

Abstract
A number of lower organisms (bacteria, fungi, and parasites) produce glycoconjugates that contain furanose rings. Of particular interest to our group are cell wall polysaccharides from mycobacteria, including the human pathogen, Mycobacterium tuberculosis, which contain a large number of arabinofuranose resides. As part of a larger project on the conformational analysis of these molecules, we report here molecular dynamics simulations on methyl α-D-arabinofuranoside (1) using the AMBER force field and the GLYCAM carbohydrate parameter set. We initially studied the ability of this method to predict rotamer populations about the hydroxymethyl group (C4–C5) bond. Importantly, we show that simulation times of up to 200 ns are required in order to obtain convergence of the rotamer populations for this ring system. We also propose a new charge derivation approach that accounts for the flexibility of the furanoside ring by taking an average of the charges from a large number of conformers across the psuedorotational itinerary. The approach yields rotamer populations that are in good agreement with available NMR data and, in addition, provides insight into the nature of the puckering angle and amplitude in 1.
Introduction
Furanose rings are important components of a number of glycoconjugates, with the most well-known examples being the nucleic acids, which contain either D-ribofuranose or 2-deoxy-D-erthyro-pentofuranose (2-deoxy-D-ribose).1 It is less widely appreciated that a number of bacteria, fungi, and parasites also biosynthesize furanoside-containing polysaccharides.2,3 Glycans composed of furanosyl moieties are typically found on the surfaces of the organisms that produce them, and thus they play important roles in the interaction of these species with their environment. Furanosyl residues are also key components of natural products other than polysaccharides, including plant opines,4 glycopeptides,5 and the aminoglycoside antibiotics.6
Among the most elaborate examples of these glycoconjugates are two polysaccharides, arabinogalactan (AG) and lipoarabinomannan (LAM), that are found in the cell wall of mycobacteria.7 Notable among these are the pathogenic organisms Mycobacterium tuberculosis, M. leprae, and M. avium, which, respectively, cause tuberculosis, leprosy, and a tuberculosis-like disease common in HIV-positive individuals. The AG, a polysaccharide containing approximately 100 monosaccharide units, is composed entirely of arabinofuranose and galactofuranose residues, except for two pyranose moieties, which serve as the linker between the glycan and peptidoglycan.8 Similarly, a significant component of LAM is an arabinan domain, representing approximately half the molecular weight, which contains only arabinofuranose residues.9
The AG is esterified at its nonreducing end with mycolic acids, C70–C90 branched lipids, yielding a glycolipid named the mycolyl-arabinogalactan (mAG) complex, which is the major structural component of the cell wall. In the accepted model for the macrostructure of the mycobacterial cell wall,10 the mycolic acids pack perpendicular to the plasma membrane thus forming a lipid layer at the periphery of the assembly. This layer of tightly packed mycolic acids serves as a major permeability barrier to the passage antibiotics. Thus, the AG serves as a scaffold by which the organism attaches an additional permeability barrier to peptidoglycan and, in turn, the plasma membrane.
The essentially exclusive presence of furanosyl rings in the AG is curious as they are of higher energy than their pyranose counterparts.11 It has been hypothesized12 that the AG is composed of furanose residues because the resulting glycan has greater flexibility relative to a pyranose-containing species. A more malleable scaffold would be expected to facilitate optimal packing of the mycolic acids, which in turn would provide the organism with great protection against its environment. This “flexible-scaffold hypothesis” is plausible considering that while pyranose rings typically adopt single well-defined chair conformations, furanose rings can exist in a variety of twist and envelope conformations that are separated by typically low-energy barriers. Therefore, a polysaccharide composed of furanose residues would be more flexible than one made of pyranose residues. Although intriguing, there are scant data to support the flexible scaffold hypothesis. As part of a program dedicated to understanding the conformation of mycobacterial AG (and LAM), we have carried out a series of NMR studies on the arabinofuranose-containing oligosaccharides13,14 and coupled these experimental studies with high-level ab initio and density functional theory calculations on methyl α-D-arabinofuranoside (1, Figure 1)15,16 and related analogs.17–20
Figure 1.

Structure of methyl α-D-arabinofuranoside.
Given their inherent flexibility, the conformational analysis of furanosides is more complicated than comparable studies with pyranosides as more than one ring conformer must be considered. For a given furanoside, the ten unique envelope (E) and twist (T) forms can be identified on the pseudorotational wheel (Figure 2), each with a unique psuedorotational phase angle, P.21 In solution, there is a dynamic equilibrium between ring conformers, usually dominated by two major species between which the interconversion barrier is low (typically <5 kcal/mol).17 One of these conformers is generally found in the northern hemisphere of the pseudorotational wheel, and the other in the southern hemisphere,21 which are termed the North and South conformers, respectively. Conformational investigations of furanoside rings by NMR spectroscopy most commonly involve analysis using PSEUROT,22 a program that assumes this two-state equilibrium and which fits the experimental 1H–1H coupling constant data to two conformers and their populations.
Figure 2.

Pseudorotational itinerary for a D-aldofuranose ring.
Other key conformational features of importance include rotamer populations about the glycosidic C1–O1 and C4–C5 bonds. The preferred rotamer about the C1–O1 bond is the one in which the aglycone (e.g., the methyl group in 1) is oriented anti with respect to the C1–C2 bond, as this is favored by the exo-anomeric effect.23 For the C4–C5 bond (ω angle), three rotamers are typically present, gt, tg, and gg (Figure 3), with the distribution being influenced by a combination of steric and stereoelectronic (gauche) effects.24–27
Figure 3.
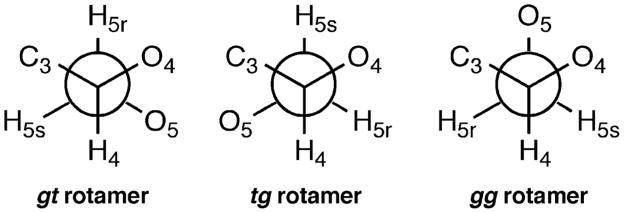
Definition of gg, gt, and tg rotamers about the C4–C5 bond.
Having studied the conformation of 1 using both experimental and high-level computational methods, we are interested in looking at larger oligomers of D-arabinofuranose, for which we have NMR data.13,14 However, given the size of these molecules, their treatment with ab initio or density functional theory methods is of limited practicality. Thus, we have begun to investigate the use of force field models to probe the conformation of these oligosaccharides. Previous molecular mechanics studies of furanosyl rings have largely been carried out using MM3 or earlier variants of this force field.28–34 However, over the past several years the use of the AMBER35 force field in conjunction with the GLY-CAM36,37 parameter set has emerged as a reliable force field for molecular mechanics studies of oligosaccharides containing pyranose rings. In this paper, we describe the results of our first investigations of the use of the GLYCAM parameters and the AMBER force field to study the conformation of furanoside rings. More specifically, we report the ability of this computational method to predict the rotamer distribution about the C4–C5 bond and pseudorotational phase angle in 1 as determined by NMR spectroscopy. In this regard, these studies are similar to recent work by Woods and Kirschner38 in which a similar analysis of hydroxymethyl groups on pyranoside rings was carried out. The notable differences here are that ring conformation is addressed and a new charge calculation procedure had to be implemented to take into account the flexibility of the furanoside ring.
Methods
Simulations
We adopted the combined AMBER/GLYCAM force field for the simulations of 1. All the MD simulations were carried out using the AMBER 9.035 suite of programs, and the electronic structure calculations were performed with Gaussian 03.39
Solution Simulations
A 200 ns MD simulation of 1 was performed in a box of 298 TIP3P40 water molecules under NPT conditions. The total box size was (25.569 × 25.372 × 25.544) (Å). The temperature was set to 300 K and the pressure to 1 atm. A cutoff of 8 Å was set for nonbonded interactions. The SCNB and SCEE scaling parameters were both set to 1.0 in accordance with the GLYCAM approach. All simulations were carried out under NPT conditions, and the SHAKE41 algorithm was used to constrain all hydrogen-containing bonds. Prior to production MD simulations, minimization of the waters was first performed, followed by minimization of the whole system, 100 ps of annealing and 150 ps of equilibration. Ewald summation was used to handle long-range electrostatics.
Gas-Phase Simulations
The temperature was set to 300 K. A cutoff of 18 Å was set for nonbonded interactions. The SCNB and SCEE scaling parameters were both set to 1.0 in accordance with the GLYCAM approach. The SHAKE41 algorithm was used to constrain all hydrogen-containing bonds.
Atomic Charges
Two charge derivation procedures were considered. The first one is the ensemble average approach proposed by Woods and workers42 and is referred to as the usual GLYCAM procedure. Following this procedure, crystallographic data43 were employed for the input geometry of methyl α-D-arabinofuranoside, and an ab initio geometry optimization was then performed at the HF/6-31G* level of theory. Based on the HF/6-31G* single point, the RESP44 approach was used to obtain an initial set of restrained partial atomic charges. A relatively short MD simulation (10 ns) based on these charges and 100 conformations were selected from the resulting trajectory. The dihedral angles of the rotatable exocyclic moieties, such as hydroxyl groups, were then determined from the 100 snapshots and transferred to the quantum mechanics optimized geometry. Single point HF/6-31G* calculations were performed for these 100 new conformations. Partial atomic charges were obtained using the RESP approach for the 100 conformations, and the final charge of each atom was obtained as an average. The value of the RESP restraint weight was set to 0.01, and fitting was performed on all of the atoms except the aliphatic hydrogen.45 The second charge derivation procedure is an important result of the current report and is described in the following section.
Results and Discussion
Atomic Charges
We present in Table 1 the atomic charges obtained from the standard GLYCAM procedure for five different ring conformers of 1, labeled A–E. It is clear from this data that the charges vary when one changes the ring conformation. While this variation is not large for all atoms, the effect is especially pronounced for atoms C3, C4, and C5. For example, for C3 the charges vary over the range 0.20–0.42. This variability will negatively impact the accuracy of the simulations, and, to remove the bias associated with the choice of a specific ring conformation, we developed a charge averaging procedure that accounts for the various furanoside ring conformations.
Table 1.
Partial Atomic Charges of 1 Obtained Using the Usual GLYCAM Procedure for Five Reference Rings (A–E) and Using the Averaged Approach Described Herea,b
| atom | A | B | C | D | E | ring averagedc |
|---|---|---|---|---|---|---|
| P = 13 | P = 13 | P = 32 | P = 139 | P = 58 | P* = 31 | |
| ϕm = 34 | ϕm = 41 | ϕm = 40 | ϕm = 35 | ϕm = 40 | ϕm* = 35 | |
| C1 | 0.38 (0.05) | 0.37 (0.06) | 0.38 (0.05) | 0.37 (0.04) | 0.38 (0.05) | 0.38 (0.04) |
| C2 | 0.35 (0.09) | 0.33 (0.09) | 0.30 (0.07) | 0.31 (0.05) | 0.28 (0.09) | 0.31 (0.07) |
| O2 | −0.72 (0.02) | −0.73 (0.02) | −0.69 (0.02) | −0.70 (0.02) | −0.70 (0.03) | −0.69 (0.02) |
| OH2 | 0.42 (0.01) | 0.43 (0.02) | 0.42 (0.01) | 0.43 (0.02) | 0.42 (0.02) | 0.42 (0.01) |
| C3 | 0.34 (0.1) | 0.42 (0.09) | 0.24 (0.09) | 0.20 (0.08) | 0.39 (0.10) | 0.30 (0.12) |
| O3 | −0.73 (0.03) | −0.76 (0.04) | −0.71 (0.03) | −0.73 (0.03) | −0.74 (0.02) | −0.72 (0.03) |
| OH3 | 0.43 (0.01) | 0.43 (0.02) | 0.43 (0.02) | 0.44 (0.03) | 0.43 (0.02) | 0.43 (0.02) |
| C4 | 0.19 (0.05) | 0.12 (0.05) | 0.33 (0.1) | 0.40 (0.1) | 0.18 (0.05) | 0.26 (0.11) |
| O4 | −0.49 (0.04) | −0.47 (0.04) | −0.49 (0.05) | −0.46 (0.04) | −0.45 (0.04) | −0.47 (0.05) |
| C5 | 0.32 (0.03) | 0.31 (0.04) | 0.22 (0.05) | 0.20 (0.05) | 0.28 (0.04) | 0.24 (0.04) |
| O5 | −0.72 (0.03) | −0.67 (0.02) | −0.67 (0.02) | −0.69 (0.03) | −0.70 (0.02) | −0.67 (0.03) |
| OH5 | 0.42 (0.03) | 0.41 (0.02) | 0.42 (0.02) | 0.43 (0.03) | 0.42 (0.02) | 0.42 (0.02) |
Partial atomic charges for the ring-averaged procedure are shown in the last column; numbers in parentheses correspond to standard deviations.
Puckering angles, P, and amplitudes, ϕm, are calculated according to the Altona-Sundaralingam method.21
For the ring-averaged results, P* and ϕm* indicate the most probable values based on the distribution shown in Figure 6.
Ring-Averaged Charges
Our modification of the usual GLYCAM approach, which incorporates the effects of the ring flexibility, is now described. Two hundred conformations were selected from a 50 ns simulation, and a constrained ab initio geometry optimization (HF/6-31G*) was performed for each. During those constrained optimizations, the dihedral angles involving hydroxyl protons were held to the values obtained from the MD simulation. For each of the 200 new conformations, single point HF/6-31G* calculations were performed for the RESP fit. Note that the ring geometry and the dihedral angles involving hydroxyl protons are different in each of the 200 geometries. The same RESP approach as the one used in the usual GLYCAM procedure was then followed to obtain partial atomic charges.
The charges obtained from our new procedure, where charges are ensemble averaged over several exocyclic torsions and ring conformations, are presented in Table 1. We note that the new charges differ from those of the standard GLYCAM approach most notably for carbon atoms C3, C4, and C5. An average rmsd of the carbon atoms of the ring based on the 200 conformations used in the ring averaging was calculated, and a value of 0.09 with a fluctuation of 0.08 was obtained. This parameter is a convenient measure of the ring flexibility of the system. Along with the calculation of the rmsd, a correlation study between rmsd and puckering was carried out to quantify the magnitude of the rmsd in terms of puckering. In essence, this correlation study will indicate what change in ring puckering corresponds to a certain value of rmsd. However, this correlation study cannot be performed accurately on 200 conformations. It is necessary to consider many more conformations to get a statistically meaningful estimate. Therefore, we selected 100 000 conformations from the simulation based on our new ring-averaged atomic charges, whose results will be shown and discussed below. Based on that study, the current average rmsd of 0.09 corresponds to a change of about 60° in the puckering angle, P.
In the development of our ring average procedure, an alternate approach was attempted where one freezes not only the dihedral angles involving hydroxyl protons (as in our final average ring procedure) but also the dihedral angles of the ring (essentially fixing the ring puckering) in the geometry optimization of the 200 conformations selected from the simulation. In this way, the shape or puckering of the ring from the MD will be preserved, and our ring average will be more consistent with the simulation and, therefore, with the flexibility of the system. However, the geometry optimization of the 200 conformations with all these constraints did not converge. The conformations were over constrained, and all attempts to make them converge failed. The conformations extracted from the simulation seem to be very far from the ab initio minimum, and many constraints render convergence impossible.
Solution Simulations
Having determined the average atomic charges for 1, we next set to establish the length of simulation required to achieve convergence. As a criteria for evaluating convergence we used the populations of rotamers about the C4–C5 bond. Shown in Figure 4 are the results of a convergence study of these rotamer populations in 1 as a function of simulation time. Charges obtained with the new ring-averaged procedure were used. From these results, it is clear that a 200 ns simulation is required to converge the populations of all the rotamers to reasonable uncertainties (a few units of percentage). Of particular note, simulations of less than 50 ns produced rotamer populations differing substantially from those present after 200 ns.
Figure 4.

Convergence of the rotamer populations of 1. Lines are a guide to the eye, and the gg, gt, and tg populations are given by the top, middle, and bottom lines, respectively.
We next compared the C4–C5 rotamer populations obtained from the simulations with those derived from experimental results.13 A histogram of the behavior of this torsion is shown in Figure 5. All three rotamers are populated, but the tg rotamer (180°) is visited infrequently. When the conformers from the three peaks in the histogram are integrated, it is possible to quantitate rotamer populations, which are presented in Table 2. In addition to the results based on our ring-averaged charge derivation procedure and the experimental values, the results of simulations based on the five charge sets of the standard (fixed ring) GLYCAM procedure are also presented. Clearly, the new ring-averaged charge calculation procedure leads to a good agreement with experiment, which is better than the fixed ring method. While both charge derivation approaches yield the correct ordering of the rotamer populations, the results based on the usual GLYCAM approach can sometime lead to a worse agreement with experiment because of the intrinsic ring bias of that procedure. These results validate the ring-averaging method for obtaining charges in these flexible rings, and, encouraged by these results, we considered other ring parameters in 1, in particular P and ϕm.
Figure 5.

Time dependence of the C4–C5 torsion angle (left panel) and its associated distribution (right panel) for 1.
Table 2.
Rotamer Populations of 1 Obtained Using the Various Approaches
| rotamer population (%) | gt | tg | gg |
|---|---|---|---|
| experiment13 | 38 | 14 | 48 |
| ring average charges | 37(3) | 7(1) | 56(3) |
| fixed ring charges A | 29(2) | 8(1) | 63(3) |
| fixed ring charges B | 29(2) | 8(1) | 63(3) |
| fixed ring charges C | 39(3) | 7(1) | 54(3) |
| fixed ring charges D | 33(3) | 8(1) | 59(3) |
| fixed ring charges E | 27(2) | 8(2) | 65(3) |
| gas phase | 7(1) | 40(3) | 53(3) |
Figure 6 contains the variation in P, which describes ring puckering; the inset shows the variation in puckering amplitude, ϕm. The distribution in ϕm is centered about 35°, which corresponds well to earlier ab initio, density functional theory, and molecular calculations15–19 on 1 as well as to the puckering amplitude of this molecule in the crystal structure.43 With regard to P, conformations with values in the northern hemisphere of the pseudorotational itinerary (Figure 2) are clearly favored although a small fraction of the conformers are also present in the southern hemisphere. The area of conformational space centered about P = 45° corresponds well to the N conformer determined for 114 using the PSEUROT22 procedure, which identified two conformers: a N conformer at P = 44° (39%) and an S conformer at P = 123° (61%). However, while there is good agreement with the identify of the N conformer, the conformer populations obtained from the simulation do not correspond well with experiment nor with previous ab initio and density functional theory calculations on 1.15–19 Indeed, the distribution shown in Figure 7 suggests that a while a small population of the S conformer (centered around P = 180) is present, the equilibrium is heavily biased to the N conformer. These results suggest that the two-state model inherent in the PSEUROT approach may not be valid for 1.
Figure 6.

Time dependence of the Altona-Sundaralingam P angle (left panel) and its associated distribution (right panel) for 1. The distribution of puckering amplitude, ϕm, is given in the inset of the right panel (ϕm* = 35°).
Figure 7.

Joint probability distribution of the puckering angle (in degrees), P, and the rmsd (in Å) of the ring carbon atoms.
Figure 7 illustrates the correlation study mentioned earlier where we calculate the joint probability distribution of the puckering angle, P, and the rmsd of the ring atoms. The graph shows that a change of 180° in ring puckering, which is the maximum possible, represents a variation of approximately 0.25 in rmsd. The figure also reveals that an rmsd value of 0.09 as obtained in the ring-averaged charge derivation procedure of the preceding section corresponds to a 60° change in the puckering angle, P. If the fluctuation magnitude of 0.08 is taken into account, the change in ring puckering will be more than 100°. Obviously, this result lends weight to our modification to the standard GLYCAM procedure to derive the set of atomic charges. The current solvated molecular system is very flexible, and the charge derivation cannot be based on only one ring but has to be based on an average over numerous rings to represent all the conformations accessible to the system.
Gas-Phase Simulations
Although we anticipated that the inclusion of explicit water molecules to simulate solvent effects would be essential to obtain results consistent with experiment, as a test of this we performed a simulation of 1 in the gas phase. We present in Figure 8 the analysis of the C4–C5 torsion angle and, in Figure 9, pseudorotation behavior in the gas phase. As expected, these gas-phase results differ from those obtained with explicit solvent inclusion. This is presumably due, in large part, to the fact that in the absence of water, the possibility of intermolecular hydrogen bond competition with the solvent is no longer possible.
Figure 8.

Time dependence of the C4–C5 torsion angle (left panel) and its associated distribution (right panel) for 1 in the gas phase.
Figure 9.
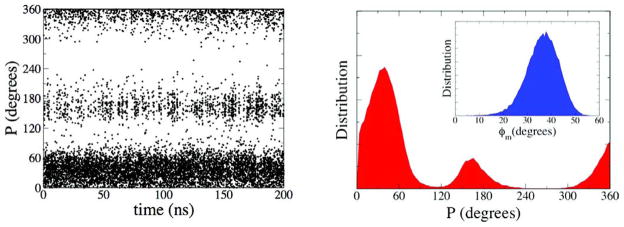
Time dependence of the P angle (left panel) and its associated distribution (right panel) for 1 in the gas phase (PN*= 38 and PS* = 165). The distribution of puckering amplitude, ϕm, is given in the inset of the right panel (ϕm* = 38).
We see from Figure 8 that the ordering of the rotamer populations is reversed compared to the solution and experimental cases. The population of the tg rotamer is now greatly enhanced at the expense of the gt rotamer. Figure 9 in turn reveals that the pseudorotation distribution now shows more distinct north (N) and south (S) populations. The most populated values of the two puckering states are PN* = 38 and PS* = 165 for the north and south regions, respectively, which agrees well with previous ab initio and density functional theory calculations on 1.15–19 This result differs significantly for the simulation done in the presence of water, where two distinct puckering states did not exist and instead a single region in the northern hemisphere of the pseudorotational itinerary was favored. As expected, these results underscore the importance of using an explicit solvent model to correctly describe solution behavior.
An ab initio and density functional theory study of several conformers of 1 in the gas phase16 showed high correlation between the rotamer and the ring puckering distributions. In other words, the rotamer population depends on the ring puckering and vice versa.
Motivated by this study, we carried out a correlation study between the C4–C5 torsion and the puckering angle. Figure 10 shows the joint probability distribution of the C4–C5 torsion and puckering angle, P, for both gas- and solution-phase simulations. The gas-phase results reveal the presence of north and south hemispheres of the pseudorotational wheel, and different trends of C4–C5 torsion distribution are obtained for each hemisphere. For example, conformations with P values between 0 and 50° (North) exhibit the trend in rotamers of tg > gg > gt, whereas for conformations with P values around 180° (South), the trend is gg > tg = gt. The favoring of the gg rotamer in the S conformers would be expected given the ability of conformers with this C4–C5 torsion to form transannular hydrogen bonds between OH2 and OH5. Similarly, in the N conformers, the tg rotamer is stabilized by hydrogen bonding between OH3 and OH5. There is therefore a marked correlation between C4–C5 torsion and ring puckering in the gas phase, as concluded from an earlier ab initio study16 although the trends in rotamers for the respective values of ring puckering do not coincide. The ab initio study shows gg > gt > tg for P ≈ 30 and gg > tg > gt for P ≈ 180. These differences may arise from the fact that in the ab initio study a full sampling of conformational space was not undertaken. Instead the energy-minimized structures were obtained by full optimization of a family of 30 ring-constrained conformers15 that had been partially optimized to probe the effect of ring conformation on various molecular parameters, e.g., bond lengths and bond angles. In solution, this strong correlation between the C4–C5 rotamer and the furanose ring conformation is not observed. As seen in Figure 10, the north hemisphere of the pseudorotational wheel is mostly populated, regardless of the C4–C5 rotamer. We propose that the effect is due to the lack of intramolecular hydrogen bonding in the solution simulations.
Figure 10.

Joint probability distribution of the puckering angle, P, and the C4–C5 torsion for 1 in the gas (left panel) and solution (right panel) phases. The units of the angles P and ω are in degrees.
Conclusions
In this paper, we have shown that the AMBER/GLYCAM model is applicable to furanoside rings, specifically methyl α-D-arabinofuranoside, 1, provided that precautions are taken to account for the inherent flexibility of these five-membered rings. In particular, it is critical to use averaged atomic charges obtained from a large number of conformations (200). This approach leads to less charge variability and, in turn, more reproducible results. The usual GLYCAM procedure, in which a single ring conformer is used to derive atomic charges, appears to be valid for the more rigid pyranoside rings but not the conformationally mobile furanosides. Furthermore, long simulation times (200 ns) are required for convergence. Simulations in which these approaches were implemented showed good agreement with rotamer populations about the C4–C5 bond and the puckering amplitude of the ring (ϕm) as determined from NMR spectroscopic data.17 In contrast, the results of the simulations in water demonstrated a single low-energy region of conformational space thus suggesting that the two-state model most frequently used to describe furanose ring conformation17,22 may not be valid for 1. In the gas-phase simulations, results consistent with the two-state model and earlier ab initio and density-functional theory calculations15–19 were found.
The differences between ring conformer populations in the gas and aqueous phases are noteworthy and, while not necessarily unexpected, underscore both the profound influence of water on these flexible rings as well as the potential danger of consistently applying the two-state model in the conformational analysis of furanose moieties. In light of the present success of this approach to model these flexible rings, future studies will involve the extension of this method to the study of other commonly occurring furanoside monosaccharides (e.g., β-D-arabinofuranoside and β-D-galactofuranoside) as well as more complex oligomeric and polymeric structures related to mycobacterial arabinogalactan and lipoarabinomannan. Other issues such as the role of the water model or polarization will also be explored in forthcoming work.
Acknowledgments
The authors acknowledge the Natural Sciences and Engineering Research Council of Canada and the Alberta Ingenuity Centre for Carbohydrate Science for funding, and the WestGrid for access to computational resources.
References
- 1.Saenger W. Principles of Nucleic Acid Structure. Spring-Verlag; Berlin: 1988. [Google Scholar]
- 2.Lowary TL. Curr Opin Chem Biol. 2003;7:749–756. doi: 10.1016/j.cbpa.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Houseknecht JB, Lowary TL. Curr Opin Chem Biol. 2001;5:677–682. doi: 10.1016/s1367-5931(01)00265-4. [DOI] [PubMed] [Google Scholar]
- 4.Ryder MH, Tate ME, Jones GP. J Biol Chem. 1984;259:9704–9710. [PubMed] [Google Scholar]
- 5.Komatsu K, Shigemori H, Kobayashi J. J Org Chem. 2001;66:6189–6192. doi: 10.1021/jo0156767. [DOI] [PubMed] [Google Scholar]
- 6.Gallego J, Varani G. Acc Chem Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 7.Brennan PJ, Nikaido H. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 8.Crick DC, Mahaptra S, Brennan PJ. Glycobiology. 2001;11:107R–118R. doi: 10.1093/glycob/11.9.107r. [DOI] [PubMed] [Google Scholar]
- 9.Briken V, Porcelli SA, Besra GS, Kremer L. Mol Microbiol. 2004;53:391–403. doi: 10.1111/j.1365-2958.2004.04183.x. [DOI] [PubMed] [Google Scholar]
- 10.Minnikin DE. In: The Biology of the Mycobacteria. Ratledge C, Standford JL, editors. Vol. 1. Academic; London: 1982. pp. 95–184. [Google Scholar]
- 11.Angyal SJ. Adv Carbohydr Chem Biochem. 1984;42:15–68. [Google Scholar]
- 12.Connell ND, Nikaido H. In: Tuberculosis: Pathogenesis, Protection and Control. Bloom BR, editor. American Society for Microbiology; Washington, DC: 1994. pp. 333–352. [Google Scholar]
- 13.D’Souza FW, Ayers JD, McCarren PR, Lowary TL. J Am Chem Soc. 2000;122:1251–1260. [Google Scholar]
- 14.Houseknecht JB, Altona C, Hadad CM, Lowary TL. J Org Chem. 2002;67:4647–4651. doi: 10.1021/jo025635q. [DOI] [PubMed] [Google Scholar]
- 15.Gordon MT, Lowary TL, Hadad CM. J Am Chem Soc. 1999;121:9682–9692. [Google Scholar]
- 16.McCarren PR, Gordon MT, Lowary TL, Hadad CM. J Phys Chem A. 2001;105:5911–5922. [Google Scholar]
- 17.Houseknecht JB, Lowary TL, Hadad CM. J Phys Chem A. 2003;107:5763–5777. [Google Scholar]
- 18.Gordon MT, Lowary TL, Hadad CM. J Org Chem. 2000;65:4954–4963. doi: 10.1021/jo000426w. [DOI] [PubMed] [Google Scholar]
- 19.Houseknecht JB, Lowary TL, Hadad CM. J Phys Chem A. 2003;107:372–378. [Google Scholar]
- 20.Houseknecht JB, McCarren PR, Lowary TL, Hadad CM. J Am Chem Soc. 2001;123:8811–8824. doi: 10.1021/ja003768s. [DOI] [PubMed] [Google Scholar]
- 21.Altona C, Sundaralingam M. J Am Chem Soc. 1972;94:8205–8212. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- 22.Deleeuw F, Altona C. J Comput Chem. 1983;4:428–437. [Google Scholar]
- 23.Lemieux RU, Koto S. Tetrahedron. 1974;30:1933–1944. [Google Scholar]
- 24.Wolfe S. Acc Chem Res. 1972;5:102–111. [Google Scholar]
- 25.Devries NK, Buck HM. Carbohydr Res. 1987;165:1–16. [Google Scholar]
- 26.Bock K, Duus JO. J Carbohydr Chem. 1994;13:513–543. [Google Scholar]
- 27.Tvaroska I, Carver JP. J Phys Chem B. 1997;101:2992–2999. [Google Scholar]
- 28.Cros S, Dupenhoat CH, Perez S, Imberty A. Carbohydr Res. 1993;248:81–93. [Google Scholar]
- 29.Cros S, Imberty A, Bouchemal N, Dupenhoat CH, Perez S. Biopolymers. 1994;34:1433–1447. [Google Scholar]
- 30.Dowd MK, French AD, Reilly PJ. J Carbohydr Chem. 2000;19:1091–1114. [Google Scholar]
- 31.Mazeau K, Perez S. Carbohydr Res. 1998;311:203–217. doi: 10.1016/s0008-6215(98)00190-6. [DOI] [PubMed] [Google Scholar]
- 32.French AD, Dowd MK. J Comput Chem. 1994;15:561–570. [Google Scholar]
- 33.French AD, Dowd MK, Reilly PJ. J Mol Struct THEOCHEM. 1997;395:271–287. [Google Scholar]
- 34.French AD, Tran V. Biopolymers. 1990;29:1599–1611. [Google Scholar]
- 35.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods RJ, Dwek RA, Edge CJ, Fraser-Reid B. J Phys Chem. 1995;99:3832–3846. [Google Scholar]
- 37.Kirschner KN, Yongye AB, Tschampel SM, Daniels CR, Foley BL, Woods RJ. J Comput Chem. 2007 doi: 10.1002/jcc.20820. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirschner KN, Woods RJ. Proc Natl Acad Sci USA. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 41.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 42.Basma M, Sundara S, Calgan D, Vernali T, Woods RJ. J Comput Chem. 2001;22:1125–1137. doi: 10.1002/jcc.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evdokimov AG, Kalb AJ, Koetzle TF, Klooster WT, Martin JML. J Phys Chem A. 1999;103:744–753. [Google Scholar]
- 44.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 45.Woods RJ, Chappelle R. J Mol Struct. 2000;527:149–156. doi: 10.1016/S0166-1280(00)00487-5. [DOI] [PMC free article] [PubMed] [Google Scholar]