

Abstract
We have developed an enantioselective, copper(I)-catalyzed addition of terminal alkynes to racemic isochroman acetals. This method is one of the first transition metal-catalyzed approaches to enantioselective additions to prochiral oxocarbenium ions. In this reaction, TMSOTf is used to form the oxocarbenium ion in situ under conditions compatible with simultaneous formation of the chiral copper acetylide. By using a bis(oxazoline) ligand, good yields and enantioselectivities are observed for a variety of enantioenriched 1-alkynyl isochromans.
Chiral substituted benzopyrans comprise a number of important molecular targets, including the natural products apo-sphaerin A and cytosporone C and synthetic U-101387, a selective dopamine D4 receptor agonist.1 Nucleophilic addition to a prochiral, cyclic oxocarbenium ion intermediate would offer a highly efficient route to such bioactive molecules.2 Li has reported the oxidative coupling of isochromans with alkynes and ketones, proposed to proceed via oxocarbenium ions.3 In addition, achiral transition metal catalysts and Lewis acid promoters have been used for the addition of silyl and boronic carbon nucleophiles to acetals.4 However, only a few enantioselective additions to such substrates have been achieved. Jacobsen reported an impressive method for the catalytic enantioselective addition of silyl ketene acetals to 1-chloroisochromans using a chiral thiourea catalyst.5 More recently, Schaus described the addition of vinyl- and arylboronic esters to chromene acetals using chiral diol catalysts with a Lewis acid co-catalyst.6 Evans has also developed an enantioselective, Ni(II)-catalyzed orthoester alkylation of N-acylthiazolidinethiones via enolate addition to achiral, acyclic oxocarbenium ion intermediates.7
In contrast, enantioselective addition of chiral metal acetylides to aldehydes and ketones is a well-developed and powerful method for the preparation of chiral alcohols.8 We envisioned that an analogous alkynylation of oxocarbenium ions would be possible if the metal acetylide could be catalytically generated under conditions compatible with oxocarbenium formation. Noting Downey’s report that trimethylsilyl triflate (TMSOTf) can be used to enable catalytic turnover in the alkynylation of aldehydes,9 we have developed an enantioselective, copper(I)-catalyzed addition of terminal alkynes to isochroman acetals in the presence of TMSOTf (Scheme 1). By using (-)-2,2′-isopropylidene[(4S)-4-benzyl-2-oxazoline] (6) as ligand, high enantioselectivities in the formation of chiral benzopyrans 5 were obtained.
Scheme 1.
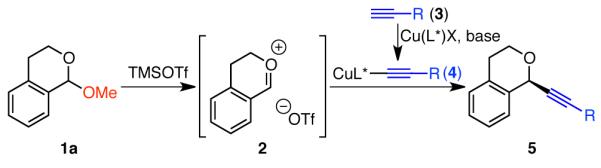
Enantioselective Alkynylation of Acetals
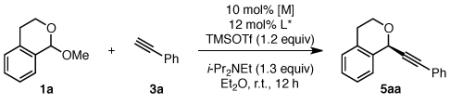
Isochroman acetal 1a, readily prepared from isochroman,5, 10 and phenylacetylene 3a were selected as model substrates for optimization of the alkynylation. We quickly found that both zinc(II) and copper(I) compounds are effective catalysts for the addition of phenylacetylene (3a) to isochroman acetals in the absence of chiral ligand (Table 1, entries 1–3). However, when chiral ligand is added, only Cu(I) catalysts provided ether 5aa in good yields (entries 4 vs. 5–7). Further, the copper counter-ion had a dramatic effect on the observed enantioselectivity with weakly coordinating PF6− being optimal (entries 5–7). Bis(oxazoline) ligands were quickly identified as promising chiral ligands, and ligand 6 was found to provide the highest selectivities (entries 6–15). By lowering the reaction temperature to −22 °C, the enantioselectivity increased to 89% ee (entry 16), but decreasing the reaction temperature further was detrimental to yield (entry 17). Reinvestigation of the Cu salt at these colder temperatures again showed [Cu(MeCN)4]PF6 gave the best enantioselectivities (entries 18–20). Notably, high enantioselectivities were only observed if the Cu salt and ligand were stirred in solvent for 30 min at room temperature prior to the addition of the other reagents.
Table 1.
Optimization of Alkynylation of Acetal 1aa
| entry | [M] | L* | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1d, e | ZnBr2 | – | 84 | – |
| 2d, e | CuI | – | (95) | – |
| 3 | [Cu(MeCN)4]PF6 | – | 12 | – |
| 4 | ZnBr2 | 6 | 15 | n.d.f |
| 5g | CuI | 6 | 78 | 26 |
| 6 | [Cu(MeCN)4]BF4 | 6 | 57 | 60 |
| 7 | [Cu(MeCN)4]PF6 | 6 | (90) | 80 |
| 8 | [Cu(MeCN)4]PF6 | 7 | 81 | 50 |
| 9 | [Cu(MeCN)4]PF6 | 8 | 70 | 7 |
| 10 | [Cu(MeCN)4]PF6 | 9 | 60 | 2 |
| 11h | [Cu(MeCN)4]PF6 | 10 | 60 | 75 |
| 12h | [Cu(MeCN)4]PF6 | 11 | 30 | 78 |
| 13 | [Cu(MeCN)4]PF6 | 12 | 70 | 36 |
| 14 | [Cu(MeCN)4]PF6 | 13 | 85 | 50 |
| 15 | [Cu(MeCN)4]PF6 | 14 | 65 | 34 |
| 16h | [Cu(MeCN)4]PF6 | 6 | (90) | 89 |
| 17i | [Cu(MeCN)4]PF6 | 6 | <5 | n.d.f |
| 18j | [CuOTf]2·PhMe | 6 | 87 | 72 |
| 19j | Cu(OTf)2 | 6 | 85 | 68 |
| 20j | CuOt-Bu | 6 | 79 | 82 |
Conditions: Acetal 1 (0.12 mmol, 1.0 equiv), [M] (0.012 mmol, 10 mol %), L* (0.014 mmol, 12 mol %), alkyne 3 (0.13 mmol, 1.1 equiv), i-Pr2NEt (0.16 mmol, 1.3 equiv), TMSOTf (0.15 mmol, 1.2 equiv), Et2O, r.t., 12 h, unless otherwise noted.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard. Numbers in parentheses are isolated yields.
Determined by chiral HPLC analysis.
NEt3 replaced i-Pr2NEt.
CH2Cl2 replaced Et2O.
n.d. = Not determined.
1.1 equiv TMSOTf, 1.2 equiv i-Pr2NEt.
Reaction was cooled to −22 °C.
Reaction was cooled to −30 °C.
Reaction was cooled to −20 °C.

Because [Cu(MeCN)4]PF6 is reported to oxidize in air, we set up these reactions in a N2-atmosphere glovebox, removing the mixtures just before additions of TMSOTf to cool them to −22 °C. However, we found that setting up the alkynylation outside the glovebox, including weighing [Cu(MeCN)4]PF6 in air, resulted in 68% isolated yield of ether 5aa in 88% ee. This benchtop procedure offers an alternative to our usual glovebox procedure.11,12
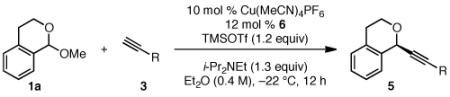
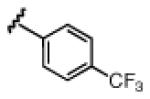
Under the optimized conditions (see Table 1, entry 16), a variety of aryl acetylenes react with acetal 1a (Table 2). In some cases, we found that mixing the Cu salt and ligand at higher concentration resulted in higher enantioselectivites.11 Increased steric bulk was well tolerated (entries 2–5). Good enantioslectivity was observed for aryl acetylenes with both electron-donating and electron-withdrawing groups at the para position (entries 4, 6–8), but a p-methoxy substituent led to diminished enantioselectivity (61% ee, entry 9), and a p-dimethylamino substituent resulted in racemic ether 5 (not shown). For substituents in the meta position, more strongly electron-withdrawing groups led to lower enantioselectivities (entries 10, 11), but the addition of m-fluorophenyl acetylene resulted in ether 5al with 88% ee (entry 12). Notably, in contrast to the result with p-methoxyphenylacetylene, the reaction of m-methoxyphenylacetylene proceeded in much better enantioselectivity (entries 9 vs. 13). Alkynes with non-aromatic substituents were less successful. Using 20 mol % Cu catalyst, cyclohexenylacetylene and cyclopropylacetylene reacted, but with lower yields and selectivities (entries 14, 15). Under the optimized conditions, the addition of 1-octyne proceeded in poor yield (entry 16).
Table 2.
Scope of Alkynea
| entry | R | 3 | 5 | yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | Ph | 3a | 5aa | 89 | 89 |
| 2 |

|
3b | 5ab | 83 | 91 |
| 3 |

|
3c | 5ac | 81 | 92 |
| 4 |

|
3d | 5ad | 72 | 85 |
| 5 |
|
3e | 5ae | 69 | 85 |
| 6 |
|
3f | 5af | 77 | 87 |
| 7d,e |

|
3g | 5ag | 78 | 81 |
| 8 |

|
3h | 5ah | 78 | 88 |
| 9 |
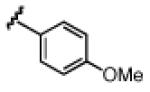
|
3i | 5ai | 64 | 61 |
| 10e,f |

|
3j | 5aj | 80 | 84 |
| 11e |

|
3k | 5ak | 73 | 85 |
| 12 |

|
3l | 5al | 79 | 88 |
| 13g |
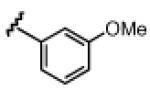
|
3m | 5am | 73 | 87 |
| 14h |
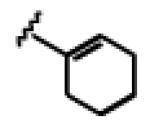
|
3n | 5an | 64 | 67 |
| 15h |

|
3o | 5ao | 28 | 42 |
| 16g,h,i | (CH2)5CH3 | 3p | 5ap | 15 | n.d.j |
Conditions: Acetal 1 (0.30 mmol, 1.0 equiv), [Cu(MeCN)]4PF6 (0.030 mmol, 10 mol %), (S,S)-6 (0.036 mmol, 12 mol %), alkyne 3 (0.34 mmol, 1.1 equiv), i-Pr2NEt (0.396 mmol, 1.3 equiv), TMSOTf (0.365 mmol, 1.2 equiv), Et2O, −22 °C, 12 h, unless otherwise noted.
Average isolated yield from duplicate experiments (±3%).
Average ee from duplicate experiments as determined by chiral HPLC analysis (±2%).
20 mol % [Cu] and 23 mol % 6, 0 °C.
(R,R)-6 was used as ligand, forming (S)-5 as product.
12 mol % [Cu], 14 mol % 6.
Results of a single experiment.
20 mol % [Cu] and 23 mol % 6, PhMe, 0 °C.
Performed on 0.1 mmol scale.
n.d. = not determined.
Good yields and high enantioselectivies were achieved in the alkynylation of a variety of acetal substrates (Table 3). In particular, the alkynylation of naphthopyranyl acetals provided ether products in high enantioselectivites (entires 1–3). Both electron-donating and electron-withdrawing substituents were tolerated on the acetal, although bromoether 5gc was formed in somewhat reduced enantioselectivity (entry 8).
Table 3.
Scope of Acetal Substratea

| entry | 1 | 3 | 5 | yield (%)b | ee (%)c | |
|---|---|---|---|---|---|---|
| 1 |
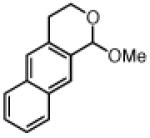
|
1b | 3a | 5ba | 73 | 94 |
| 2 |

|
1b | 3c | 5bc | 73 | 91 |
| 3 |

|
1c | 3c | 5cc | 70 | 91 |
| 4 |

|
1d | 3c | 5dc | 83 | 91 |
| 5 |

|
1e | 3a | 5ea | 88 | 87 |
| 6 |

|
1e | 3c | 5ec | 81 | 92 |
| 7 |

|
1f | 3c | 5fc | 78 | 88 |
| 8 |

|
1g | 3c | 5gc | 73 | 81 |
| 9d |

|
1h | 3c | 5hc | 50e | 85e |
Conditions: Acetal 1 (0.30 mmol, 1.0 equiv), [Cu(MeCN)]4PF6 (0.030 mmol, 10 mol %), (S,S)-6 (0.036 mmol, 12 mol %), alkyne 3 (0.34 mmol, 1.1 equiv), i-Pr2NEt (0.396 mmol, 1.3 equiv), TMSOTf (0.365 mmol, 1.2 equiv), Et2O, −22 °C, 12 h, unless otherwise noted.
Average isolated yield from duplicate experiments (±3%).
Average ee from duplicate experiments as determined by chiral HPLC analysis (±2%).
Results of a single experiment.
Yield (over two steps) and ee of 15hc.
Some of the ether products slowly decompose under ambient conditions to give lactone and benzoic acid,13 but most products are fairly stable when stored as solutions in Et2O at −26 °C. However, dimethoxybenzopyran 5hc decomposes quickly even with these precautions. Reduction of unpurified alkyne 5hc provided stable ether 15hc in 85% ee and 50% yield from acetal 1h (Scheme 2). The analogous reduction of alkyne 5aa proceeded with minimal loss of enantioselectivity, showing that the preparation of enantioenriched alkyl-substituted ethers is also possible via a two-step alkynylation/hydrogenation procedure. Reduction of 5aa also allowed assignment of the absolute configuration by comparison to ether 15aa independently prepared with known absolute configuration.11 The absolute configurations of the other propargylic ether products were assigned by analogy.
Scheme 2.

Reduction of Alkynes
Preliminary results suggest that this strategy will also enable the preparation of other classes of enantioenriched ethers. Under modified conditions, the Cu-catalyzed alkynylation of acetal 166 resulted in efficient formation of benzopyran 17 in 71% yield and 63% ee (eq 1). 11,14
 |
(1) |
As a working mechanistic hypothesis, we propose the catalytic cycle shown in Scheme 3.15 The reaction likely proceeds via initial formation of Cu acetylide 19. Although silylacetylene 20 is observed as a minor byproduct under certain conditions, we have determined that it is not a competent nucleophile in the alkynylation. In the presence of 10 mol % [Cu(MeCN)4]PF6, 12 mol % 6, i-Pr2NEt, and TMSOTf in Et2O at room temperature, only 3% yield of 5aa was observed when phenyl acetylene was replaced with trimethylsilyl acetylene 19. Further, the addition of copper phenylacetylide16 (1 equiv) to acetal 1a and TMSOTf resulted in 92% yield (determined by 1H NMR analysis), showing that Cu acetylide is a competent nucleophile.17 From the 18-electron Cu acetylide complex 19, dissociation of neutral ligand (L) likely occurs to allow approach of the oxocarbenium ion. Formation of the C–C bond may then occur either directly from trivalent 21 or via π-complexation of the oxocarbenium to Cu.18 Because π-backbonding is significant in such Cu–olefin structures, we propose that the Cu may bind either the arene (22), the C=O (23), or slip between these binding modes. At this point, it is unclear whether the C–C bond formation occurs via initial single-electron transfer or nucleophilic attack.
Scheme 3.
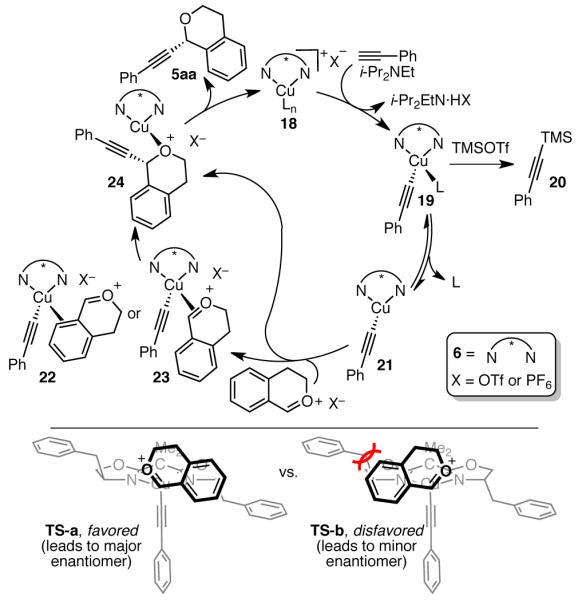
Proposed Mechanistic Hypothesis and Stereochemical Rationale
Analysis of the C–C bond forming step suggests a preliminary model for enantioinduction. Approach of Cu acetylene to the si face of the oxocarbenium ion minimizes steric interactions between the benzyl group of ligand 6 and the aromatic ring of the oxocarbenium ion (TS-a, Scheme 3). Significant steric hindrance between the aromatic ring of the oxocarbenium ion and the benzyl group destabilizes the diastereomeric transition state (TS-b). This model is consistent with the observed absolute stereochemistry of the major enantiomer and also explains the lack of enantioselectivity in the alkynylation of acyclic oxocarbenium ions (Scheme 4). For an E-configured acyclic oxocarbenium ion, there appears little difference in the stability of TS-c and TS-d. However, we must note that this steric-hindrance model does not explain why benzyl-substituted 6 is better than ligands 7 and 9 with i-Pr and t-Bu substituents, respectively. We have ruled out cation-π interactions as a possible explanation; increasing the π-donating capacity of the Bn group does not increase enantioselectivity (Table 1, entries 16 vs. 11 and 12), suggesting that this group does not participate in cation–π interactions with the oxocarbenium ion.19 Further, we do not observe a linear free-energy relationship between enantioselectivity and either the electronic or steric nature of substituents on the acetal or alkyne.11 Mechanistic investigations to explain these details are underway in our laboratory and will be reported in due course.
Scheme 4.

Alkynylation of Acyclic Oxocarbenium Ion
As described above, we have developed a highly enantioselective method for the direct alkynylation of benzopyranyl acetals to form chiral cyclic ethers. This method allows facile access to a variety of 1-alkynyl isochromans, as well as 1-alkyl isochromans via reduction. Promising results with chromene acetals suggest that this Cu-catalyzed strategy may enable efficient enantioselective alkynylation of a variety of cyclic oxocarbenium ion intermediates. Efforts to expand the scope of this alkynylation to other acetal and alkyne substrates and to determine the reaction mechanism are underway in our laboratory.
Supplementary Material
ACKNOWLEDGMENT
To Alyssa Hellreich (University of Delaware) for synthetic assistance, Prof. Joseph Fox (University of Delaware) for insightful suggestions, Kaitlin Papson (University of Delaware) for HRMS, and Ram Selvaraj (University of Delaware) for assistance with LRMS. Acknowledgement is also made to the Donors of the American Chemical Society Petroleum Research Fund, the University of Delaware Research Fund and the University of Delaware for partial support of this research. NMR and other data were acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF MIR 0421224, NSF CRIF MU CHE0840401, NIH P20 RR017716).
Footnotes
ASSOCIATED CONTENT Supporting Information. Experimental procedures, characterization data and spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Supporting Information Placeholder
REFERENCES
- (1) (a).Albrecht U, Lalk M, Langer P. Bioorg. Med. Chem. 2005;13:1531. doi: 10.1016/j.bmc.2004.12.031. [DOI] [PubMed] [Google Scholar]; (b) TenBrink RE, Bergh CL, Duncan JN, Harris DW, Huff RM, Lahti RA, Lawson CF, Lutzke BS, Martin IJ, Rees SA, Schlachter SK, Sih JC, Smith MW. J. Med. Chem. 1996;39:2435. doi: 10.1021/jm960084f. [DOI] [PubMed] [Google Scholar]; (c) Xu J, Kjer J, Sendker J, Wray V, Guan H, Edrada R, Muller WEG, Bayer M, Lin W, Wu J, Proksch P. Bioorg. Med. Chem. 2009;17:7362. doi: 10.1016/j.bmc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- (2).For an alternative strategy employing conjugate addition, see Hoveyda A, Hird A, Kacprzynski M. Chem. Commun. 2004:1779. doi: 10.1039/b401123f.
- (3) (a).Correia CA, Li C-J. Heterocycles. 2010;82:555. [Google Scholar]; (b) Zhang Y, Li C-J. J. Am. Chem. Soc. 2006;128:4242. doi: 10.1021/ja060050p. [DOI] [PubMed] [Google Scholar]
- (4) (a).Hayashi M, Inubushi A, Mukaiyama T. Bull. Chem. Soc. Jpn. 1988;61:4037. [Google Scholar]; (b) Mitchell TA, Bode JW. J. Am. Chem. Soc. 2009;131:18057. doi: 10.1021/ja906514s. [DOI] [PubMed] [Google Scholar]; (c) Vieira AS, Fiorante PF, Hough TLS, Ferreira FP, Ludtke DS, Stefani HA. Org. Lett. 2008;10:5215. doi: 10.1021/ol8022177. [DOI] [PubMed] [Google Scholar]; (d) Vo C-VT, Mitchell TA, Bode JW. J. Am. Chem. Soc. 2011;133:14082. doi: 10.1021/ja205174c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Reisman SE, Doyle AG, Jacobsen EN. J. Am. Chem. Soc. 2008;130:7198. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moquist PN, Kodama T, Schaus SE. Angew. Chem. Int. Ed. 2010;49:7096. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Evans DA, Thomson RJ. J. Am. Chem. Soc. 2005;127:10506. doi: 10.1021/ja053386s. [DOI] [PubMed] [Google Scholar]
- (8) (a).Cozzi, Pier G, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2004;2004:4095. [Google Scholar]; (b) Pu L. Tetrahedron. 2003;59:9873. [Google Scholar]; (c) Frantz DE, Fässler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806. [Google Scholar]; (d) Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; (e) Boyall D, Frantz DE, Carreira EM. Org. Lett. 2002;4:2605. doi: 10.1021/ol026282k. [DOI] [PubMed] [Google Scholar]; (f) Takita R, Yakura K, Ohshima T, Shibasaki M. J. Am. Chem. Soc. 2005;127:13760. doi: 10.1021/ja053946n. [DOI] [PubMed] [Google Scholar]; (g) Asano Y, Hara K, Ito H, Sawamura M. Org. Lett. 2007;9:3901. doi: 10.1021/ol701534n. [DOI] [PubMed] [Google Scholar]; (h) Koyuncu H, Dogan Ö. Org. Lett. 2007;9:3477. doi: 10.1021/ol701535y. [DOI] [PubMed] [Google Scholar]; (i) Yang F, Xi P, Yang L, Lan J, Xie R, You J. J. Org. Chem. 2007;72:5457. doi: 10.1021/jo0707535. [DOI] [PubMed] [Google Scholar]; (j) Asano Y, Ito H, Hara K, Sawamura M. Organometallics. 2008;27:5984. [Google Scholar]; (k) Ito J-I, Asai R, Nishiyama H. Org. Lett. 2010;12:3860. doi: 10.1021/ol1015338. [DOI] [PubMed] [Google Scholar]; (l) Trost BM, Chan VS, Yamamoto D. J. Am. Chem. Soc. 2010;132:5186. doi: 10.1021/ja910656b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Chen C, Hong L, Xu Z-Q, Liu L, Wang R. Org. Lett. 2006;8:2277. doi: 10.1021/ol060526+. [DOI] [PubMed] [Google Scholar]; (n) Zhou S, Chen C-R, Gau H-M. Org. Lett. 2010;12:48. doi: 10.1021/ol902454n. [DOI] [PubMed] [Google Scholar]; (o) Reynolds T, Bharadwaj A, Scheidt K. J. Am. Chem. Soc. 2006;128:15382. doi: 10.1021/ja0653674. [DOI] [PubMed] [Google Scholar]; (p) Unger R, Weisser F, Chinkov N, Stanger A, Cohen T, Marek I. Org. Lett. 2009;11:1853. doi: 10.1021/ol9004036. [DOI] [PubMed] [Google Scholar]; (q) Li F-Q, Zhong S, Lu G, Chan A. Adv. Synth. Catal. 2009;351:1955. [Google Scholar]; (r) Mukaiyama T, Suzuki K, Soai K, Sato T. Chem. Lett. 1979:447. [Google Scholar]; (s) Mukaiyama T, Suzuki K. Chem. Lett. 1980:255. [Google Scholar]; (t) Huffman M, Yasuda N, DeCamp A, Grabowski E. J. Org. Chem. 1995;60:1590. [Google Scholar]; (u) Thompson A, Corley E, Huntington M, Grabowski EJJ. Tetrahedron Lett. 1995;36:8937. [Google Scholar]; (v) Tan L, Chen C-Y, Tillyer R, Grabowski E, Reider P. Angew. Chem. Int. Ed. 1999;38:711. doi: 10.1002/(SICI)1521-3773(19990301)38:5<711::AID-ANIE711>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]; (w) Cozzi P. Angew. Chem. Int. Ed. 2003;42:2895. doi: 10.1002/anie.200351230. [DOI] [PubMed] [Google Scholar]; (x) Lu G, Li X, Jia X, Chan W, Chan A. Angew. Chem. Int. Ed. 2003;42:5057. doi: 10.1002/anie.200352013. [DOI] [PubMed] [Google Scholar]; (y) Niwa S, Soai K. J. Chem. Soc., Perkin Trans. 1. 1990:937. [Google Scholar]; (z) Corey E, Cimprich K. J. Am. Chem. Soc. 1994;116:3151. [Google Scholar]; (aa) Usanov DL, Yamamoto H. J. Am. Chem. Soc. 2011;133:1286. doi: 10.1021/ja1102822. [DOI] [PubMed] [Google Scholar]; (bb) Liu L, Wang R, Kang Y-F, Cai H-Q, Chen C. Synlett. 2006:1245. [Google Scholar]
- (9) (a).Downey C, Johnson M, Tracy K. J. Org. Chem. 2008;73:3299. doi: 10.1021/jo8001084. [DOI] [PubMed] [Google Scholar]; (b) Downey C, Mahoney B, Lipari V. J. Org. Chem. 2009;74:2904. doi: 10.1021/jo900102w. [DOI] [PubMed] [Google Scholar]
- (10) (a).DeNinno M, Perner R, Morton H, DiDomenico S. J. Org. Chem. 1992;57:7115. [Google Scholar]; (b) Xu Y-C, Lebeau E, Gillard J, Attardo G. Tetrahedron Lett. 1993;34:3841. [Google Scholar]
- (11).See Supporting Information for full experimental details.
- (12).We have not determined how long [Cu(MeCN4)]PF6 can be stored outside a glovebox. It has been reported to oxidize when stored under atmospheric conditions. See: Kubas GJ. Inorg. Syn. 1990;28:68.
- (13).Das A, Chaudhuri R, Liu R-S. Chem. Commun. 2009:4046. doi: 10.1039/b908338c. [DOI] [PubMed] [Google Scholar]
- (14).The absolute configuration of ether 14 has not been determined.
- (15) (a).For studies of Cu acetylide additions to aldehydes or imines, see: Asano Y, Ito H, Hara K, Sawamura M. Organometallics. 2008;27:5984. Gommermann N, Koradin C, Polborn K, Knochel P. Angew. Chem. Int. Ed. 2003;42:5763. doi: 10.1002/anie.200352578.
- (16).Shi W, Luo Y, Luo X, Chao L, Zhang H, Wang J, Lei A. J. Am. Chem. Soc. 2008;130:14713. doi: 10.1021/ja8049436. [DOI] [PubMed] [Google Scholar]
- (17).Due to the insolubility of copper phenylacetylide, this reaction required CH2Cl2 as solvent.
- (18) (a).Because π-backbonding is significant in these complexes, we propose that the Cu may bind either the C=O or the arene (or slip between these binding modes). For examples of Cu-arene and Cu-olefin complexes, see: Wang X-S, Zhao H, Li Y-H, Xiong R-G, You X-Z. Top. Catal. 2005;35:43. Badiei YM, Warren TH, Chiang KP, Holland PL. Inorg. Synth. 2010;35:50. Pérez-Galán P, Delpont N, Herrero-Gómez E, Maseras F, Echavarren AM. Chem. Eur. J. 2010;16:5324. doi: 10.1002/chem.200903507.
- (19).For an example of 1-naphthyl giving significant improvement over phenyl in a reaction that likely involves cation–π interactions, see: Birrell JA, Desrosiers J-N, Jacobsen EN. J. Am. Chem. Soc. 2011;133:13872. doi: 10.1021/ja205602j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.