (Page 1996). 16S rRNA sequences were obtained from the Ribosomal Database Project II (Maiden et al. 2001), and phylograms were created using HKY85 distances with Phylip 3.5 (Felsenstein 1989). Sequences of RpoA (RNA polymerase subunit A), RecA (recombination protein A), and GroE (HSP60 family chaperonin) were obtained from the COG database (Tatusov et al. 2001), and phylograms created using mean distances with Phylip 3.5 (Felsenstein 1989).
Analysis of Congruence Among Phylogenetic Trees
Analysis of symmetrical distances among phylogenetic trees was performed using the method of Penny and Hendy (1985). Briefly, 100 phylograms were created for 16S rRNA, RecA, GroE, RpoA, or tetranucleotides by bootstrapping, and 100 phylograms with random topology also were created. Each set of phylograms was compared using Paup 4.0b8 (Swofford 1998). Analysis of congruence among the gene phylograms was performed on consensus trees, and 200 trees were created with random topology. A maximum likelihood method, similar to that used by Feil et al. (2001), was used to determine the extent of congruence among phylograms; differences in log likelihood (Δ-ln L) were computed between phylograms based on 16S rRNA and phylograms based on RecA, RpoA, GroE, tetranucleotides, dinucleotides, and random topology. Differences in Δ-ln L for random phylograms can be considered as the null distribution, which would be obtained when there is no more similarity in topology than that expected by chance. If the Δ-ln L values for comparisons among the phylograms fall within the 99th percentile of the null distribution, then the topologies are significantly different, and thus incongruent (Feil et al. 2001).
Figure a.

Appendix Figure 1 Phylograms of 27 selected organisms for which genomic sequences are available. Organisms were grouped by using distance matrices based on the sums of the differences from the other organisms for the frequencies of the 64 codons, and phylogenies created by neighbor-joining analysis. Bootstrap values based on 100 replicates are represented at each node, and branch length index is indicated in each panel.
WEB SITE REFERENCES
ftp://ncbi.nlm.nih.gov/genbank/genomes/bacteria/; GenBank Web site which offers bacterial and archaeal genome sequences.
http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/phg.html; GenBank Web site which offers bacteriophage genome sequences.
http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/eub_p.html; GenBank Web site which offers bacterial and archaeal plasmid sequences.
http://www.bacteriamuseum.org/SWAAP/SwaapPage.htm; Web site which offers Swaap 1.0.0 and Swaap PH 1.0.
Table a.
Appendix. Linear Regression Analysisa of DNA Tetranucleotide Usage Profiles Among Selected Prokaryotes.b
 |
R2 values from linear regression analysis displayed. R2 values ≥ 0.50 in bold.
Markov chain analysis displayed in top half of matrix. Zero-order Markov analysis displayed in bottom half of matrix.
Ae, Aquifex aeolicus; Ap, Aeropyrnum pernix; Bh, Bacillus halodurans; Bs, Bacillus subtilis; Cj, Campylobacter jejuni; Cp, Chlamydia pneumoniae; Ct, Chlamydia trachomatis; Dr,Deinococcus radiodurans chromosomes 1 and 2; EcK,Escherichia coli K12; EcO, Escherichia coli O157:H7; Hi, Haemophilus influenzae; HpJ, Helicobacter pylori J99; Hp2, Helicobacter pylori 26695; Ll,lactococcus lactis; Mth, Methanobacterium thermoautotrophicum; Mj, Methanococcus janaschii; Ml, Mycobacterium leprae; Mtb, Mycobacterium tuberculosis; Mg, Mycoplasma genitalium; Mp, Mycoplasma pneumoniae; Nm, Neisseria meningitidis serotypes A and B; Pa, Pyrococcus abyssi; Ph,Pyrococcus horikoshii; Rp, Ricketsia prowazekii; St, Salmonella typhi; Ss, Synechocystis species; Tm, Thermotoga maritima; Vc, Vibrio cholerae chromosomes 1 and 2; Yp, Yersenia pestis.
Acknowledgments
Supported in part by the Medical Scientist Training Program, the National Institutes of Health (RO1DK53707, RO1GM63270, and the Cancer Center Core grant CA68485), the UNCF-Merck Science Initiative, the Foundation for Bacteriology, and the Gates Millennium Scholars Program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL Prided01@med.nyu.edu; FAX (212) 252–7164.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.335003. Article published online before print in January 2003.
REFERENCES
- 1.Almagor H. 1983. A Markov analysis of DNA sequences. J. Theor. Biol. 104: 633-645. [DOI] [PubMed] [Google Scholar]
- 2.Bhagwat A.S. and McClelland, M. 1992. DNA mismatch correction by very short patch repair may have altered the abundance of oligonucleotides in the E. coli genome. Nucleic Acids Res. 20: 1663-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burge C., Campbell, A.M., and Karlin, S. 1992. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci. 89: 1358-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cardon L.R. and Karlin, S. 1994. Computational DNA sequence analysis. Annu. Rev. Microbiol. 48: 619-654. [DOI] [PubMed] [Google Scholar]
- 5.Doolittle W.F. 1999. Phylogenetic classification and the universal tree. Science 284: 2124-2128. [DOI] [PubMed] [Google Scholar]
- 6.Eisen J.A. 2000a. Assessing evolutionary relationships among microbes from whole-genome analysis. Curr. Opin. Microbiol. 3: 475-480. [DOI] [PubMed] [Google Scholar]
- 7.Eisen J.A. 2000b. Horizontal gene transfer among microbial genomes: New insights from complete genome analysis. Curr. Opin. Genet. Dev. 10: 606-611. [DOI] [PubMed] [Google Scholar]
- 8.Feil E.J., Holmes, E.C., Bessen, D.E., Chan, M.-S., Day, N.P.J., Enright, M.C., Goldstein, R., Hood, D.W., Kalia, A., Moore, C.E., et al. 2001. Recombination within natural populations of pathogenic bacteria: Short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. 98: 182-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felsenstein J. 1989. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5: 164-166. [Google Scholar]
- 10.Fitz-Gibbon S.T. and House, C.H. 1999. Whole-genome based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 27: 4218-4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gelfand M.S. and Koonin, E.V. 1997. Avoidance of palindromic words in bacterial and archaeal genome: A close connection with restriction enzymes. Nucleic Acids Res. 25: 2430-2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grantham R., Gautier, C., Guoy, M., Jacobzone, M., and Mercier, R. 1981. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 9: R43-R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosjean H. and Freirs, W. 1982. Preferential codon usage in prokaryotic genes: The optimal codon-anti-codon interaction energy and the selective codon usage in efficiently expressed genes. Gene 18: 199-209. [DOI] [PubMed] [Google Scholar]
- 14.Heidelberg J.F., Eisen, J.A., Nelson, W.C., Clayton, R.A., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Umayam, L., et al. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406: 477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henaut A., Rouxel, T., Gleizes, A., Moszer, I., and Danchin, A. 1996. Uneven distribution of GATC motifs in the Escherichia coli chromosome, its plasmids and its phages. J. Mol. Biol. 257: 574-585. [DOI] [PubMed] [Google Scholar]
- 16.Holmes E.C., Urwin, R., and Maiden, M.C.J. 1999. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitides. Mol. Biol. Evol. 16: 741-749. [DOI] [PubMed] [Google Scholar]
- 17.Karlin S., Mrazek, J., and Campbell, A.M. 1997. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 179: 3899-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlin S., Campbell, A.M., and Mrazek, J. 1998. Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 32: 185-225. [DOI] [PubMed] [Google Scholar]
- 19.Koonin E.V., Aravind, L., and Kondrashov, A.S. 2000. The impact of comparative genomics on our understanding of evolution. Cell 101: 573-576. [DOI] [PubMed] [Google Scholar]
- 20.Kong H., Lin, L.-F., Porter, N., Stickel, S., Byrd, D., Posfai, J., and Roberts, R.J. 2000. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 28: 3216-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence J.G. and Ochman, H. 1997. Amelioration of bacterial genomes: Rates of change and exchange. J. Mol. Evol. 44: 383-397. [DOI] [PubMed] [Google Scholar]
- 22.Leung M.Y., Marsh, G.M., and Speed, T.P. 1996. Over- and underrepresentation of short DNA words in herpesvirus genomes. J. Comp. Biol. 3: 345-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin L.-F., Posfai, J., Roberts, R. J., and Kong, H. 2001. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc. Natl. Acad. Sci. 98: 2740-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiden B.L., Cole, J.R., Lilburn, T.G., Parker, C.T., Jr., Saxman, P.R., Farris, R.J., Garrity, G.M., Olsen, G.J., Schmidt, T.M., and Tiedje, J.M. 2001. The RDP-II (ribosomal database project). Nucleic Acids Res. 29: 173-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maynard Smith J. and Smith, N.H. 1998. Detecting recombination from gene trees. Mol. Biol. Evol. 15: 590-599. [DOI] [PubMed] [Google Scholar]
- 26.Muto A. and Osawa, S. 1987. The guanine and cytosine content of genomic DNA and bacterial-evolution. Proc. Natl. Acad. Sci. 84: 166-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson K.E., Clayton, R.A., Gill, S.R., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Nelson, W.C., Ketchum, K.A., et al. 1999. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritime. Nature 399: 323-329. [DOI] [PubMed] [Google Scholar]
- 28.Olsen G.J., Woese, C.R., and Overbeek, R. 1994. The winds of (evolutionary) change: Breathing new life into microbiology. J. Bacteriol. 176: 1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace N.R. 1997. A molecular view of microbial diversity and the biosphere. Science 276: 734-740. [DOI] [PubMed] [Google Scholar]
- 30.Page R.D.M. 1996. TREEVIEW: An application to display phylogenetic trees on personal computers. Comp. Appl. Biosci. 12: 357-458. [DOI] [PubMed] [Google Scholar]
- 31.Parkhill J., Wren, B.W., Mungall, K., Ketley, J.M., Churcher, C., Basham, D., Chillingworth, T., Davies, R.M., Feltwell, T., Holroyd, S., et al. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665-668. [DOI] [PubMed] [Google Scholar]
- 32.Penny D. and Hendy, M.D. 1985. The use of tree comparison metrics. Systematic Zoology 34: 75-82. [Google Scholar]
- 33.Perna N.T., Plunkett, G., III, Burland, V., Mau, B., Glasner, J.D., Rose, D.J., Mayhew, G.F., Evans, P.S., Gregor, J., Kirkpatrick, H.A., et al. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409: 529-533. [DOI] [PubMed] [Google Scholar]
- 34.Pride D.T. and Blaser, M.J. 2002. Identification of horizontally acquired genetic elements in Helicobacter pylori and other prokaryotes using oligonucleotide difference analysis. Genome Letters 1: 2-15. [Google Scholar]
- 35.Rocha E.P.C., Viari, A., and Danchin, A. 1998. Oligonucleotide bias in Bacillus subtilis: General trends and taxonomic comparisons. Nucleic Acids Res. 26: 2971-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sankoff D., Leduc, G., Antoine, N., Paquin, B., Lang, B.F., and Cedergren, R. 1992. Gene order comparisons for phylogenetic inference: Evolution of the mitochondrial genome. Proc. Natl. Acad. Sci. 89: 6575-6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schbath S., Prum, B., and de Turckheim, E. 1995. Exceptional motifs in different Markov chain models for a statistical analysis of DNA sequences. J. Comp. Biol. 2: 417-437. [DOI] [PubMed] [Google Scholar]
- 38.Sharp P.M., Stenico, M., Peden, J.F., and Lloyd, A.T. 1993. Codon usage: Mutational bias, translational selection, or both? Biochem. Soc. Trans. 21: 835-841. [DOI] [PubMed] [Google Scholar]
- 39.Snel B., Bork, P., and Huynen, M.A. 1999. Genome phylogeny based on gene content. Nature Genetics 21: 108-110. [DOI] [PubMed] [Google Scholar]
- 40.Suoeka N. 1988. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. 85: 2653-2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swofford, D.L. 1998. Paup 4.0b2. Phylogenetic analysis using parsimony (* and other methods). Version 4. Sinauer Associates, Sunderland, Massachusetts.
- 42.Tatusov R.L., Natale, D.A., Garkavtsev, I.V., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The Cog Database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29: 22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomb J.-F., White, O., Kervalage, A.R., Clayton, R.A., Sutton, G.G., Fleischman, R.D., Ketchum, K.A., Klenk, H.P., Gill, S., Dougherty, B.A., et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388: 539-547. [DOI] [PubMed] [Google Scholar]
- 44.Woese C.R. and Fox, G.E. 1977. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. 74: 5088-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woese C.R., Kandler, O., and Wheelis, M.L. 1990. Towards a natural system of organisms: Proposal for the domains archaea, bacteria, and eukarya. Proc. Natl. Acad. Sci. 87: 4576-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Q., Morgan, R.D., Roberts, R.J., and Blaser, M.J. 2000. Identification of type II restriction and modification systems in Helicobacter pylori reveals their substantial diversity among strains. Proc. Natl. Acad. Sci. 97: 9671-9676. [DOI] [PMC free article] [PubMed] [Google Scholar]