

Abstract
In many eukaryotic species, gene order is not random. In humans, flies, and yeast, there is clustering of coexpressed genes that cannot be explained as a trivial consequence of tandem duplication. In the worm genome this is taken a step further with many genes being organized into operons. Here we analyze the relationship between gene location and expression in Caenorhabditis elegans and find evidence for at least three different processes resulting in local expression similarity. Not surprisingly, the strongest effect comes from genes organized in operons. However, coexpression within operons is not perfect, and is influenced by some distance-dependent regulation. Beyond operons, there is a relationship between physical distance, expression similarity, and sequence similarity, acting over several megabases. This is consistent with a model of tandem duplicate genes diverging over time in sequence and expression pattern, while moving apart owing to chromosomal rearrangements. However, at a very local level, nonduplicate genes on opposite strands (hence not in operons) show similar expression patterns. This suggests that such genes may share regulatory elements or be regulated at the level of chromatin structure. The central importance of tandem duplicate genes in these patterns renders the worm genome different from both yeast and human.
[Supplemental material is available online at http://www.genome.org.]
It is often presumed that, aside from clusters of tandem duplicates, gene order within the eukaryotic genome is random. However, there is increasing evidence that this is not always the case. In species as diverse as humans (Lercher et al. 2002), flies (Spellman and Rubin 2002), and yeast (Cohen et al. 2000), neighboring genes show similar expression patterns, even when accounting for the coexpression of duplicated genes.
In the worm Caenorhabditis elegans, the tendency for similarly expressed genes to be linked is taken one step further, in that ∼15% of genes are incorporated into bacterial-like operons (Blumenthal 1998; Blumenthal et al. 2002). Genes located within the same operon are transcribed together, and thus coregulated, that is, they share regulatory elements. Below, we examine coexpression, which is a statistical statement about observable expression patterns. Direct coregulation is only one possible cause of coexpression. Other possible causes are chromatin-level regulation of gene expression (Lercher et al. 2002; Roy et al. 2002), or conserved expression patterns after the duplication of regulatory elements together with coding regions. Is coregulation of genes within operons the dominant cause of any regional coexpression in C. elegans? Does tandem gene duplication, which is especially common in the worm genome (Semple and Wolfe 1999), contribute to the formation of clusters of coexpressed genes? Based on the observation that muscle-expressed genes are clustered in the C. elegans genome after accounting for operon and tandem duplication effects, Roy et al. (2002) suggested that chromatin-level regulation also plays an important role. What is the range of any such effect? Is it comparable to the range seen in human, where analogous effects (Bortoluzzi et al. 1998) have been attributed to the clustering of housekeeping genes (Lercher et al. 2002)?
The worm genome is also potentially unusual in that it has been considered to be broken into genomic compartments (The C. elegans Sequencing Consortium 1998). The autosome arms of C. elegans differ from the central regions in several genomic properties, such as gene density, density of repetitive sequences, and number of EST matches (which may be a surrogate of expression rate). Do these compartments represent groupings of genes with comparable expression profiles, or is gene distribution between compartments random?
The availability of both sequence (The C. elegans Sequencing Consortium 1998) and expression data (Kim et al. 2001) allows us to address these issues to better understand the special genomic architecture of C. elegans. Below, we analyze the spatial patterns of coexpression, paying particular attention to operons and to the distribution of duplicate genes.
RESULTS
Compartmental Heterogeneity
When comparing microarray expression profiles for genes located in different autosomal genomic compartments (autosome arms or central regions), we find that genes are not randomly distributed relative to their expression patterns. We observe significant heterogeneity (P < 0.05) for 31 out of the 44 clusters (‘mounts’) of coexpressed genes defined by Kim et al. (2001). This includes all 24 mounts with n > 50 (detailed results available as supplemental data). A similar result is obtained when we define expression by sorting genes into functional classes: We find significant compartmental heterogeneity for almost all such classes (Kim et al. 2001) with large enough sample sizes (31 out of 55, including 23 of 25 classes with n > 50; see supplemental data). In these analyses, we independently assess statistical significance for n = 44 mounts (or n = 55 classes) simultaneously. As this increases the probability of finding at least one ‘significant’ result even under the null hypothesis of an equal distribution, we repeated the tests with a more stringent significance level of P < 0.05 / n (Bonferroni correction). After this correction, the heterogeneity is still significant for 21 mounts and for 18 functional classes.
Level of Coexpression
We defined distance-dependent indices of coexpression, which measure the degree of similarity in expression of all autosomal genes that are a distance d ± 10 kb apart. Figure 1 shows the level of coexpression from microarray data (A) and from functional classes (B). For microarray data, the local similarity (rd) is significant for all distances up to 4.20 Mb (P < 0.05 from randomization), and appears to extend over whole genomic compartments. For distances above ∼200 kb, the similarity measure rd decreases linearly (R2 = 0.88 between 0.5 and 5 Mb). When accounting for the unequal distribution of expression classes to genomic compartments by randomizing genes only within compartments, local similarity is significant only up to 1.16 Mb. A similar signal is found for coexpression in terms of functional classification (ICFd). Here the similarity is significant for all distances up to 3.4 Mb. Again, the similarity seems to extend over whole compartments. Accordingly, when accounting for the nonrandom distribution of functional classes to genomic compartments, the range of significant similarity is reduced to 160 kb. For distances above ∼200 kb, the measure of similar function ICFddecreases approximately linearly (R2 = 0.21 between 0.5 and 5 Mb).
Figure 1.


Level of coexpression for 20-kb distance bins, for microarray data (A) and for functional classes (B). (Insets) Data for 2-kb distance bins. The solid lines are linear regressions (microarray data: R2 = 0.88, functions: R2 = 0.21 for 0.5–5 Mb).
Coexpression of Genes in Operons
We measured the level of coexpression of neighboring genes in operons as Pearson's correlation coefficient r of the normalized microarray data (Kim et al. 2001). In operons, the distance between the 3′ end of one open reading frame (ORF) and the 5′ end of the next ORF is on average 672 bp (median 446 bp). To test whether coexpression in operons is just a consequence of the proximity of genes, we also analyzed all neighboring gene pairs closer than 500 bp that were not identified as part of operons. As shown in Table 1, the level of coexpression r for close neighbors either on the same or on opposing strands is significantly lower compared to gene pairs sharing the same operon. For all three classes of close neighbor pairs, we found strong coexpression compared to randomly paired genes. All classes in Table 1 are significantly different from each other (including Bonferroni correction for multiple tests; P < 10−19 from one-sided t-tests), except when comparing close neighbors on the same and on opposing strands (P = 0.066 from two-sided t-test).
Table 1.
Level of coexpression (correlation coefficient of microarray data r ± standard error) for neighbouring genes in operons, on the same strand, on opposing strands, and for random gene pairs
| All genes | Exc. duplicate pairs | P | |||
| N | r | N | r | ||
| Operons | 1058 | 0.229 ± 0.007 | 747 | 0.210 ± 0.008 | 0.033 |
| <500 bp on the same strand | 1105 | 0.143 ± 0.007 | 586 | 0.097 ± 0.008 | 8 × 10−6 |
| <500 bp on opposing strands | 2091 | 0.128 ± 0.005 | 1513 | 0.116 ± 0.006 | 0.053 |
| Random pairs | 10,000 | 0.006 ± 0.002 | 10,000 | 0.001 ± 0.002 | 0.011 |
The P values (for the difference between all gene pairs and gene pairs exc. duplicates) are from one-sided t-tests (significance at 0.05/4 = 0.013).
If genes organized into the same operon are always 100% coexpressed (and the low r value in Table 1 is due to random error in the microarray experiments), then we would expect no distance dependence of r within operons. To test this, we calculated the level of coexpression for all possible gene pairs within operons (Fig. 2). Contrary to the prediction, rdecreases significantly with increasing physical gene distance (R2 = 0.79 from linear regression for 0–13 kb, with P = 0.00003 from 106 random data pairings). This is qualitatively unchanged when measuring distance by counting (0–3) intervening genes, or when restricting the analysis to operons (Blumenthal et al. 2002) whose structure has been confirmed by microarray data or cDNA clones (data not shown).
Figure 2.

Distance dependence of coexpression for gene pairs within operons, including all gene pairs and after the exclusion of duplicate gene pairs. In both cases, we find a significant negative correlation (all genes: R2 = 0.79, P = 0.00003; excl. duplicates: R2 = 0.67, P = 0.0005).
Coexpression of Duplicated Gene Pairs
It is known that the genome of C. elegans contains many pairs of duplicated genes (Semple and Wolfe 1999), with a strong bias towards intrachromosomal duplication, and with an excess of duplicates that are located close to the original gene (distance ≤ 30 kb). It appears likely that in many gene duplication events, control regions are duplicated together with the coding sequence. Initially, many duplicated gene pairs will then show similar expression patterns, although mutations in the control regions will cause a divergence over time (C. Pal, pers. comm.). Are then the patterns of local coexpression mainly a consequence of the nonrandom distribution of duplicated gene pairs? If this is the case, we expect two consequences: (1) when removing duplicate gene pairs, the level of coexpression (Table 1; Fig. 1) should be greatly reduced; and (2) the probability of finding duplicates and/or the degree of similarity of duplicated gene pairs should show a similar distance dependence as the level of coexpression (Fig. 1).
We tested prediction (1) by removing one gene of each pair of duplicated genes. Table 1 shows the resulting level of coexpression for gene pairs in operons, close gene pairs not in operons, and for random gene pairs. While coexpression is reduced in each case compared to the inclusion of all genes, this reduction is nonsignificant (after Bonferroni correction for multiple tests) for genes in operons and for neighboring pairs on opposing strands (Table 1). The reduction is significant for nonoperon neighbors on the same strand and for random gene pairs. As before, all classes of pairs are significantly different from each other (P < 10−21 from one-sided t-tests) except for the comparison of close neighbors on the same to those on opposing strands (P = 0.059 from two-sided t-test). After the exclusion of duplicates, random gene pairs show no correlation of expression, as is expected.
The removal of duplicate gene pairs does not change the negative correlation of coexpression with distance when comparing genes within operons (Fig. 2; R2 = 0.67, P = 0.0005). This suggests that some local coregulation occurs even within operons.
The level of coexpression beyond operons after excluding duplicate gene pairs is shown in Figure 3 for microarray data (A) and for functional classes (B). For both measures, the level of local coexpression is greatly reduced compared to Figure 1, and is significant only for the closest neighbors (distance < 20 kb) when taking the nonrandom distribution across compartments into account. This is not due to the reduced sample size: When calculating our similarity measures (rd and ICFd) for random subsets of all genes of the same sample sizes, we find values similar to the full data sets (see Fig. 3).
Figure 3.


Level of coexpression for 20-kb distance bins after removal of duplicate genes, for microarray data (A) and for functional classes (B). For comparison, the open symbols show data before the removal of duplicates [circles: all genes; triangles: random subsets of 2679 genes (A) and 1959 genes (B)].
Distribution of Duplicated Gene Pairs
The above results suggest that the local similarity in expression beyond operons is largely due to the nonrandom distribution of duplicated gene pairs. How then does the distribution of such gene pairs compare to the distance dependence of the level of coexpression (see prediction [2] above)? Figure 4A shows the probability of finding duplicate gene pairs at distance d ± 10 kb on the same chromosome (corrected for the total number of gene pairs in this distance bracket; this analysis is for 19,325 genes in Wormbase WS78). In Figure 4B, we plot a measure of the degree of sequence similarity (BLAST expect value) of duplicate gene pairs for different distance brackets. Both the fraction of duplicate genes and the degree of sequence similarity decrease exponentially for distances up to ∼20 kb, and decrease linearly above ∼200 kb (duplicate fraction: R2 = 0.70, log(expect): R2 = 0.60, between 0.5 and 5 Mb). Thus, there are not only a higher number of similar genes at close distances, but they are also much more similar than duplicates further apart. In summary, all features of the local similarity in expression (Fig. 1) are compatible with the distribution of duplicate genes beyond operons: the extreme degree of similarity at very small distances (d < 20 kb), the steep decrease for small distances (d < 200 kb), and the linear decrease for larger distances.
Figure 4.


Distribution of duplicate genes in 20-kb distance bins. (A) Fraction of duplicate gene pairs relative to all pairs at this distance; (B) mean log(Expect) as a measure of similarity. The insets show data for 2-kb distance bins. The solid lines are linear regressions (duplicate fraction: R2 = 0.70, log(Expect): R2 = 0.66 for 0.5–5 Mb). The functional form seems to change at ∼30 kb and at ∼200 kb.
DISCUSSION
The worm genome, like other closely analyzed eukaryotic genomes, is not the random array of genes that is often supposed. It is remarkable, for example, that although genes in operons show the strongest coexpression, neighboring genes on the same strand, as well as on opposing strands, show much stronger coexpression compared to genes that are paired at random. This feature remains after the exclusion of duplicate gene pairs. Although we may not have identified some operons with genes expressed at very low levels, this cannot explain the coexpression of neighbors located on opposing strands. This would be consistent either with a chromatin-based level of gene expression, with certain chromosomal regions being accessible to transcription factors in any given tissue, or with gene pairs sharing regulatory elements (Fig. 5). Chromatin-level regulation has also been proposed as an explanation for the clustering of C. elegans muscle-expressed genes (Roy et al. 2002). The latter analysis included at most one gene from each operon and each family of duplicates. Consistent with what we found here, significant clustering of such coexpressed genes was restricted to distances <25 kb.
Figure 5.

Simplified representation of three putative modes of coexpression in C. elegans. The indicated distances are meant for rough guidance only. The fourth putative mode, chromatin-level regulation, is not depicted.
The putative existence of shared regulatory elements could explain the variation of coexpression level within operons shown in Figure 2. Alternatively, this variation could be caused by common errors in operon transcription. Ideally, all genes within an operon are transcribed together, and the individual transcripts are then separated by trans-splicing. Coregulation affecting only part of an operon must then happen after transcription, but before the trans-splicing which separates the individual genes. In the absence of such coregulation, all genes in the same operon should be perfectly coexpressed. Sometimes however, transcription may terminate prematurely, or trans-splicing may not be achieved successfully. In this situation, the probability of two genes to be coexpressed will decrease with increasing distance between them. Furthermore, genes in operons sometimes may appear not to be coregulated because their mRNAs may be subject to differential mRNA destabilization.
Longer-range patterns of similarity of expression are also found. At the grossest level, genomic compartments themselves differ in their expression patterns. Most strikingly however, we have shown a general correlation between pair distance and coexpression level, with the similarity in expression decreasing linearly over the range of whole genomic compartments. This pattern can be explained by a simple model of tandem duplication of both coding and regulatory regions (Fig. 5), followed by divergence in sequence and expression over time. We may suppose that at the point of tandem duplication the sequences (both coding and regulatory), and consequently their expression, are near identical. Over time, the sequences diverge, the expression pattern diverges and—owing to random rearrangements—the physical distance between the duplicates tends to increase. As support for this model, we find that local coexpression beyond ∼20 kb seems indeed to stem from the nonrandom distribution of duplicate genes (Fig. 3).
A nonrandom distribution of duplicates was first demonstrated by Semple and Wolfe (1999). We have extended their work by showing that the distribution of duplicate gene pairs shows striking behaviors over different length scales, and that this behavior is compatible with the distance dependence of the level of coexpression. As gene pairs from more recent duplications are expected to show stronger sequence similarity, Figure 4B suggests that many duplicated genes begin their life at very close distances (<2 kb) to the original sequence, and then gradually move away. The three length scales over which different behaviors are apparent in Figure 4, that is, 20 kb, 200 kb, and 5 Mb, may be characteristic for different mechanisms affecting the movement of genes along chromosomes. Inversions are most likely responsible for the shortest of these scales. Indeed, it has been estimated that two-thirds of all inversions in C. elegans are <25 kb (Coghlan and Wolfe 2002). The larger-scale movement of duplicate genes is likely to be caused by the insertion of intervening DNA through transpositions. These are about twice as frequent in C. elegans as both inversions and translocations; most transposed DNA segments are <30 kb (Coghlan and Wolfe 2002).
Although the above four reasons for local similarity—tandem duplicates, operons, putative chromatin-level regulation, and shared regulatory elements—present a complex portrait of the nonrandom relationship between gene location and gene expression (Fig. 5), it is perhaps surprising how weak some of the local similarity in expression appears to be. Most notably, for confirmed operons, it is striking that the correlation of expression is not higher, that is, why is r in Table 1 not close to 1? To some extent, this may represent noise in the data. Indeed, in comparable experiments in yeast, the correlation between the results of identical assays is often even lower than this figure. Thus, a correlation coefficient of r = 0.23 may in fact be relatively large. However, the demonstrated distance dependence of coexpression within operons indicates that coexpression in operons is not perfect, and is affected by additional, distance-dependent factors.
The results presented here contrast with those found in humans (Lercher et al. 2002) and flies (Spellman and Rubin 2002) in one important regard. In all three genomes, linked genes show similar expression profiles. For C. elegans, we found no significant coexpression beyond 20 kb after the exclusion of operon and duplication effects (see also Roy et al. 2002). In contrast, significant coexpression of neighboring genes over substantial distances was found in humans (∼500 kb) and flies (∼200 kb), even when duplicate genes were excluded. These two genomes also lack any structures resembling operons. If we consider the action of a shared regulatory element on genes located several 100 kb apart to be unlikely, this leaves chromatin-level gene regulation as the most likely explanation for regional coexpression in these species. Consistent with this hypothesis, the local similarity in humans has been attributed to the clustering of housekeeping genes (Lercher et al. 2002). Any selective force on chromatin-level regulation should only depend on where and when the genes contained in the region are expressed. Thus, it is not required that the gene products interact directly or perform related functions, consistent with reports that clustered coexpressed genes in Drosophila are not functionally related (Spellman and Rubin 2002). Why does the worm not show such signs of chromatin-based expression regulation beyond regions of ∼20 kb size? It seems likely that operons in C. elegans, like bacterial operons, function to ensure the concerted expression of genes that are needed at the same time in the same cell. In some sense, operons may thereby perform the same function as chromatin-level regulation in other studied eukaryotes. In addition, genes in the worm genome are much more tightly packed compared to, for example, humans (median gene distance ∼2.8 kb in C. elegans, compared to ∼28 kb in humans). Thus, C. elegans chromatin regions of one-tenth the size of analogous human regions contain approximately the same amount of genes, and may thereby represent comparable targets for selection.
Taken together, our findings suggest that the genome organization of C. elegans differs from the genomes of other eukaryotes not only by the existence of operons, but also by the relative role played by recent gene duplications. Could there be a link between these two genomic characteristics? Interestingly, we found that duplicated genes are located outside of operons more often than expected if there was no correlation between the two features. Of all duplicate pairs, 29,089 had both genes outside of operons, whereas only 2101 pairs (6.7%) had one or both genes within an operon. Using the finding that 2208 of 19,325 genes in our data set (=11.4%) are found in operons, we would have predicted this number to be 1 − (1 − 0.114)2 = 21.5%. These figures are for a definition of “duplicates” as having pairwise BLAST expect value <10−50; qualitatively similar results are obtained for other cut-off values (data not shown).
If the duplication of genes is selectively favorable by allowing the evolution of new functions, then the underrepresentation of operonic genes may not be unexpected. Such new functions may require a changed expression pattern of individual genes. However, genes organized in operons are effectively “frozen” in the expression pattern of their operon; to be expressed, such genes need to be duplicated together with the operon's 5′ regulatory elements, and thus with all intervening genes. Conversely, genes outside of operons can easily be duplicated individually together with their control regions.
This places a constraint on the evolution of new functions in C. elegans. Due to the “frozen operons,” only a subset of all genes are available both for individual changes in expression pattern, and for individual duplication together with their control elements. The worm may thus be forced to choose new metabolic pathways that are more complex than they would be if it could recruit all genes equally. It is noteworthy that this speculative argument not only suggests an explanation for the many recent gene duplications found in C. elegans (Semple and Wolfe 1999) compared to the human, fly, or yeast genome. It also suggests an explanation for the high total gene number (∼19,000) compared to more complex animals, for example, Drosophila melanogaster (∼14,000). In principle, this idea could be tested by comparing metabolic pathways that evolved in the ancestors of C. elegans after the widespread use of operons, to corresponding pathways in other species.
METHODS
Expression and Genome Data
We took expression data from a recent meta-analysis of 553 microarray experiments (Kim et al. 2001). Certain experiments (expt463, expt546, expt547, expt548, expt549) were targeted specifically at operons (Blumenthal et al. 2002), and were thus excluded to avoid potential bias. In addition to the raw data, we use two definitions of genic expression profiles by Kim et al. (2001): (1) the expression in 55 nonexclusive functional groups, and (2) 44 exclusive coexpression clusters (coined “mounts”) that are automatically built from correlations of the raw data.
We located 15,924 genes with raw expression profile, 5630 genes with known functional profile, and 15,262 genes with unambiguous mount assignment on the genome map of Wormbase (WS78, available at ftp://ftp.wormbase.org). Of these, 13,511, 5015, and 12,949, respectively were positioned on autosomes. Gene position was defined as the midpoint between the 3′ and 5′ ends of the unspliced coding sequence. A list of approximately 2500 genes organized in operons was obtained from Blumenthal et al. (2002).
Identification of Tandem Duplicates
As duplicated genes may be coexpressed for trivial reasons, we performed part of our analysis excluding such genes. We used a criterion previously developed for the identification of duplicated genes in vertebrates (Lercher et al. 2002). Removing one gene of each pair with expect value E < 0.2 from pairwise protein BLAST (word size 2) identifies ∼93% of even distantly related genes, while at the same time removing ∼10% of unrelated genes. Protein sequences were obtained from the Wormpep database (Wormpep78, available at http://www.sanger.ac.uk). Applying this criterion to all gene pairs within 200 kb of each other reduced sample sizes to 3315 (microarray data) and 1959 (function) genes. For the analysis of the distribution of duplicate gene pairs, we blasted all genes in our data set against all other genes on the same chromosome; all gene pairs with E < 0.2 were regarded as putative duplicates.
Compartmental Heterogeneity
To test whether coexpressed genes tend to be located on the same genomic compartment, we calculated the test functions
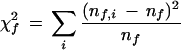 |
with total fraction nf of genes expressed in functional class (or mount) f, and nf,i the fraction of these genes on compartment i. χf2 was compared to the corresponding values from 10,000 random genomes, each obtained by permuting the compartmental assignments of all genes.
Level of Coexpression/Cofunction
Following Davidson et al. (2001), we first normalized the raw microarray data for each experiment by subtracting the median and dividing by the interquartile distance. We then defined the level of coexpression ra,b between two genes a,b as Pearson's correlation coefficient of the normalized microarray expression data of the genes (Kim et al. 2001). When assessing the coexpression in terms of functional classifications, we defined an index of common function (ICFa,b) as the number of shared functions, weighted by the geometric mean of the two expression breadths (c runs over all functional classes, fa,c ∈ {0,1} indicates not expressed/expressed):
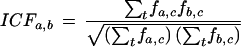 |
From ra,b and ICFa,b, we calculated distance-based indices rd and ICFd as the mean over all gene pairs that lie within a physical distance bracket [d − 10 kb, d + 10 kb] of each other. rd and ICFd were compared to expectations under the null hypothesis (no spatial pattern in coexpression), by recalculating them for 10,000 data sets with randomly permuted gene positions. To test whether any local coexpression is a secondary effect caused by the nonrandom distribution of genes to genomic compartments, we repeated these randomization procedures, this time permuting gene positions only within each compartment. The ranges of significant coexpression were defined as the largest distances so that all rd (ICFd) values for smaller distances are significantly larger (P < 0.05) compared to random data.
WEB SITE REFERENCES
http://www.sanger.ac.uk; The Wellcome Trust Sanger Institute.
ftp://ftp.wormbase.org; Wormbase database.
Acknowledgments
We thank Csaba Pal for discussions, and two anonymous referees for suggestions on the manuscript. We acknowledge support by the Wellcome Trust (M.J.L.) and the Biotechnology and Biological Sciences Research Council (L.D.H.). The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL M.J.Lercher@bath.ac.uk; FAX 44-1225-386779.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.553803.
REFERENCES
- 1.Blumenthal T. 1998. Gene clusters and polycistronic transcription in eukaryotes. BioEssays 20: 480-487. [DOI] [PubMed] [Google Scholar]
- 2.Blumenthal T., Evans, D., Link, C.D., Guffanti, D., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W.L., Duke, K., Kiraly, M., et al. 2002. A global analysis of the Caenorhabditis elegans operons. Nature 417: 851-854. [DOI] [PubMed] [Google Scholar]
- 3.Bortoluzzi S., Rampoldi, L., Simionati, B., Zimbello, R., Barbon, A., dAlessi, F., Tiso, N., Pallavicini, A., Toppo, S., Cannata, N., et al. 1998. A comprehensive, high-resolution genomic transcript map of human skeletal muscle. Genome Res. 8: 817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The C. elegans Sequencing Consortium 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012-2018. [DOI] [PubMed] [Google Scholar]
- 5.Coghlan A. and Wolfe, K.H. 2002. Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 12: 857-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen B.A., Mitra, R.D., Hughes, J.D., and Church, G.M. 2000. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 26: 183-186. [DOI] [PubMed] [Google Scholar]
- 7.Davidson G.S., Wylie, B.N., and Boyack, K.W., 2001. Cluster stability and the use of noise in the interpretation of clustering. In Proc. IEEE Information Visualization 2001, pp. 23–30. IEEE, New York, NY.
- 8.Kim S.K., Lund, J., Kiraly, M., Duke, K., Jiang, M., Stuart, J.M., Eizinger, A., Wylie, B.N., and Davidson, G.S. 2001. A gene expression map for Caenorhabditis elegans. Science 293: 2087-2092. [DOI] [PubMed] [Google Scholar]
- 9.Lercher M.J., Urrutia, A.O., and Hurst, L.D. 2002. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Gen. 31: 180-183. [DOI] [PubMed] [Google Scholar]
- 10.Roy P.J., Stuart, J.M., Lund, J., and Kim, S.K. 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975-979. [DOI] [PubMed] [Google Scholar]
- 11.Semple C. and Wolfe, K.H. 1999. Gene duplication and gene conversion in the Caenorhabditis elegans genome. J. Mol. Evol. 48: 555-564. [DOI] [PubMed] [Google Scholar]
- 12.Spellman P.T. and Rubin, G.M. 2002. Evidence for large domains of similarly expressed genes in the Drosophila genome. J. Biol. 1: 5.1-5.8. [DOI] [PMC free article] [PubMed] [Google Scholar]