

Abstract
It is a well-established scientific observation that mammalian cells contain fidelity proteins that appear to protect against and adapt to various forms of endogenous and exogenous cellular conditions. Loss of function or genetic mutation of these fidelity proteins has also been shown to create a cellular environment that is permissive for the development of tumors, suggesting that these proteins also function as tumor suppressors (TSs). While the first identified TSs were confined to either the nucleus and/or the cytoplasm, it seemed logical to hypothesize that the mitochondria may also contain fidelity proteins that serve as TSs. In this regard, it now appears clear that at least two mitochondrial sirtuins function as sensing, watchdog, or TS proteins in vitro, in vivo, and in human tumor samples. In addition, these new results demonstrate that the mitochondrial anti-aging or fidelity/sensing proteins, SIRT3 and SIRT4, respond to changes in cellular nutrient status to alter the enzymatic activity of specific downstream targets to maintain energy production that matches energy availability and ATP consumption. As such, it is proposed that loss of function or genetic deletion of these mitochondrial genes results in a mismatch of mitochondrial energy metabolism, culminating in a cell phenotype permissive for transformation and tumorigenesis. In addition, these findings clearly suggest that loss of proper mitochondrial metabolism, via loss of SIRT3 and SIRT4, is sufficient to promote carcinogenesis.
Keywords: SIRT3, SIRT4, Acetylome, Acetylation, Carcinogenesis
Review
Mammalian cells express proteins that protect against endogenous and exogenous forms of cellular damage to both monitor and maintain the integrity of a cell [1-3]. An extension of this observation would be that the loss of function or genetic mutation of these genes creates a cellular environment that is permissive for the development and/or accumulation of cellular damage that can put the cell at a significantly increased risk for several human illnesses, including cancer [4-7]. Since it is unlikely that evolutionary pressure selected for proteins in mammalian cells to prevent carcinogenesis, these proteins are more likely fidelity proteins that have evolved over time to protect specific organelles from damage by agents that induce genotoxic stress [8]. These proteins are often referred to as tumor suppressors (TSs) as mice lacking these genes tend to develop tumors, and in many cases, these TS genes are deleted or mutated in human tumors [9,10]. While the first identified TSs were confined to either the nucleus and/or the cytoplasm, it seems logical to hypothesize that the mitochondria would also contain fidelity proteins that would serve as TSs.
It now seems clear that the cellular processes that govern or oversee aging, perhaps better defined as longevity, are directed by a combination of complex genetic, biochemical, and cellular pathways that appear to be regulated, at least in part, by a relatively new gene family referred to as sirtuins [11,12]. The sirtuin family genes are the human and murine homologs of the Saccharomyces cerevisiae Sir2 that have been shown to directly regulate both replicative and overall lifespan [13,14] as well as longevity in Caenorhabditis elegans and D. melanogaster[11,12]. In these more primitive species, it appears that these genes direct longevity, at least in some significant part, by silencing telomeres and sub-telomeric regions, silent mating type loci, and, crucially, the rDNA, suppressing formation of rDNA circles [12,15].
In contrast, mammalian sirtuin members are associated with numerous physiological roles including stress response, regulation of metabolism, gene silencing, and aging [16,17]. While it has not been shown that these genes determine longevity in mammals, they do appear to regulate critical signaling networks, and following stress, several mice lacking one of the sirtuin genes develop illnesses that mimic those observed in older humans [12,18]. Sirtuins were initially discovered to function as deacetyltransferases; however, it now appears that several of the proteins also function as ribosyltransferases [19-21] under specific conditions. Based on these results, it has been proposed that the mammalian sirtuins play a significant role, at least in part, in directing the acetylome signaling network that has recently been shown to be critical in regulating multiple cellular processes [22]. While this represents only a part of the overall role of sirtuins in mammalian biology, it now appears clear that this protein family, in some significant way, directs the activity of downstream targets via post-translational modifications involving protein acetylation (Figure 1A) to maintain cellular metabolic homeostasis [23].
Figure 1.

Overview of sirtuin biology. (A) Cellular localization of the nuclear, cytoplasmic, and mitochondrial sirtuins. (B) Schematic of the enzymatic function of mitochondrial sirtuins using SIRT3.
Mammalian sirtuins are classified as class III histone deacetylases, which are different than traditional class I and II histone deacetylases (HDACs) [24,25]. Unlike conventional HDACs, sirtuins have a variety of non-histone substrates ranging from metabolic enzymes to structural proteins as well as histones [15,19]. The function of sirtuins is very well conserved via a common 275-amino acid catalytic domain, and these proteins are localized to the nucleus (SIRT1, 6, and 7), mitochondria (SIRT3, 4, and 5), and cytoplasm (SIRT2) (Figure 1B) [13]. Sirtuins are NAD+-dependent deacetylases, and early on in the investigation of sirtuin biology, it was assumed that their requirement for NAD+ implied that their mechanistic activity was connected to cellular metabolism, providing a link between sirtuin activity, energy, and stress responses [26,27]. For instance, the mitochondrial sirtuins, SIRT3 and SIRT4, appear to respond to changes in cellular and nutrient stress, resulting in the activation of their deacetylase or ribosyltransferase activity and hence in post-translational modifications of downstream target proteins [27-31]. It is now well established that SIRT3 deacetylation activity is activated by caloric restriction (CR) and fasting [29-31], and this induction of deacetylation activity also appears to protect against the development of age-related human pathology, including tumorigenesis [29,32]. While these results do not, a priori, directly connect sirtuins to longevity, they do strongly suggest that sirtuins, including mitochondrial sirtuins, play a role, at least in some significant part, in the complex process of aging.
Inflection points, aging, and human carcinogenesis
One fundamental observation in cancer etiology is that the rate of malignancies in any mammalian population increases exponentially as a function of age, suggesting a mechanistic link between the cellular processes governing longevity and carcinogenesis [33,34]. This observation is even more stark when only considering human somatic solid cancers [35]. When the data for human solid tumors are presented as a function of increasing age, an intriguing phenomenon is observed: a clear inflection point that occurs at just after 50 years of age [36,37] (shown by a circle in Figure 2A). This inflection point is preceded by an initially gradual slope (referred to as the early or E-slope) but is followed by a very steep curve (referred to as the S-slope), indicating a significant increase in human cancer incidence after age 50 (Figure 2A).
Figure 2.

Cancer incidence rises with age. (A) The incidence of solid tumor cancers derived from somatic cells increases exponentially with age. The circle marks the inflection point at the transition between the early (E) slope and steep (S) slope. (B) The effect of sirtuin gene expression on lifespan. This is a graphical summary of data obtained from increased or decreased sirtuin expression in C. elegans. Overexpression of sirtuin genes results in increased lifespan (curve C), whereas underexpression of these genes shortened lifespan (curve B). The time of the inflection point (circle) is shifted, but the general shape of the survival curve remains unchanged.
These results strongly suggest that some change in, or dysregulation of, critical biological processes and/or cellular reparative pathways occurs at this inflection point, putting us at an increased risk for somatic tumors [34]. However, while it is tempting to suggest this is due to loss of a specific protein family, that seems unlikely. It is more reasonable to assume that the inflection point is due to a large host of proteins and signaling pathways that maintain a cell's homeostatic poise. As such, our laboratory, as well as many others, is interested in the changes that occur in the cell at this critical inflection point that marks the transition to the tumor-permissive phenotype.
This unique and potentially informative inflection point that is observed in human somatic solid tumors is also observed in almost all species, including mice [38,39]. Analysis of this longevity data in multiple species, C. elegans being a common example, shows an initial long, flat slope, followed by an inflection point, and finally a steep curve [40-42], similar to the curve observed in humans. While these results clearly suggest that both aging and somatic human tumors share similar curves, the more interesting question is whether there is a correlative and/or mechanistic relationship between these two curves that involves, at least in some part, the biology of sirtuin proteins.
In this regard, several research groups, most notably the Guarente laboratory, suggest that the time to the inflection point of the longevity curve is directed, at least in some part, by the sirtuin protein family. This suggestion is based on the observation that overexpression of the sirtuin proteins in C. elegans increased overall lifespan, while in contrast, deletion of these genes resulted in the opposite effect [40-42]. Perhaps the most interesting aspect of these seminal studies is that loss of and/or enforced expression of the C. elegans sirtuins altered the length of the early slope while the slope of the curve after the inflection point remained unchanged (Figure 2B). This result suggests two obvious possibilities: (1) the inflection point is, to some degree, directed by sirtuin activity or (2) there may be a threshold of cellular repair directed by sirtuins, and at some point, cellular damage exceeds repair, potentially playing a role in the appearance of the inflection point. However, it is safe to assume that there are likely many more plausible explanations as well as other proteins that direct this cellular process.
Caloric restriction, mitochondrial energy metabolism, aging, and human carcinogenesis
It is a well-established observation that animals on a CR diet exhibit significant health-related effects, including an increase in overall lifespan, which is, of course, also dependent upon other nuanced factors [14,43,44]. However, if one carefully analyzes these results, the closer a diet is to the maximum CR level (i.e., 70% of ad libitum), the greater the increase in murine longevity (Figure 3A) [45]. In addition, it also appears to some significant extent that the increase in lifespan is due to increasing the length of time to reach the inflection point (Figure 3A) and not the other portions of the longevity curve [45]. Furthermore, it is well established that murine models genetically designed to induce specific types of tumors also exhibit a decrease in spontaneous disease when placed on a CR diet [46], as shown by an example of a murine model of mutant Kras-driven carcinogenesis (Figure 3B). Similarly, a decrease in spontaneous pancreas tumors was also observed in a rat model for pancreas malignancies [47]. While there are no rigorous data in humans that definitively link either an increase in lifespan or decreased incidence of malignancies to CR, there are multiple reports of soft data to suggest this [33,34].
Figure 3.

Effects of caloric restriction on murine survival and carcinogenesis. (A) Overall survival or longevity in mice on a standard ad libitum diet or CR diets consisting of 85, 50, or 40 kcal/week. The black circles highlight the inflection points of the survival curves on the ad libitum and 40 kcal/week diets. (B) The incidence of pancreatic cancers in an LSL-KrasG12D genetic knock-in mouse model on either an ad libitum diet or a CR diet. Results are presented as %survival or %tumor free, respectively, as a function of mouse age.
It is also well established that there is a strong relationship between aging and mitochondrial function [48-51], suggesting that the accumulation of mitochondrial damage results in cellular damage that may include that due to reactive oxygen species (ROS), mtDNA damage, etc. as well as a decrease in lifespan. In this regard, three of the seven mammalian sirtuins are found in the mitochondria, including SIRT3 and SIRT4 [52]. These results suggest that the mitochondrial sirtuins (or at least SIRT3 and SIRT4) may respond to changes in cellular and nutrient stress by modification of downstream target proteins [27-31]. While this has not been clearly shown for SIRT4, it has been shown that SIRT3 activity is activated by CR and fasting [29-31].
If sirtuins, including the mitochondrial sirtuins, sense nutrient status (i.e., fasting), it seems reasonable to propose that one function of these proteins is to match energy production to cellular need as well as energy consumption. In addition, it would also suggest that the regulation of the mitochondrial acetylome would play a role, at least in part, in matching cellular energy need to availability [17,53]. Reversible acetylation of lysine is a post-translational modification that neutralizes the positive charge of this amino acid, potentially altering the 3-dimensional structure of a protein as well as its enzymatic function [54,55]. Thus, it has been proposed that at least one function of the sirtuin gene family is the regulation and maintenance of the metabolome via the deacetylation of specific downstream target proteins that direct the specific pathways in the mitochondria that direct energy productions [15,56].
These results imply that sirtuins in general, and mitochondrial sirtuins specifically, are nutrient status sensing proteins that transmit a signal to downstream target genes, as well as critical mitochondrial processes, via protein deacetylation. We would propose that the mitochondrial sirtuins respond to what has often been described in most physiology courses as the fed versus the fasting organismal/cellular state [57]. In our adaptation of this model, it is proposed that in a fed state, the cells sense that energy packets (i.e., food) are readily abundant and as such, cellular processes are activated. This favors a pro-metabolism, pro-aging, and pro-carcinogenic phenotype. The activation of these processes, potentially induced by insulin secretion from the pancreas, would inactivate sirtuins, resulting in a general cellular state of increased protein acetylation (Figure 4). In addition, insulin would also activate a series of kinases that would activate pro-metabolism, and it is suggested that these pathways would result over time in aging and a carcinogenesis-permissive phenotype. Finally, a pro-metabolism status may drive aging at the organismal levels while different degrees of aging may occur at the cellular and/or tissue/organ level, and it is this combination of forces that results in the more complex signs and symptoms of increasing age on a species.In contrast, when an organism is a fasting state, which was likely quite often for evolutionarily primitive man, it seems reasonable to propose that a metabolic state would be established that maximizes the efficiency of energy generation as well as energy use. In this environment, is seems reasonable to propose that the activity of the mitochondrial sirtuins, as well as perhaps the entire sirtuin family, would be activated, resulting in a generalized deacetylated status (Figure 4). This would result in a cellular phenotype that would be energy conserving, anti-metabolism, anti-aging, as well as anti-carcinogenic. However, this is presented as a very simplistic model that may apply mostly to SIRT3 and the mitochondrial, and it is clear that changes in both acetylation and phosphorylation are much more complex as well as the interplay between this PTM on specific proteins. Finally, does this provide some insight as to why sirtuins might be fidelity or TS proteins? In this regard, it could be proposed that one consequence of making cells adapt to become more efficient is the induction of pathways that might also result in a cellular and/or organellar reparative phenotype.
Figure 4.
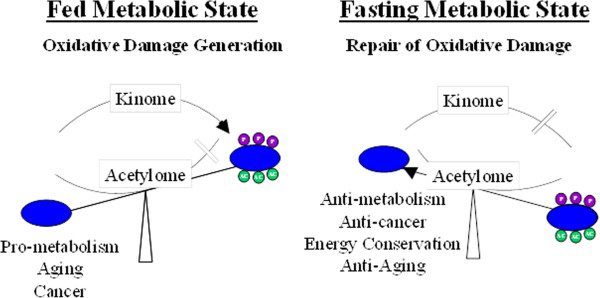
Schema outlining the opposing effects of the kinome and the acetylome on metabolism in response to energy availability. Fed conditions favor oxidative damage due to the induction of pro-metabolism pathways that are induced by insulin and other cytokines that signal a high-energy availability status that would inactivate sirtuins. A fasting state is proposed to activate sirtuins and should induce cellular pathways that conserve or increase cellular efficiency, resulting in energy conservation and preservation of cellular integrity.
Mitochondrial SIRT3 acts as a tumor suppressor
Lysine acetylation appears to be not only important but also perhaps the primary post-translational modification used to adapt cells to periods of fasting and to direct the activity of specific mitochondrial proteins [58-60]. In this regard, several proteomic surveys have identified a disproportionately high number of acetylated proteins in the mitochondria, which contain reversible acetyl-lysines associated with energy homeostasis [23,61]. SIRT3 is the primary mitochondrial deacetylase, and genetic knockout of Sirt3 alters a significant number of mitochondrial protein acetylation sites [62], including proteins involved in the generation of ATP [26]. Therefore, it seems reasonable to propose that lysine acetylation of mitochondrial proteins may serve to both maintain and regulate overall mitochondrial metabolism and function. Therefore, we believe that it is logical to hypothesize that SIRT3 acts as a metabolic sensing protein to direct the regulation of mitochondrial energy metabolism and ATP synthesis, the detoxification of mitochondrial ROS, and other biological processes essential for proper mitochondrial function. In addition, as discussed above (Figure 4), it is likely that SIRT3 senses decreased nutrient availability and responds by increasing the efficiency of mitochondrial pathways that generate ATP, shifting energy away from glycolysis and towards oxidative phosphorylation.
It is a well-established observation that there is a link between dysregulation of mitochondrial function in cancer cells, which exhibit a significant increase in glycolysis, and aberrant cellular metabolism. This link is commonly referred to as the “Warburg Effect” [26,63]. This has also been observed to be a function of age, suggesting a potential mechanistic link between the cellular processes governing mitochondrial function, longevity, and the development of cancers [34]. Finally, tumor cells also appear to have increased ROS levels that may be due to aberrant metabolism, either through increased production or decreased enzymatic detoxification, either of which may result in oxidative stress and persistent oxidative cell damage, adversely affecting genome stability. Increased ROS levels are considered an early event in carcinogenesis and, under specific cellular conditions, can further affect cell dedifferentiation, tumor initiation, and progression. These results provide strong evidence to support the hypothesis that mitochondrial dysregulation plays a significant role in the multi-hit process of cell transformation and ultimately carcinogenesis. Based on these results, it has been proposed that mitochondrial proteins, such as SIRT3 and SIRT4, may function as critical regulators at the crossroads between metabolism, aging, and aging-related human illnesses such as cancer [64]. Finally, the logical extension of this idea is that the loss of SIRT3 activity, by one of several potential mechanisms, would create a cellular environment permissive for age-related cancers [29].
Therefore, mice lacking the mitochondrial Sirt3 gene were established to determine whether SIRT3 is a TS protein, in which case, cells or mice lacking Sirt3 would likely display a tumor-permissive phenotype. In this regard, our laboratory has shown that mice lacking Sirt3 do not exhibit an obvious or early in vivo phenotype or other physiological abnormalities; however, the livers of these mice exhibit a significant increase in acetylated mitochondrial proteins, as compared to the wild-type mice [62]. Moreover, when these mice or primary tissue cultures derived from them were treated with various stress factors, such as oxidative stressors, chemical-hormonal, or ionizing radiation, they displayed physiological phenotypes consistent with increasing age, including cardiac hypertrophy [28,65], carcinogenesis [29,66,67], fatty liver [27,68], radiation-induced liver damage [31], and age-related hearing loss [32,64]. Interestingly, a common observation in each of these studies showed loss of Sirt3-induced higher steady-state levels of ROS as well as oxidative stress.
While fidelity proteins, whether in the nucleus, cytoplasm, or the mitochondria, appear to have multiple downstream targets, it seems reasonable to suggest that the observed increase in ROS levels in cells deficient in SIRT3 may contribute to the development of age-associated pathologies. Thus, loss of Sirt3 may induce aberrant mitochondrial metabolism, and when the cells are exposed to additional endogenous and exogenous insults that also result in stress, there may be resultant intracellular redox imbalance that may have deleterious biological effects.
Therefore, there are two questions that must be addressed: (1) how does SIRT3 regulate mitochondrial metabolic homeostasis? (2) What are the downstream targets involved in this regulatory process?
Recent studies have demonstrated that Sirt3 regulates the tricarboxylic acid cycle by deacetylating isocitrate dehydrogenase [32], glutamate dehydrogenase (GDH) [69], and acetyl-CoA synthetase [61,70]. SIRT3 also regulates the fatty acid cycle by deacetylating long-chain acyl-coenzyme A dehydrogenase and 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 [26,60,70]. Our group and others have observed that subunits of the electron transport chain (complexes I–III and ATP synthase) are also the substrates of SIRT3 [71,72]. In addition, SIRT3 deacetylates manganese superoxide dismutase (MnSOD), altering its activity of superoxide removal [31]. Combined, these results strongly suggest that SIRT3 is the primary mitochondrial deacetylase serving to direct mitochondrial energy production as well as to limit the accumulation of mitochondrial ROS.
In vivo loss of Sirt3 in mice exhibit dysregulation of mitochondrial functions including increased mitochondrial DNA damage in the liver, reduced ATP production, an increase in mitochondrial ROS (including superoxide), as well as increased ER/PR positive breast malignancies [29]. Similarly, when Sirt3 knockout mouse embryonic fibroblasts (MEFs) were challenged with various stress factors, these cells had loss of contact inhibition and were subsequently immortalized/transformed by infection with a single oncogene, suggesting that SIRT3 may function as a TS [29,31].
Interestingly, there is a significant decrease in SIRT3 levels in human tumors compared to normal tissue controls. While these results appear to suggest that SIRT3 is a genomically expressed, mitochondrially localized TS, the mechanism through which SIRT3 protects against tumorigenesis is unclear. As discussed, many substrates of SIRT3 are tightly linked with energy homeostasis and ROS production. Therefore, it has been suggested that increased mitochondrial oxidative stress contributes to human carcinogenesis. Our data support this hypothesis, as murine breast tissue lacking Sirt3 exhibited an increase in steady-state ROS. Similarly, human breast tissue samples also displayed increased mitochondrial superoxide levels coinciding with decreased Sirt3 expression. Sirt3-/- mouse hepatocytes [28,29] and cardiomyoctes [28,65] also presented with significantly higher basal superoxide levels, which were observed to further increase when exposed to different types of exogenous cellular stress.
In recent years, three seminal papers have been published that demonstrated that deacetylation of MnSOD by SIRT3 directs its enzymatic activity [30,31,73]. Furthermore, in several tissue culture experiments, co-infection of lenti-MnSOD not only decreased mitochondrial superoxide levels but also prevented immortalization of Sirt3-/- MEFs by a single oncogene [31]. These experiments were confirmed using a MnSOD construct in which lysine 122 was mutated to arginine (MnSOD122K-R), resulting in a constitutively active, dominant positive protein [31]. Co-infection of lenti-MnSOD122K-R also prevented immortalization of Sirt3-/- MEFs by a single oncogene. In contrast, co-infection with dominant negative mutant MnSOD gene (lenti-MnSOD122K-Q) mimicking a constitutively acetylated lysine failed to prevent immortalization by infection with a single oncogene [31]. Finally, it has also been shown that infection with the lenti-MnSOD122K-R gene prevented tissue culture transformation with exogenous agents, including ionizing radiation and stress-induced increases in cellular ROS [31]. These experiments strongly suggest that aberrant mitochondrial superoxide metabolism plays a significant role in the tumor-permissive phenotype (Figure 5) observed in cells lacking Sirt3.
Figure 5.
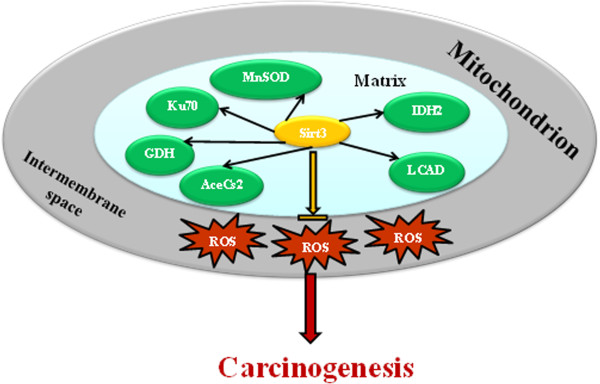
Schema outlining the multiple mechanisms by which SIRT3 blocks ROS production, thereby preventing carcinogenesis. Loss of SIRT3 results in mitochondrial dysregulation as well as increased ROS, due in part to increased mitochondrial protein acetylation, including that in MnSOD, and decreased MnSOD detoxification activity as well as other downstream target proteins deacetylated by SIRT3. The increase in ROS is thought to be an early event in the in vivo carcinogenesis observed in mice lacking Sirt3.
Many studies also suggest that changes in the steady state level of ROS may play a critical role in how the mitochondria communicate with other parts of the cell and further lead to changes in gene expression, cell proliferation, and apoptosis [29,74-78]. In support of this hypothesis, Venkataraman et al. showed that overexpression of MnSOD in PC-3 cells resulted in a delay of the G1-S phase transition. This delay was mediated in part by modulation of the redox status of the cell through the increased levels of H2O2[79]. In addition, Karawajew et al. demonstrated that mitochondrial ROS serve as second messengers by guiding p53 translocation to the mitochondria, leading to the activation of apoptosis and p53 target gene expression [75]. They also showed that treatment of cells with oligomycin, an inhibitor of ATP synthase, prevents stress-induced mitochondrial accumulation of p53 and abrogates p53-dependent apoptosis by reducing mitochondrial ROS levels [75]. These results strongly support the hypothesis that the alteration of mitochondrial ROS production, via changes in MnSOD enzymatic activity (Figure 5) or mitochondrial metabolic homeostasis, represents a potential mechanism for inter-compartmental cellular communication and may play a role in SIRT3 deficiency-induced aging-related cancers.
Finally, several studies also indicate that the acetylation statuses of SIRT3 substrates like acetyl-CoA synthetase, GDH, long-chain acyl-CoA dehydrogenase (LCAD), succinate dehydrogenase, and mitochondrial ribosome subunit MRPL10 are frequently altered in human cancers. Interestingly, SIRT3 has also been shown to have pro-apoptotic or anti-apoptotic effects on different cell types, and at least one mechanism involves deacetylating Ku70, preventing the release of BAX into mitochondria [80]. Although the detailed mechanism of the connection between these mitochondrial protein acetylation and carcinogenesis events is still unclear, these results provide evidence that the mitochondrial acetylome may play an important role in the cellular damage and tumor-permissive phenotype (Figure 5).
SIRT4 functions as tumor suppressor by directing glutamine metabolism
Recent evidence suggests that SIRT4 may also have a role in cell metabolism and carcinogenesis. Like SIRT3, SIRT4 regulates metabolic function through variety of mechanisms. While SIRT3 directs post-translational modifications via protein deacetylation, SIRT4 affects its targets largely through NAD-dependent ADP-ribosylation (Figure 6) [20]. SIRT4 is expressed in several cell types including liver, kidney, testis, striated muscle, and vascular smooth muscle as well as the insulin-producing β cells in the islets of Langerhans [81].
Figure 6.

Schema outlining the proposed pathway by which SIRT4 regulates proliferation. It is proposed that under nutrient rich conditions mTORC1 inhibits CREB2, decreasing the expression of SIRT4. When SIRT4 activity is decreased, which is observed in the Sirt4 knockout mice, and what might be expected with increasing age, the glutamate/αketoglutarate and TCA cycles are dysregulated. As such, it is suggested that this plays a role, at least in some part, in the tumor-permissive phenotype in mice lacking Sirt4.
SIRT4 is activated in response to genotoxic stress and is required for the block in glutamate metabolism allowing for a proper DNA damage response [82]. While SIRT4 exhibits no deacetylase activity on histones or serum albumin [81], recent findings suggest SIRT4 deacetylates malonyl CoA decarboxylase (MCD) under low nutrient conditions. MCD produces acetyl CoA from malonyl CoA, the latter providing a carbon skeleton for lipogenesis under nutrient-rich conditions [83]. When deacetylated by SIRT4, MCD functions less efficiently, and animals lacking SIRT4 present with increased MCD activity, dysregulated lipid metabolism, and protection against diet-induced obesity (Figure 6). Therefore, SIRT4 opposes fatty acid oxidation, promoting lipid anabolism by regulating MCD function/malonyl CoA levels [83]. Similarly, in both myocytes and hepatocytes, loss of SIRT4 increased fatty acid oxidation gene expression and cell respiration [84].
In pancreas β cell mitochrondria, SIRT4 serves to ADP-ribosylate GDH, a mitochondrial enzyme that converts glutamate to α-ketoglutarate, the activity of which is also modulated by ADP-ribosylation [85]. GDH promotes glutamine/glutamate metabolism, facilitating ATP production and insulin secretion. Once ADP-ribosylated, the enzymatic function of GDH is repressed, leading to \reduced ATP synthesis and less effective insulin secretion in response to exogenous amino acids [86,87].
Clinically, SIRT4 mRNA expression is reduced in several malignancies, including breast, colon, bladder, gastric, ovarian, and thyroid cancers, though SIRT4 loss was particularly pronounced in lung cancer patients (Figure 6). Accordingly, mice with whole body knockout of Sirt4 present with a variety of solid tumors, though most frequently lung tumors [86,88]. In addition, loss of SIRT4 corresponds with increased aggressiveness in women with breast cancer. Furthermore, overexpression of SIRT4 opposes cell proliferation, transformation, and tumor progression as shown in an in vivo murine model [89]. Similarly, loss of SIRT4 accelerates Myc-induced B cell lymphomagenesis in mice lacking Sirt4, and SIRT4 overexpression sensitizes cells to apoptosis induced by glycolysis inhibitors [88].
Combined, these observations strongly suggest that SIRT4 has tumor-suppressive effects and that its downregulation may serve to facilitate the progression of several human cancers. Loss of SIRT4 appears to be a result of mammalian target of rapamycin complex 1 (mTORC1), a complex consisting of mTOR, Raptor, and mLST8 that is dysregulated in human cancers and activated under nutrient-rich conditions [90]. mTORC1 leads to proteasome-mediated destabilization of cAMP-responsive element binding 2 (CREB2), a key transcriptional regulator of SIRT4. By destabilizing CREB2, mTORC1 reduces SIRT4 expression, thereby increasing GDH activity and glutamine/glutamate metabolism [89].
Conclusions
The results discussed above suggest that loss of a single mitochondrial protein leads to the aberrant regulation of the mitochondrial acetylome signaling network that responds to metabolic demands and deacetylates downstream target proteins, resulting in a phenotype permissive for human illnesses associated with aging. In this regard, it is proposed that SIRT3 and SIRT4 respond to changes in cellular nutrient status to alter the enzymatic activity of specific downstream targets to maintain energy production that matches energy availability and ATP consumption. As such, it is proposed that loss of function or genetic deletion of these mitochondrial genes results in a mismatch of mitochondrial energy metabolism, culminating in a cell phenotype permissive for transformation and tumorigenesis. As such, we believe that the Sirt3 and Sirt4 knockout mice represent a new paradigm that mechanistically links mitochondrial metabolism, the acetylome post-translational signaling network, and age-related disease including carcinogenesis.
Abbreviations
CR: calorie restriction; CREB2: cAMP-responsive element binding 2; GDH: glutamate dehydrogenase; HDAC: histone deacetylases; LCAD: long-chain acyl-CoA dehydrogenase; MCD: malonyl CoA decarboxylase; MEF: mouse embryonic fibroblast; MnSOD: manganese superoxide dismutase; mTORC1: mammalian target of rapamycin complex 1; NAD+: nicotinamide adenine dinucleotide; ROS: reactive oxygen species; SIRT: sirtuin; TS: tumor suppressors.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YZ reviewed the Sirt3 studies, participated in the preparation of the figures, and drafted the manuscript. YY participated in manuscript design and coordination and helped to edit the manuscript. DP reviewed the Sirt4 studies, participated in the preparation of the figures, and drafted the manuscript. XZ and AV participated in the design of the manuscript and edited the manuscript. DG conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yueming Zhu, Email: yueming.zhu@northwestern.edu.
Yufan Yan, Email: yufan.yan@northwestern.edu.
Daniel R Principe, Email: principe.danny@gmail.com.
Xianghui Zou, Email: xianghuizou2017@u.northwestern.edu.
Athanassios Vassilopoulos, Email: athanasios.vasilopoulos@northwestern.edu.
David Gius, Email: david.gius@northwestern.edu.
Acknowledgements
DG is supported by NCI-1R01CA152601-01, 1R01CA152799-01A1, 1R01CA168292-01A1, and BC093803 from the DOD and a Hirshberg Foundation for Pancreatic Cancer Research Seed Grant Award.
References
- Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, Choudhary M, Hawkins GA, D'Agostino RB Jr, Willingham MC, Inoue K. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell. 2007;12(4):381–394. doi: 10.1016/j.ccr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, Lagcher W, Sie D, Tanger E, Cox T, Reinders M, Hubbard TJ, Rogers J, Jonkers J, Wessels L, Adams DJ, van Lohuizen M, Berns A. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133(4):727–741. doi: 10.1016/j.cell.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet. 1989;83(2):155–158. doi: 10.1007/BF00286709. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249(4971):912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Johnson LA, Law DJ, Kuehn SE, Steenman M, Williams BR, Thomas G, Boland CR, Rainier S, Koi M. Multiple tumor suppressor genes in multistep carcinogenesis. Tohoku J Exp Med. 1992;168(2):149–152. doi: 10.1620/tjem.168.149. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. doi: 10.1016/S1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Hunter T. Oncoprotein networks. Cell. 1997;88(3):333–346. doi: 10.1016/S0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- Tlsty TD. Functions of p53 suppress critical consequences of damage and repair in the initiation of cancer. Cancer Cell. 2002;2(1):2–4. doi: 10.1016/S1535-6108(02)00088-0. [DOI] [PubMed] [Google Scholar]
- Blackburn AC, Jerry DJ. Knockout and transgenic mice of Trp53: what have we learned about p53 in breast cancer? Breast Cancer Res. 2002;4(3):101–111. doi: 10.1186/bcr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101(45):15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430(7000):686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Albaugh BN, Denu JM. Where in the cell is SIRT3?—functional localization of an NAD+-dependent protein deacetylase. Biochem J. 2008;411(2):e11–e13. doi: 10.1042/BJ20080336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–152. doi: 10.1146/annurev.med.54.101601.152156. [DOI] [PubMed] [Google Scholar]
- Guarente L, Partridge L, Wallace DC. Molecular biology of aging (Cold Spring Harbor Laboratory Press) N.Y: Cold Spring Harbor; 2008. [Google Scholar]
- Haigis MC, Guarente LP. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20(21):2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. Embo J. 2007;26(13):3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7(2):104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26(37):5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- Li X, Kazgan N. Mammalian sirtuins and energy metabolism. Int J Biol Sci. 2011;7(5):575–587. doi: 10.7150/ijbs.7.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KT, Workman JL. Introducing the acetylome. Nat Biotechnol. 2009;27(10):917–919. doi: 10.1038/nbt1009-917. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Saunders LR, Verdin E. Cell biology. Stress response and aging. Science. 2009;323(5917):1021–1022. doi: 10.1126/science.1170007. [DOI] [PubMed] [Google Scholar]
- Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9(2):285–290. doi: 10.1111/j.1474-9726.2010.00548.x. [DOI] [PubMed] [Google Scholar]
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku-70. Mol Cell Biol. 2008;28(20):6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17(1):41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12(6):662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40(6):893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143(5):802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ershler WB, Longo DL. Aging and cancer: issues of basic and clinical science. J Natl Cancer Inst. 1997;89(20):1489–1497. doi: 10.1093/jnci/89.20.1489. [DOI] [PubMed] [Google Scholar]
- Ershler WB, Longo DL. The biology of aging: the current research agenda. Cancer. 1997;80(7):1284–1293. doi: 10.1002/(SICI)1097-0142(19971001)80:7<1284::AID-CNCR14>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126(2):257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20(10):1646–1654. doi: 10.1089/ars.2013.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gius D, Cao XM, Rauscher FJ 3rd, Cohen DR, Curran T, Sukhatme VP. Transcriptional activation and repression by Fos are independent functions: the C terminus represses immediate-early gene expression via CArG elements. Mol Cell Biol. 1990;10(8):4243–4255. doi: 10.1128/mcb.10.8.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom DL, Gottschling DE. The mother enrichment program: a genetic system for facile replicative life span analysis in Saccharomyces cerevisiae. Genetics. 2009;183(2):413–22. doi: 10.1534/genetics.109.106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turturro A, Witt WW, Lewis S, Hass BS, Lipman RD, Hart RW. Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J Gerontol. 1999;54(11):B492–501. doi: 10.1093/gerona/54.11.B492. [DOI] [PubMed] [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408(6809):255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4(2):e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Mitochondria—a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132(2):171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126(9):987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, de Jonge L, Frisard MI, DeLany JP, Larson-Meyer DE, Rood J, Nguyen T, Martin CK, Volaufova J, Most MM, Greenway FL, Smith SR, Deutsch WA, Williamson DA, Ravussin E. Pennington CALERIE Team. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA. 2006;295(13):1539–1548. doi: 10.1001/jama.295.13.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N Engl J Med. 1997;337(14):986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza-Jacoby S, Yan G, Radice G, LePhong C, Baliff J, Hess R. Calorie restriction delays the progression of lesions to pancreatic cancer in the LSL-KrasG12D; Pdx-1/Cre mouse model of pancreatic cancer. Exp Biol Med (Maywood, NJ) 2013;238(7):787–797. doi: 10.1177/1535370213493727. [DOI] [PubMed] [Google Scholar]
- Roebuck BD, Baumgartner KJ, MacMillan DL. Caloric restriction and intervention in pancreatic carcinogenesis in the rat. Cancer Res. 1993;53(1):46–52. [PubMed] [Google Scholar]
- Singh KK. Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann N Y Acad Sci. 2004;1019:260–264. doi: 10.1196/annals.1297.043. [DOI] [PubMed] [Google Scholar]
- Afanas'Ev IB. Mechanism of superoxide-mediated damage relevance to mitochondrial aging. Ann N Y Acad Sci. 2004;1019:343–345. doi: 10.1196/annals.1297.058. [DOI] [PubMed] [Google Scholar]
- Berneburg M, Kamenisch Y, Krutmann J. Repair of mitochondrial DNA in aging and carcinogenesis. Photochem Photobiol Sci. 2006;5(2):190–198. doi: 10.1039/b507380d. [DOI] [PubMed] [Google Scholar]
- Singh KK. Mitochondria damage checkpoint, aging, and cancer. Ann N Y Acad Sci. 2006;1067:182–190. doi: 10.1196/annals.1354.022. [DOI] [PubMed] [Google Scholar]
- Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. J Intern Med. 2008;263(2):128–141. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306(5704):2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cimen H, Han MJ, Shi T, Deng JH, Koc H, Palacios OM, Montier L, Bai Y, Tong Q, Koc EC. NAD + -dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem. 2010;285(10):7417–7429. doi: 10.1074/jbc.M109.053421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta. 2009;1789(1):45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JE, Guyton AC. Guyton and Hall textbook of medical physiology. 12. Philadelphia, PA: Saunders/Elsevier; 2011. p. xix, 1091. [Google Scholar]
- Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, Kim HS, Mishra M, Sun L, Nguyen P, Ahn BH, Leclerc J, Deng CX, Spitz DR, Gius D. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4(5):291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Hirschey MD, Shimazu T, Ho L, Verdin E. Mitochondrial sirtuins. Biochim Biophys Acta. 2010;1804(8):1645–1651. doi: 10.1016/j.bbapap.2009.12.021. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Huang JY, Verdin E. Acetylation of mitochondrial proteins. Methods Enzymol. 2009;457:137–147. doi: 10.1016/S0076-6879(09)05008-3. [DOI] [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103(27):10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27(24):8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Sebastian C, Mostoslavsky R. SIRT3 in calorie restriction: can you hear me now? Cell. 2010;143(5):667–668. doi: 10.1016/j.cell.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19(3):416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D, Sack MN, Jing E, Kahn CR, Friedman JE, Jonscher KR. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433(3):505–514. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382(3):790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103(27):10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE. 2011;6(8):e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, Kim HS, Deng CX, Walker JE, Gius D. SIRT3 deacetylates ATP synthase F complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal. 2014;21(4):551–64. doi: 10.1089/ars.2013.5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12(6):534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozden O, Park SH, Kim HS, Jiang H, Coleman MC, Spitz DR, Gius D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging (Albany NY) 2011;3(2):102–107. doi: 10.18632/aging.100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karawajew L, Rhein P, Czerwony G, Ludwig WD. Stress-induced activation of the p53 tumor suppressor in leukemia cells and normal lymphocytes requires mitochondrial activity and reactive oxygen species. Blood. 2005;105(12):4767–4775. doi: 10.1182/blood-2004-09-3428. [DOI] [PubMed] [Google Scholar]
- Wang F, Fu X, Chen X, Chen X, Zhao Y. Mitochondrial uncoupling inhibits p53 mitochondrial translocation in TPA-challenged skin epidermal JB6 cells. PLoS One. 2010;5(10):e13459. doi: 10.1371/journal.pone.0013459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, St Clair DK, Gu X, Zhao Y. Blocking mitochondrial permeability transition prevents p53 mitochondrial translocation during skin tumor promotion. FEBS Lett. 2008;582(9):1319–1324. doi: 10.1016/j.febslet.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Zhong W, Oberley TD. Reversible modulation of cell cycle kinetics in NIH/3 T3 mouse fibroblasts by inducible overexpression of mitochondrial manganese superoxide dismutase. Antioxid Redox Signal. 2004;6(3):489–500. doi: 10.1089/152308604773934251. [DOI] [PubMed] [Google Scholar]
- Venkataraman S, Jiang X, Weydert C, Zhang Y, Zhang HJ, Goswami PC, Ritchie JM, Oberley LW, Buettner GR. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene. 2005;24(1):77–89. doi: 10.1038/sj.onc.1208145. [DOI] [PubMed] [Google Scholar]
- Marfe G, Tafani M, Indelicato M, Sinibaldi-Salimei P, Reali V, Pucci B, Fini M, Russo MA. Kaempferol induces apoptosis in two different cell lines via Akt inactivation, Bax and SIRT3 activation, and mitochondrial dysfunction. J Cell Biochem. 2009;106(4):643–650. doi: 10.1002/jcb.22044. [DOI] [PubMed] [Google Scholar]
- Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007;282(46):33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, Xu X, Li C, Wang RH, Lee J, Csibi A, Cerione R, Blenis J, Clish CB, Kimmelman A, Deng CX, Haigis MC. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, Lovitch SB, Sharpe AH, Kurland IJ, Steegborn C, Gygi SP, Muoio DM, Ruderman NB, Haigis MC. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell. 2013;50(5):686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrin N, Wu X, Fortier E, Feng Y, Bare OC, Chen S, Ren X, Wu Z, Streeper RS, Bordone L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J Biol Chem. 2010;285(42):31995–32002. doi: 10.1074/jbc.M110.124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Yraola A, Bakhit SM, Franke P, Weise C, Schweiger M, Jorcke D, Ziegler M. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J. 2001;20(10):2404–2412. doi: 10.1093/emboj/20.10.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126(5):941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Argmann C, Auwerx J. Insulin secretion: SIRT4 gets in on the act. Cell. 2006;126(5):837–839. doi: 10.1016/j.cell.2006.08.031. [DOI] [PubMed] [Google Scholar]
- Jeong SM, Lee A, Lee J, Haigis MC. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J Biol Chem. 2014;289(7):4135–4144. doi: 10.1074/jbc.M113.525949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl 2):S43–S51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]