

Abstract
The presence of Ca2+-activated Cl– currents (ICl(Ca)) in vascular smooth muscle cells (VSMCs) is well established. ICl(Ca) are supposedly important for arterial contraction by linking changes in [Ca2+]i and membrane depolarization. Bestrophins and some members of the TMEM16 protein family were recently associated with ICl(Ca). Two distinct ICl(Ca) are characterized in VSMCs; the cGMP-dependent ICl(Ca) dependent upon bestrophin expression and the ‘classical’ Ca2+-activated Cl– current, which is bestrophin-independent. Interestingly, TMEM16A is essential for both the cGMP-dependent and the classical ICl(Ca). Furthermore, TMEM16A has a role in arterial contraction while bestrophins do not. TMEM16A’s role in the contractile response cannot be explained however only by a simple suppression of the depolarization by Cl– channels. It is suggested that TMEM16A expression modulates voltage-gated Ca2+ influx in a voltage-independent manner and recent studies also demonstrate a complex role of TMEM16A in modulating other membrane proteins.
Keywords: Ca2+-activated Cl– channels, intracellular Ca2+, membrane potential, siRNA, smooth muscle cells, voltage-gated Ca2+ channels
Introduction
For more than two decades, the functional role of Ca2+-activated Cl– channels (CaCCs) in vascular smooth muscle cells (VSMCs) has been actively investigated.34,41,42,47,95 This research focus was initiated by the first demonstration in 1987 13 of a Ca2+-activated Cl– current (ICl(Ca)) in smooth muscle cells, which naturally led to the suggestion that CaCCs are also present in VSMCs. Although ICl(Ca) has subsequently been well-characterized in patch clamp studies of isolated VSMCs11,48,49,59 these studies have not linked the ICl(Ca) to its function in the vascular wall. Since Cl– is actively accumulated in VSMCs,1,71 opening of CaCCs at resting membrane potentials will lead to Cl– efflux and membrane depolarization. These CaCC properties integrate intracellular calcium ([Ca2+]i) increases with membrane potential changes and suggest an amplifying link for agonist-induced contraction32,40,46,62,87 as well as several other important properties of the vascular wall, e.g., vascular rhythmicity9,37 and myogenic tone.12,64 Uncertainties in the mechanistic explanation of this amplifying link are, at least in part, due to the lack of specific pharmacological tools and uncertainty about the molecular structure of CaCCs.30
Significant progress in this field has occurred over the last years as putative CaCC proteins have been identified.14,83,88,97,103 Four members of the bestrophin family can produce ICl(Ca),83,97 although their direct association with the endogenous ICl(Ca) has been questioned.30 TMEM16A and some other members of the TMEM16 protein family are also shown to be essential for ICl(Ca).14,88,103 There is no universal profile for the biophysical and pharmacological properties of ICl(Ca) reported in VSMCs.54 The CaCCs responsible for these currents are suggested to vary in their Ca2+-sensitivity and the mechanism of Ca2+ activation, voltage- and time-dependency, halide permeability and sensitivities to blockers. These variable characteristics could partially be attributable to differences in the methods of registration, protocols for current activation, solutions and origins of VSMCs. It is, however, most probable that the CaCCs represent a heterologous group of channel proteins unified by their anion sensitivity and Ca2+-dependence.77 Several subgroups of CaCCs have been suggested based upon their activation mechanism, e.g., the CaCCs activated directly by [Ca2+]i and those that need additional activators such as CaMKII or cGMP-dependent protein kinase (PKG).24,25,30 The current, general classification of ICl(Ca) is based on biophysical and pharmacological characteristics obtained under variable experimental conditions. There is a substantial need to improve this classification based on an understanding of the molecular structures that are associated with the specific characteristics.54 Evidence for the association between ICl(Ca) with different properties55,75,76 and different putative CaCC proteins16,17,33,52,58,93 is needed. Such associations are beginning to appear, e.g., in the vasculature, where the ‘classical’ ICl(Ca) co-exists with another ICl(Ca), which has an absolute requirement for cyclic GMP for its activation and several other biophysical and pharmacological characteristics distinct from the classical ICl(Ca).55,75,76 The cGMP-dependent ICl(Ca) depends upon the expression of bestrophins21,58 while the classical ICl(Ca) does not. It is therefore of interest to know whether TMEM16A is important for the classical ICl(Ca) and what relation this protein has to the cGMP-dependent ICl(Ca) in VSMCs. Interestingly, TMEM16A is essential for both the classical ICl(Ca)17,52,93 and the cGMP-dependent ICl(Ca).16 This complicates the description of ICl(Ca) with a certain characteristic to a specific protein or protein family.
Although the association of bestrophins and TMEM16A with CaCCs has been originally suggested in heterologous expression experiments, detailed molecular-based studies of the functional importance of CaCCs in the vascular wall in vitro and in vivo have been made possible by novel approaches to studying vascular tissue; siRNA-induced downregulation of the protein of interest,58 tissue specific knockdown in mice,33 molecular cloning and mutagenesis14 and drug-screening strategies.68 An siRNA approach suggests that the bestrophin-associated cGMP-dependent ICl(Ca) has no significance for tonic contractile response in small arteries.10 These findings are further supported by a comprehensive study suggesting minor or little importance of the Cl– conductance for arterial contraction in rat mesenteric small arteries.9,33 However, suppressing TMEM16A expression reduces arterial contraction12,16,17 and lowers arterial blood pressure.33 A detailed analysis of the functional consequences of TMEM16A expression changes suggests that TMEM16A is more than a channel-forming protein and likely has several other modulatory and expression-related cellular functions.16
In spite of significant progress in our understanding the role CaCC has in the vasculature, several significant incongruities between various reports as well as between working hypotheses and experimental results remain to be resolved. Some of these questions will be discussed in this review.
1) the role of Cl– conductances for arterial contraction;
2) whether the two CaCCs are represented by a single channel-forming protein family;
3) how TMEM16A and bestrophins interact with each other to produce different Ca2+-activated Cl– conductances;
4) other putative functions of CaCC-associated proteins in the vascular wall.
Two Functionally Distinct ICl(Ca) in VSMCs
The existence of a depolarizing conductance that can be activated by cGMP and [Ca2+]i was previously hypothesized based on the model for generation of vasomotion.37,72 A depolarizing current dependent on or activated by cGMP is unexpected as cGMP normally leads to hyperpolarization in VSMCs. However, the cGMP-dependent ICl(Ca) in VSMCs in rat mesenteric small arteries has been reported by two independent laboratories.55,75,76 The functional characterization of this conductance clearly indicates that it is a unique current in comparison with the well-described classical ICl(Ca).30,48 In addition to an absolute cGMP-dependence of only one of these two ICl(Ca), these conductances differ in their Ca2+- and voltage-dependence as well as in their pharmacological profile.54 The inhibitors which have been shown to be effective, at least in patch clamp experiments, for the classical ICl(Ca), e.g., niflumic acid, 5-isothiocyanato-2-[2-(4-isothiocyanato-2-sulfophenyl)ethenyl]benzene-1-sulfonic acid (DIDS) and indanyloxyacetic acid 94 (IAA94),28 affected the cGMP-dependent ICl(Ca) only at high concentrations.55 In contrast, the cGMP-dependent ICl(Ca) was shown to be sensitive to extracellular Zn2+ in concentrations that are without effect on the classical ICl(Ca).55,56,76 Differences in blocker sensitivity and distinction in cGMP-activity make it possible to differentiate these two ICl(Ca) in VSMCs during voltage-clamp experiments.9,16,17,55,56,58,75,76 Until recently this was the approach to isolate and study the different ICl(Ca) in VSMCs and pharmacological differentiation was limited in situ due to off-target effects of the blockers.9,28 When inhibitors are tested in patch clamp experiments, the experimental conditions are created to emphasize the current of interest, e.g., the ICl(Ca). In contrast, in situ experiments do not often provide such possibility and several conductances can therefore be affected by the same blocker. Thus, a commonly used CaCC inhibitor, niflumic acid, is also known to activate the big conductance Ca2+-activated K+ channels (BKCa) and to inhibit L-type Ca2+ channel (LTCC).27,35,56,70,74 These effects would have the same consequences for VSMC membrane potential and vascular tone as inhibition of the ICl(Ca). This limited specificity of the conventional CaCC inhibitors limits their use for studies of the importance of CaCCs for regulation of vascular tone.28
The pharmacological overlap between ICl(Ca) and cation currents has led to the suggestion of molecular interaction between different groups of channels and even that some molecules may be shared in the molecular complexes important for these currents.28 The overlapping pharmacological profile does not in itself prove protein-protein interaction nor molecular sharing. Although the observation that CaCC blockers can modulate BKCa and LTCC, and that the modulation of CaCCs by BKCa and LTCC can also occur,28 together with localization of these channels in the same membrane microdomains, does suggest a close physical interaction.
Greenwood and co-authors have addressed this question suggesting the potential co-localization of BKCa and CaCCs in cholesterol-rich lipid rafts in VSMC membrane.90 While BKCa are shown to localize in cholesterol-rich lipid rafts and their properties are modulated by interaction with caveolin2 and cholesterol depletion,89 the pharmacological overlap between CaCCs and BKCa cannot be explained by a simple co-localization of these channels. The putative CaCC TMEM16A is shown to be distributed more evenly in the VSMC plasma membrane having only slight overlap with BKCa localization.90 This is consistent with the finding that cholesterol depletion omits the effects of BKCa blockers on ICl(Ca) but has no major consequences for the effects of niflumic acid. This suggests that the interaction between BKCa and CaCCs is more subtle or complex than simple consolidation in a microdomain.90
In contrast, a close proximity of CaCCs to voltage-gated Ca2+ channels (VGCCs) in the membrane of hippocampal neurons was previously shown.36 This co-localization is, however, tissue specific as CaCCs were shown to be functionally and structurally separate from VGCCs in other types of neurons: CaCCs were instead shown to interact specifically with ryanodine receptors in avian sensory neurons100 and with Ins(1,4,5)P3 receptors in dorsal root ganglia neurons.39 This variation in the interacting partner for CaCCs could be the reason for variability of side-effects for different pharmacological agents in different types of cells.
An interesting approach to differentiate ICl(Ca) in VSMCs was recently used where inhibitory antibodies against two putative CaCC proteins – TMEM16A and bestrophin – were applied to suppress the membrane currents associated with these proteins.17 Importantly, antibodies raised against bestrophin or TMEM16A inhibited the cGMP-dependent ICl(Ca) and the classical ICl(Ca), respectively. Moreover, anti-bestrophin antibody was without effect on the classical ICl(Ca). While the authors did not test whether the anti-TMEM16A antibody affects the cGMP-dependent ICl(Ca) it is tempting to suggest that these two Ca2+-activated Cl– conductances have different molecular identities.17
Knowledge of molecular candidates significantly improves the efficiency for drug-screening that has led to the generation of novel, small molecule inhibitors.98 The recently developed inhibitor T16Ainh-A0163 effectively inhibits the TMEM16A-associated ICl(Ca) without effect on the bestrophin-associated cGMP-dependent ICl(Ca) in VSMCs.17 This inhibitor is now widely used for pharmacological identification of TMEM16A-associated ICl(Ca) and its functions,23,38,60,69,101 including VSMC contraction.17 Drug-screening also generated another inhibitor, CaCCinh-A01, which has been shown to inhibit TMEM16A-associated ICl(Ca).18 This inhibitor has, however, some peculiar properties, since it was shown to affect TMEM16A in cancer cells in two distinct ways: in addition to its inhibitory action on ICl(Ca), CaCCinh-A01 reduces protein level in the membrane by facilitating reticulum-associated, proteasomal turn-over of TMEM16A.8 The latter action of CaCCinh-A01 is complex because after prolonged exposure of a cell culture to CaCCinh-A01 a pool of CaCCinh-A01 resistant cells developed, where the inhibitor could still block the ICl(Ca) but TMEM16A remained in the membrane. Importantly, the initial CaCCinh-A01-induced retrieval of TMEM16A from the membrane is associated with inhibition of cell proliferation and the authors provided evidence that the anti-proliferative effect could be ascribed to the disappearance of TMEM16A. This interpretation was consistent with observation that T16Ainh-A01, which only blocks the ICl(Ca), has no effect on proliferation. These findings strongly suggest a role for TMEM16A in proliferation, which is only partially dependent upon its channel function.8 This conclusion is further complicated, however, by the lack of direct evidence for binding of T16Ainh-A01 and CaCCinh-A01 to TMEM16A8 and by the lack of knowledge about the mechanism of their inhibitory action. A structurally different inhibitor, MONNA, was recently shown to have high potency and selectivity for the TMEM16A-associated ICl(Ca), while it was ineffective toward inhibition of bestrophin-1 associated ICl(Ca).68 The fact that these novel small molecular inhibitors can inhibit the TMEM16A-associated ICl(Ca) without an effect on the bestrophin-associated ICl(Ca) is supportive evidence for different proteins forming these two conductances.17,68 Unfortunately, a specific inhibitor for the bestrophin-associated ICl(Ca) is still lacking. Importantly, although the effects of these inhibitors on cation membrane conductances have not been analyzed in detail, T16Ainh-A01 has been shown to relax different types of vasculature17 in accordance with the hypothesized role of CaCCs in agonist-induced arterial contraction.48,49 An in-depth analysis of the effects of T16Ainh-A01 on voltage-gated Ca2+ currents and K+ currents in VSMC membrane is, however, necessary to substantiate this conclusion. The other small molecular inhibitors, CaCCinh-A01 and MONNA, have not been evaluated on vascular tissue.
Is Bestrophin still a Putative CaCC Protein?
The cGMP-dependent ICl(Ca) in rat VSMCs is absent after knockdown of both TMEM16A16 and bestrophin-358 proteins. However, expression of bestrophin-3 is required only for the cGMP-dependent ICl(Ca).16 Is it possible that bestrophin is a channel subunit that confers the cGMP-dependence? In fact, a cGMP-dependence has previously been shown for human bestrophin-1 but the mechanism behind it has not been analyzed.20,21
The data from rat VSMCs are inconsistent with the results from a heterologously expressed mouse heart variant of bestrophin-3, which can produce the ICl(Ca) of significant amplitude without any presence of cGMP under whole-cell configuration.66,91 Importantly, bestrophins are known to be expressed in various splice variants, sometimes present in the same cell type,43,91 and this could explain the inconsistent experimental findings. This possibility is supported by a study showing that expression of the full-length clone of mouse bestrophin-3 does not produce any significant Cl– current due to autoinhibition by the C-terminus.29,78,85 This autoinhibitory cytoplasmic C-region has been predicted to contain sites for PKG-dependent phosphorylation58 as well as for other protein-protein interactions.61 Moreover, mouse bestrophin-3 with the C-terminus deleted loses its autoinhibitory capacity and produces a large Cl– current when expressed heterologously.78,85 These studies indirectly suggest that the cGMP-dependency of bestrophin-3-associated ICl(Ca) could be a consequence of the splice variant expressed. Although the presence of the autoinhibitory C-terminal domain has been suggested for all members of the bestrophin family,85 the exact autoinhibitory phosphorylation site remains to be determined. There is no evidence for direct PKG-phosphorylation of the bestrophin protein: this effect might be indirect and involve several additional phosphorylation and dephosphorylation steps. Thus, the C-terminal autoinhibitory domain of mouse bestrophin-3 has been suggested to interact directly with negatively-charged membrane phospholipids and this interaction dynamically regulates the bestrophin-3-associated CaCC activation via the phosphatidyl inositol 3-kinase.80 However, this modulation is not essential for channel activation and other regulatory sites have been suggested.29 Indeed, it has been previously shown that [Ca2+]i might activate human bestrophin-3 without going through a freely diffusible messenger or through protein phosphorylation.96 This conclusion was, however, reached based upon very slow activation and deactivation kinetics of the CaCC and cannot exclude the involvement of membrane-associated messenger(s) acting on the autoinhibitory C-terminal.96 Some reports suggest the presence of potent phosphorylation sites on bestrophin.6,58,85 Accordingly, bestrophin-1 activation was shown to be dependent on a CaMKII-dependent phosphorylation.19 This phosphorylation is further regulated by a modulatory action of protein phosphatase 2A, which has been found physically-associated with bestrophin.53 Finally, the regulation of bestrophin-associated CaCCs could involve the phosphorylation of an accessory subunit, which is as yet unidentified. Very recently, however, novel evidence has been presented to suggest that bestrophins can also form homotetrameric channels and perform highly-regulated CaCC activity independently of other proteins.7
The Expression of Bestrophins in Vascular Wall is Modulated by TMEM16A: A Challenge for Structure-Function Analyses
A recent study of siRNA-induced downregulation of TMEM16A demonstrated that both the bestrophin-associated cGMP-dependent ICl(Ca) and the classical ICl(Ca) disappeared.16 Based on these results it is tempting to suggest that TMEM16A might be the channel-forming protein and association with bestrophin generates the cGMP-dependent ICl(Ca) (Fig. 1). This suggestion is, however, complicated by the demonstrated ability of TMEM16A to modulate the expression of other membrane proteins including bestrophins and L-type Ca2+ channels.16 It is therefore not clear how the expression of bestrophins is essential for the cGMP-dependent ICl(Ca) and whether TMEM16A downregulation affects this current indirectly via reduction in bestrophin expression. Other experimental models, e.g., TMEM16A knockout mice,33 will be helpful in addressing this question.
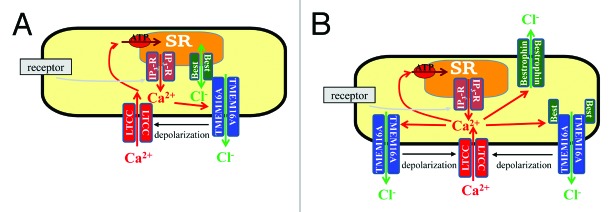
Figure 1. Agonist-induced Ca2+-release activates the ICl(Ca) which in term depolarises VSMCs leading to influx of Ca2+ through voltage-gated L-type Ca2+ channels (LTCC) and consequent force development. It has been suggested that TMEM16A is the pore-forming subunit responsible for ICl(Ca). Bestrophin has been suggested to act as sarcoplasmic reticulum (SR)-located channel that serves as a countercurrent for Ca2+ movement (A). Other data suggest that bestrophin may be either a subunit of a TMEM16A-formed channel, which modifies the biophysical and pharmacological characteristics of the TMEM16A-associated CaCCs or forms the CaCCs with characteristics distinct from the TMEM16A-associated CaCCs (B). Further studies are needed to test for these possibilities.
The mechanism by which TMEM16A affects expression of other genes and proteins is unknown but might involve protein kinase-dependent signaling pathways. In fact, bestrophin-3 expression in renal epithelial cells has been shown to be modulated by extracellular-signal-regulated kinases (ERK1/2).50 This ERK1/2 signaling is in turn known as the mechanism involved in TMEM16A-induced cancer cell proliferation.22 Interestingly, the small-molecular inhibitors (T16Ainh-A0122 and CaCCinh-A018) have been shown to abrogate the tumorigenic action of TMEM16A, but their mechanism of action is not fully understood, as discussed above. While the effect of CaCCinh-A01is independent from CaCC activity,8 the pore-forming region is necessary for successful inhibition of ERK1/2 signaling by T16Ainh-A01.22 The involvement of ERK1/2 signaling for the control of bestrophin expression by TMEM16A remains to be shown; nevertheless, ERK1/2 signaling is known to be active in VSMCs. It is tempting to consider that similar mechanism(s) have a place in regulation of the function and growth of the vascular wall. Moreover, TMEM16A has also been shown to activate nuclear factor–kB (NF-kB), which mediates the expression of numerous genes.51 Whether bestrophins are also modulated by NF-kB is unknown. Moreover, whether NF-kB signaling in VSMCs is under control of TMEM16A is also unknown.
There is no molecular evidence to date that TMEM16A can work directly as a transcription regulator but it is possible that proteolytically processed part(s) of TMEM16A may act as gene transcription factor. A similar function has previously been suggested for a C-terminal segment of voltage-gated L-type Ca2+ channels in neurons and VSMCs.3,26 Recent evidence from siRNA-mediated downregulation of TMEM16A suggests that TMEM16A can modulate the expression of L-type Ca2+ channels.16 This is inconsistent with a recently reported tamoxifen-induced VSMC-specific TMEM16A knockout where voltage-dependent Ca2+ influx was unaffected.33 However, in this knockout study the ICl(Ca) was abolished suggesting a defective CaCC function but a truncated TMEM16A protein was still present in VSMCs and this could potentially be sufficient for gene transcription regulation.
Finally, it is possible that the expression-related effect of TMEM16A is indirectly mediated via changes in intracellular spatio-temporal Ca2+ signaling, affecting the Ca2+-dependent transcription factors in VSMCs.57 Although it remains to be found which gene(s) are modulated by TMEM16A and the mechanism behind this modulation, lessons from cancer cell proliferation studies and the recent demonstration of TMEM16A regulation of gene expression in VSMCs16 clearly indicate that TMEM16A serves as a modulator for gene transcription.
Although the role of TMEM16A in modulation of gene expression is completely unexplored, the importance of this mechanism can be highlighted by the involvement of TMEM16A expression in several physiological and pathophysiological changes in the vascular wall. Thus, it has been shown that hypoxia-induced pulmonary artery hypertrophy and experimental pulmonary hypertension are associated with an increase in TMEM16A-associated ICl(Ca) and TMEM16A protein expression.23,92 Whether these changes are primary or secondary to the remodeling-associated changes in gene expression is not known, but TMEM16A has been suggested to be a negative regulator of VSMC proliferation in hypertension-induced cerebrovascular remodeling.99 This importance of TMEM16A for arterial structure can vary between different vascular beds in addition to being a stimulus-specific response. Interestingly, no structural changes in peripheral arteries have been reported for the recently published TMEM16A knockout mice.33 Further studies are needed to clarify the role of TMEM16A for arterial structure.
TMEM16A and Bestrophins can Interact in the Vascular Wall
There are several potential modes of interaction between TMEM16A and bestrophins (Fig. 1). One possibility is that bestrophin are intracellular channels in the sarcoplasmic reticulum membrane providing countercurrent for Ca2+ release, which in turn stimulates TMEM16A-formed CaCCs.6,44 However, voltage-clamp studies where bestrophin-associated ICl(Ca) was measured do not provide experimental support for this model.4,5,15,19,29,55,58,66,73,78,79,81-85,96,97,102 Indeed, the bestrophin-associated cGMP-dependent ICl(Ca) was elicited independently from the sarcoplasmic reticulum by elevating [Ca2+]i via patch pipette solution and via membrane permeabilization for Ca2+.58 Although it cannot entirely be ruled out that the high cytosolic Ca2+ leads to increased release of Ca2+ from the sarcoplasmic reticulum6 these experiments58 together with single channel recordings,17 suggest that bestrophin is directly associated with a plasma membrane conductance. Whether this is also the case for all the different bestrophin isoforms is unclear.
There are two possibilities for bestrophin functions in the cell membrane (Fig. 1). Bestrophins could be responsible for atypical ICl(Ca) currents, i.e., the cGMP-dependent ICl(Ca), while TMEM16A proteins could be responsible for the classical ICl(Ca). This possibility cannot be tested easily as the expression of TMEM16A modulates bestrophin expression,16 as discussed above. Moreover, heterologous expression of bestrophins may affect endogenous currents. The bestrophin expression profile has not yet been compared between wild-type and TMEM16A knockout mice.33 Another possibility is that bestrophin forms a regulatory subunit modifying the biophysical and pharmacological properties of a TMEM16A-formed channel (Fig. 1): similar modulation has been shown for the inward-rectifying K+ channels, which change their electrophysiology and pharmacology substantially after binding to the sulfonylurea receptor subunit to form ATP-dependent K+ channels.86 Testing of this possibility for interaction between TMEM16A and bestrophin is difficult due to technical difficulties in independently modulating the expression of TMEM16A and bestrophin proteins. The majority of cells used for heterologous expression studies possess an endogenous CaCC and express either bestrophins or TMEM16A or both.30,31 A CaCC-free expressional system, such as axolotl (Ambystoma Mexicanum) oocytes,88 would be suitable for addressing this question. Importantly, recent single-molecule subunit analysis suggested that bestrophins are preferentially homotetrameric7 channels which have little or no interaction with each other and TMEM16A. Similarly, homotypic self-assembly into CaCCs was shown for TMEM16A.94 Whether these proteins interact with each other remains to be studied.
What is the Role of the Ca2+-Activated Cl- Conductance for Arterial Contraction?
Since Cl– is actively pumped into VSMCs and opening of Cl– channels consequently produces membrane depolarization, the CaCCs have been suggested to participate in the excitation-contraction coupling where they link [Ca2+]i and membrane potential changes.48,49 An agonist-induced Ca2+ increase leads to CaCC-dependent membrane depolarization, opening of voltage-gated Ca2+ channels and further increases in [Ca2+]i. This amplifying mechanism has been suggested to be important for agonist-induced contraction of VSMCs. If the Cl– conductance is important for contraction, the contraction should depend on the Cl– gradient across the plasma membrane. Several studies have shown that replacement of extracellular Cl– with impermeable anions acutely amplifies agonist-induced depolarization and contraction45,46,64 but this is not always the case. Substitution of extracellular Cl– was shown to have only mild effect on both resting membrane potential and membrane potential in noradrenaline-stimulated rat mesenteric small arteries65 and no significant effect on contraction.9 Nevertheless, a variable significance of Cl– for VSMC contraction depending on the type of vasculature and stimulation can be suggested.46 In fact, this is consistent with recent results from TMEM16A knockout mice showing a lack of effect of knockout in mesenteric arteries, while in other blood vessels, e.g., aorta and carotid artery, the contractile response was significantly diminished by TMEM16A knockout.33
The Functional Significance of TMEM16A in the Vascular Wall
The lack of the effect of downregulation of bestrophin-associated cGMP-dependent ICl(Ca) on force development supported the suggestion that Cl– conductance has little role for agonist-induced contraction of rat mesenteric small arteries.10 However, when the TMEM16A-associated classical ICl(Ca) and cGMP-dependent ICl(Ca) were downregulated, the arteries depolarized significantly less to noradrenaline, and the [Ca2+]i increase and contraction were reduced16 as was the response to vasopressin in these arteries. If this effect was due to lower CaCC-dependent depolarization, it should be rescued by depolarizing the arterial wall but this was not the case: KCl-induced depolarization and contraction were significantly reduced in the TMEM16A-downregulated arteries.16 Furthermore, in the same experimental series, when the membrane potential was similar, [Ca2+]i was lower in the arteries where TMEM16A was downregulated. Taken together these functional results suggest that TMEM16A may modify voltage-dependent Ca2+ influx independently of the membrane potential.16 Importantly, these results do not exclude CaCCs from having a role in VSMC depolarization but rather suggest that this role is not exclusive, at least in rat mesenteric arteries.
In contrast, the agonist-induced contractions of other arteries have been shown to be strongly dependent on CaCC depolarizations with no indication of the involvement of voltage-gated Ca2+ channels. Thus, in functional studies on mouse thoracic aorta and mesenteric artery as well as human abdominal visceral adipose artery a novel blocker of TMEM16A-associated CaCCs, e.g., T16Ainh-A01, relaxed preconstricted arteries in vitro.17 T16Ainh-A01 was without significant effect on arterial contraction induced by 60 mM KCl.17 This contrasts with the findings in rat mesenteric arteries discussed above.16 This could be due to a chronic effect of siRNA-induced downregulation that reduces voltage-gated Ca2+ channels vs. an acute pharmacological effect of the blocker. However, siRNA-induced downregulation of TMEM16A in rat cerebral artery organ culture does not support this suggestion.12 Rat cerebral arteries downregulated for TMEM16A with siRNA demonstrate a reduction in pressure-induced depolarization and myogenic vasoconstriction, but the contraction to 60 mM KCl depolarization was unaffected. However, the authors suggested that Ca2+ influx via mechanosensitive nonselective cation channels stimulates TMEM16A-associated channels to induce myogenic vasoconstriction and demonstrated no importance of voltage-dependent Ca2+-influx or Ca2+-release for activation of TMEM16A-associated channels.12
The reason for this inconsistency is unclear and could reflect different roles of CaCC in different vascular beds. In fact, it has recently been shown that mouse mesenteric small arteries have a very weak expression of TMEM16A and an ICl(Ca) of low amplitude.33 Consistently, an inducible VSMC-specific knockout of TMEM16A in mice was without any effect on mesenteric small artery contractility. In contrast, larger blood vessels, e.g., aorta and carotid artery from TMEM16A knockout mice, had reduced contractility to angiotensin II and U46619 (thromboxane analog) compared with their wild type counterparts.33 These larger arteries from wild type mice have a large TMEM16A-associated ICl(Ca). It is of interest to determine whether the responses to adrenergic agonists are also reduced in the larger vessels. Knockout of TMEM16A had no effect on the contraction to 60 mM KCl suggesting that the expression of voltage-gated Ca2+ channels was unaffected in this model.33 However, as mentioned above, although the ICl(Ca) was gone a significant part of the truncated TMEM16A protein was still present in the VSMCs which could be relevant for function.
Concluding Remarks
The current evidence indicates that TMEM16A is essential for generation of ICl(Ca) suggesting that this protein is a channel-forming subunit of CaCCs.12,16,17,23,52,67,93 It remains to be determined whether TMEM16A is associated with different forms of CaCCs and whether other ICl(Ca)-associated proteins, e.g., bestrophins, serve as regulatory proteins that modify the channel’s properties. This challenge is hampered by the surprising finding that TMEM16A expression is important for the expression of other genes, such as bestrophins, which are associated with some forms of the ICl(Ca), and voltage-gated Ca2+ channels.16 These non-contractile functions of TMEM16A protein in VSMCs further complicate the analysis of CaCC function in the vascular wall. It seems that the expression and the function (either membrane depolarization or modulation of voltage-gated Ca2+ channels) of TMEM16A vary in different vascular beds and thus may greatly influence experimental results. There is however a general consensus that the TMEM16A-associated ICl(Ca) is an important component of VSMC function and is important for maintenance of total peripheral resistance and, thus, blood pressure.12,16,17,33
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study was supported by the Danish Research Council, the Danish Heart Foundation and the Lundbeck Foundation.
Glossary
Abbreviations:
- CaCC
Ca2+-activated Cl– channel
- cGMP
cyclic guanosine monophosphate
- BKCa
Ca2+-activated K+ channel
- ICl(Ca)
Ca2+-activated Cl– current
- LTCC
L-type Ca2+ channel
- PKG
protein kinase G
- siRNA
small interference RNA
- SR
sarcoplasmic reticulum
- VSMC
vascular smooth muscle cell
References
- 1.Aickin CC, Vermuë NA. Microelectrode measurement of intracellular chloride activity in smooth muscle cells of guinea-pig ureter. Pflugers Arch. 1983;397:25–8. doi: 10.1007/BF00585163. [DOI] [PubMed] [Google Scholar]
- 2.Alioua A, Lu R, Kumar Y, Eghbali M, Kundu P, Toro L, Stefani E. Slo1 caveolin-binding motif, a mechanism of caveolin-1-Slo1 interaction regulating Slo1 surface expression. J Biol Chem. 2008;283:4808–17. doi: 10.1074/jbc.M709802200. [DOI] [PubMed] [Google Scholar]
- 3.Bannister JP, Leo MD, Narayanan D, Jangsangthong W, Nair A, Evanson KW, Pachuau J, Gabrick KS, Boop FA, Jaggar JH. The voltage-dependent L-type Ca2+ (CaV1.2) channel C-terminus fragment is a bi-modal vasodilator. J Physiol. 2013;591:2987–98. doi: 10.1113/jphysiol.2013.251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barro-Soria R, Schreiber R, Kunzelmann K. Bestrophin 1 and 2 are components of the Ca(2+) activated Cl(-) conductance in mouse airways. Biochim Biophys Acta. 2008;1783:1993–2000. doi: 10.1016/j.bbamcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Barro Soria R, Spitzner M, Schreiber R, Kunzelmann K. Bestrophin-1 enables Ca2+-activated Cl- conductance in epithelia. J Biol Chem. 2009;284:29405–12. doi: 10.1074/jbc.M605716200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barro-Soria R, Aldehni F, Almaça J, Witzgall R, Schreiber R, Kunzelmann K. ER-localized bestrophin 1 activates Ca2+-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Arch. 2010;459:485–97. doi: 10.1007/s00424-009-0745-0. [DOI] [PubMed] [Google Scholar]
- 7.Bharill S, Fu Z, Palty R, Isacoff EY. Stoichiometry and specific assembly of Best ion channels. Proc Natl Acad Sci U S A. 2014;111:6491–6. doi: 10.1073/pnas.1400248111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bill A, Hall ML, Borawski J, Hodgson C, Jenkins J, Piechon P, Popa O, Rothwell C, Tranter P, Tria S, et al. Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J Biol Chem. 2014;289:11029–41. doi: 10.1074/jbc.M114.549188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boedtkjer DM, Matchkov VV, Boedtkjer E, Nilsson H, Aalkjaer C. Vasomotion has chloride-dependency in rat mesenteric small arteries. Pflugers Arch. 2008;457:389–404. doi: 10.1007/s00424-008-0532-3. [DOI] [PubMed] [Google Scholar]
- 10.Broegger T, Jacobsen JC, Secher Dam V, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV. Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res. 2011;91:685–93. doi: 10.1093/cvr/cvr111. [DOI] [PubMed] [Google Scholar]
- 11.Simon Bulley SB, Jaggar JH. Cl⁻ channels in smooth muscle cells. Pflugers Arch. 2014;466:861–72. doi: 10.1007/s00424-013-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulley S, Neeb ZP, Burris SK, Bannister JP, Thomas-Gatewood CM, Jangsangthong W, Jaggar JH. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ Res. 2012;111:1027–36. doi: 10.1161/CIRCRESAHA.112.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrne NG, Large WA. Membrane mechanism associated with muscarinic receptor activation in single cells freshly dispersed from the rat anococcygeus muscle. Br J Pharmacol. 1987;92:371–9. doi: 10.1111/j.1476-5381.1987.tb11333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–4. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 15.Chien LT, Zhang ZR, Hartzell HC. Single Cl- channels activated by Ca2+ in Drosophila S2 cells are mediated by bestrophins. J Gen Physiol. 2006;128:247–59. doi: 10.1085/jgp.200609581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dam VS, Boedtkjer DM, Nyvad J, Aalkjaer C, Matchkov V. TMEM16A knockdown abrogates two different Ca(2+)-activated Cl (-) currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch. 2014;466:1391–409;. doi: 10.1007/s00424-013-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis AJ, Shi J, Pritchard HA, Chadha PS, Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP, Greenwood IA. Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) -A01. Br J Pharmacol. 2013;168:773–84. doi: 10.1111/j.1476-5381.2012.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73:758–68. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 19.Duran C, Chien LT, Hartzell HC. Drosophila bestrophin-1 currents are regulated by phosphorylation via a CaMKII dependent mechanism. PLoS One. 2013;8:e58875. doi: 10.1371/journal.pone.0058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duta V, Duta F, Puttagunta L, Befus AD, Duszyk M. Regulation of basolateral Cl(-) channels in airway epithelial cells: the role of nitric oxide. J Membr Biol. 2006;213:165–74. doi: 10.1007/s00232-006-0062-x. [DOI] [PubMed] [Google Scholar]
- 21.Duta V, Szkotak AJ, Nahirney D, Duszyk M. The role of bestrophin in airway epithelial ion transport. FEBS Lett. 2004;577:551–4. doi: 10.1016/j.febslet.2004.10.068. [DOI] [PubMed] [Google Scholar]
- 22.Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72:3270–81. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forrest AS, Joyce TC, Huebner ML, Ayon RJ, Wiwchar M, Joyce J, Freitas N, Davis AJ, Ye L, Duan DD, et al. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol. 2012;303:C1229–43. doi: 10.1152/ajpcell.00044.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frings S, Reuter D, Kleene SJ. Neuronal Ca2+ -activated Cl- channels--homing in on an elusive channel species. Prog Neurobiol. 2000;60:247–89. doi: 10.1016/S0301-0082(99)00027-1. [DOI] [PubMed] [Google Scholar]
- 25.Fuller CM, Ji HL, Tousson A, Elble RC, Pauli BU, Benos DJ. Ca(2+)-activated Cl(-) channels: a newly emerging anion transport family. Pflugers Arch. 2001;443(Suppl 1):S107–10. doi: 10.1007/s004240100655. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenwood IA, Large WA. Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. Br J Pharmacol. 1995;116:2939–48. doi: 10.1111/j.1476-5381.1995.tb15948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl- and K+ channels. Trends Pharmacol Sci. 2007;28:1–5. doi: 10.1016/j.tips.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Han X, Qu Z, Xie J, Jiang H. C-terminal membrane association of Bestrophin 3 and its activation as a chloride channel. J Membr Biol. 2013;246:177–82. doi: 10.1007/s00232-012-9514-7. [DOI] [PubMed] [Google Scholar]
- 30.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–58. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 31.Hartzell HC. Physiology. CaCl-ing channels get the last laugh. Science. 2008;322:534–5. doi: 10.1126/science.1165668. [DOI] [PubMed] [Google Scholar]
- 32.He Y, Tabrizchi R. Effects of niflumic acid on alpha1-adrenoceptor-induced vasoconstriction in mesenteric artery in vitro and in vivo in two-kidney one-clip hypertensive rats. Eur J Pharmacol. 1997;328:191–9. doi: 10.1016/S0014-2999(97)83045-2. [DOI] [PubMed] [Google Scholar]
- 33.Heinze C, Seniuk A, Sokolov MV, Huebner AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, Vitzthum H, Gollasch M, Ehmke H, et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest. 2014;124:675–86. doi: 10.1172/JCI70025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helliwell RM, Wang Q, Hogg RC, Large WA. Synergistic action of histamine and adenosine triphosphate on the response to noradrenaline in rabbit pulmonary artery smooth muscle cells. Pflugers Arch. 1994;426:433–9. doi: 10.1007/BF00388307. [DOI] [PubMed] [Google Scholar]
- 35.Hogg RC, Wang Q, Large WA. Action of niflumic acid on evoked and spontaneous calcium-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol. 1994;112:977–84. doi: 10.1111/j.1476-5381.1994.tb13177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY. Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron. 2012;74:179–92. doi: 10.1016/j.neuron.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobsen JC, Aalkjaer C, Nilsson H, Matchkov VV, Freiberg J, Holstein-Rathlou NH. Activation of a cGMP-sensitive calcium-dependent chloride channel may cause transition from calcium waves to whole cell oscillations in smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;293:H215–28. doi: 10.1152/ajpheart.00726.2006. [DOI] [PubMed] [Google Scholar]
- 38.Jeon JH, Paik SS, Chun MH, Oh U, Kim IB. Presynaptic Localization and Possible Function of Calcium-Activated Chloride Channel Anoctamin 1 in the Mammalian Retina. PLoS One. 2013;8:e67989. doi: 10.1371/journal.pone.0067989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin X, Shah S, Liu Y, Zhang H, Lees M, Fu Z, Lippiat JD, Beech DJ, Sivaprasadarao A, Baldwin SA, et al. Activation of the Cl- channel ANO1 by localized calcium signals in nociceptive sensory neurons requires coupling with the IP3 receptor. Sci Signal. 2013;6:ra73. doi: 10.1126/scisignal.2004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirkup AJ, Edwards G, Green ME, Miller M, Walker SD, Weston AH. Modulation of membrane currents and mechanical activity by niflumic acid in rat vascular smooth muscle. Eur J Pharmacol. 1996;317:165–74. doi: 10.1016/S0014-2999(96)00713-3. [DOI] [PubMed] [Google Scholar]
- 41.Klöckner U. Intracellular calcium ions activate a low-conductance chloride channel in smooth-muscle cells isolated from human mesenteric artery. Pflugers Arch. 1993;424:231–7. doi: 10.1007/BF00384347. [DOI] [PubMed] [Google Scholar]
- 42.Klöckner U, Isenberg G. Endothelin depolarizes myocytes from porcine coronary and human mesenteric arteries through a Ca-activated chloride current. Pflugers Arch. 1991;418:168–75. doi: 10.1007/BF00370467. [DOI] [PubMed] [Google Scholar]
- 43.Krämer F, Stöhr H, Weber BH. Cloning and characterization of the murine Vmd2 RFP-TM gene family. Cytogenet Genome Res. 2004;105:107–14. doi: 10.1159/000078016. [DOI] [PubMed] [Google Scholar]
- 44.Kunzelmann K, Kongsuphol P, Chootip K, Toledo C, Martins JR, Almaca J, Tian Y, Witzgall R, Ousingsawat J, Schreiber R. Role of the Ca2+ -activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol Chem. 2011;392:125–34. doi: 10.1515/bc.2011.010. [DOI] [PubMed] [Google Scholar]
- 45.Lamb FS, Barna TJ. Chloride ion currents contribute functionally to norepinephrine-induced vascular contraction. Am J Physiol. 1998;275:H151–60. doi: 10.1152/ajpheart.1998.275.1.H151. [DOI] [PubMed] [Google Scholar]
- 46.Lamb FS, Kooy NW, Lewis SJ. Role of Cl(-) channels in alpha-adrenoceptor-mediated vasoconstriction in the anesthetized rat. Eur J Pharmacol. 2000;401:403–12. doi: 10.1016/S0014-2999(00)00471-4. [DOI] [PubMed] [Google Scholar]
- 47.Lamb FS, Volk KA, Shibata EF. Calcium-activated chloride current in rabbit coronary artery myocytes. Circ Res. 1994;75:742–50. doi: 10.1161/01.RES.75.4.742. [DOI] [PubMed] [Google Scholar]
- 48.Large WA, Wang Q. Characteristics and physiological role of the Ca(2+)-activated Cl- conductance in smooth muscle. Am J Physiol. 1996;271:C435–54. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- 49.Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O’Driscoll K, Britton F, Perrino BA, Greenwood IA. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–56. doi: 10.1139/y05-040. [DOI] [PubMed] [Google Scholar]
- 50.Lee WK, Chakraborty PK, Roussa E, Wolff NA, Thévenod F. ERK1/2-dependent bestrophin-3 expression prevents ER-stress-induced cell death in renal epithelial cells by reducing CHOP. Biochim Biophys Acta. 2012;1823:1864–76. doi: 10.1016/j.bbamcr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Liu Y, Ren Y, Kang L, Zhang L. Transmembrane protein with unknown function 16A overexpression promotes glioma formation through the nuclear factor-κB signaling pathway. Mol Med Rep. 2014;9:1068–74. doi: 10.3892/mmr.2014.1888. [DOI] [PubMed] [Google Scholar]
- 52.Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–14. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marmorstein LY, McLaughlin PJ, Stanton JB, Yan L, Crabb JW, Marmorstein AD. Bestrophin interacts physically and functionally with protein phosphatase 2A. J Biol Chem. 2002;277:30591–7. doi: 10.1074/jbc.M204269200. [DOI] [PubMed] [Google Scholar]
- 54.Matchkov VV. Mechanisms of cellular synchronization in the vascular wall. Mechanisms of vasomotion. Dan Med Bull. 2010;57:B4191. [PubMed] [Google Scholar]
- 55.Matchkov VV, Aalkjaer C, Nilsson H. A cyclic GMP-dependent calcium-activated chloride current in smooth-muscle cells from rat mesenteric resistance arteries. J Gen Physiol. 2004;123:121–34. doi: 10.1085/jgp.200308972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matchkov VV, Aalkjaer C, Nilsson H. Distribution of cGMP-dependent and cGMP-independent Ca(2+)-activated Cl(-) conductances in smooth muscle cells from different vascular beds and colon. Pflugers Arch. 2005;451:371–9. doi: 10.1007/s00424-005-1472-9. [DOI] [PubMed] [Google Scholar]
- 57.Matchkov VV, Kudryavtseva O, Aalkjaer C. Intracellular Ca²⁺ signalling and phenotype of vascular smooth muscle cells. Basic Clin Pharmacol Toxicol. 2012;110:42–8. doi: 10.1111/j.1742-7843.2011.00818.x. [DOI] [PubMed] [Google Scholar]
- 58.Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H. Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res. 2008;103:864–72. doi: 10.1161/CIRCRESAHA.108.178517. [DOI] [PubMed] [Google Scholar]
- 59.Matchkov VV, Secher Dam V, Bødtkjer DM, Aalkjær C. Transport and function of chloride in vascular smooth muscles. J Vasc Res. 2013;50:69–87. doi: 10.1159/000345242. [DOI] [PubMed] [Google Scholar]
- 60.Mazzone A, Eisenman ST, Strege PR, Yao Z, Ordog T, Gibbons SJ, Farrugia G. Inhibition of cell proliferation by a selective inhibitor of the Ca(2+)-activated Cl(-) channel, Ano1. Biochem Biophys Res Commun. 2012;427:248–53. doi: 10.1016/j.bbrc.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milenkovic VM, Krejcova S, Reichhart N, Wagner A, Strauss O. Interaction of bestrophin-1 and Ca2+ channel β-subunits: identification of new binding domains on the bestrophin-1 C-terminus. PLoS One. 2011;6:e19364. doi: 10.1371/journal.pone.0019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Min SA, Stapleton MP, Tabrizchi R. Influence of chloride ions on alpha1-adrenoceptor mediated contraction and Ca2+ influx in rat caudal artery. Life Sci. 1999;64:1631–41. doi: 10.1016/S0024-3205(99)00100-9. [DOI] [PubMed] [Google Scholar]
- 63.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–74. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J Physiol. 1997;502:259–64. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nilsson H, Videbaek LM, Toma C, Mulvany MJ. Role of intracellular calcium for noradrenaline-induced depolarization in rat mesenteric small arteries. J Vasc Res. 1998;35:36–44. doi: 10.1159/000025563. [DOI] [PubMed] [Google Scholar]
- 66.O’Driscoll KE, Hatton WJ, Burkin HR, Leblanc N, Britton FC. Expression, localization, and functional properties of Bestrophin 3 channel isolated from mouse heart. Am J Physiol Cell Physiol. 2008;295:C1610–24. doi: 10.1152/ajpcell.00461.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Driscoll KE, Pipe RA, Britton FC. Increased complexity of Tmem16a/Anoctamin 1 transcript alternative splicing. BMC Mol Biol. 2011;12:35. doi: 10.1186/1471-2199-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oh SJ, Hwang SJ, Jung J, Yu K, Kim J, Choi JY, Hartzell HC, Roh EJ, Lee CJ. MONNA, a potent and selective blocker for transmembrane protein with unknown function 16/anoctamin-1. Mol Pharmacol. 2013;84:726–35. doi: 10.1124/mol.113.087502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohshiro J, Yamamura H, Saeki T, Suzuki Y, Imaizumi Y. The multiple expression of Ca²⁺-activated Cl⁻ channels via homo- and hetero-dimer formation of TMEM16A splicing variants in murine portal vein. Biochem Biophys Res Commun. 2014;443:518–23. doi: 10.1016/j.bbrc.2013.11.117. [DOI] [PubMed] [Google Scholar]
- 70.Ottolia M, Toro L. Potentiation of large conductance KCa channels by niflumic, flufenamic, and mefenamic acids. Biophys J. 1994;67:2272–9. doi: 10.1016/S0006-3495(94)80712-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Owen NE. Regulation of Na/K/Cl cotransport in vascular smooth muscle cells. Biochem Biophys Res Commun. 1984;125:500–8. doi: 10.1016/0006-291X(84)90568-0. [DOI] [PubMed] [Google Scholar]
- 72.Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ Res. 2001;88:810–5. doi: 10.1161/hh0801.089603. [DOI] [PubMed] [Google Scholar]
- 73.Pifferi S, Pascarella G, Boccaccio A, Mazzatenta A, Gustincich S, Menini A, Zucchelli S. Bestrophin-2 is a candidate calcium-activated chloride channel involved in olfactory transduction. Proc Natl Acad Sci U S A. 2006;103:12929–34. doi: 10.1073/pnas.0604505103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piper AS, Greenwood IA, Large WA. Dual effect of blocking agents on Ca2+-activated Cl(-) currents in rabbit pulmonary artery smooth muscle cells. J Physiol. 2002;539:119–31. doi: 10.1113/jphysiol.2001.013270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Piper AS, Large WA. Direct effect of Ca2+-calmodulin on cGMP-activated Ca2+-dependent Cl-channels in rat mesenteric artery myocytes. J Physiol. 2004;559:449–57. doi: 10.1113/jphysiol.2004.070045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Piper AS, Large WA. Single cGMP-activated Ca(+)-dependent Cl(-) channels in rat mesenteric artery smooth muscle cells. J Physiol. 2004;555:397–408. doi: 10.1113/jphysiol.2003.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pusch M. Ca(2+)-activated chloride channels go molecular. J Gen Physiol. 2004;123:323–5. doi: 10.1085/jgp.200409053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qu Z, Cui Y, Hartzell C. A short motif in the C-terminus of mouse bestrophin 3 [corrected] inhibits its activation as a Cl channel. FEBS Lett. 2006;580:2141–6. doi: 10.1016/j.febslet.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 79.Qu Z, Fischmeister R, Hartzell C. Mouse bestrophin-2 is a bona fide Cl(-) channel: identification of a residue important in anion binding and conduction. J Gen Physiol. 2004;123:327–40. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qu Z, Han X, Cui Y, Li C. A PI3 kinase inhibitor found to activate bestrophin 3. J Cardiovasc Pharmacol. 2010;55:110–5. doi: 10.1097/FJC.0b013e3181c87c85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qu Z, Hartzell C. Determinants of anion permeation in the second transmembrane domain of the mouse bestrophin-2 chloride channel. J Gen Physiol. 2004;124:371–82. doi: 10.1085/jgp.200409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qu Z, Hartzell HC. Bestrophin Cl- channels are highly permeable to HCO3- Am J Physiol Cell Physiol. 2008;294:C1371–7. doi: 10.1152/ajpcell.00398.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qu Z, Wei RW, Mann W, Hartzell HC. Two bestrophins cloned from Xenopus laevis oocytes express Ca(2+)-activated Cl(-) currents. J Biol Chem. 2003;278:49563–72. doi: 10.1074/jbc.M308414200. [DOI] [PubMed] [Google Scholar]
- 84.Qu Z, Chien LT, Cui Y, Hartzell HC. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J Neurosci. 2006;26:5411–9. doi: 10.1523/JNEUROSCI.5500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qu ZQ, Yu K, Cui YY, Ying C, Hartzell C. Activation of bestrophin Cl- channels is regulated by C-terminal domains. J Biol Chem. 2007;282:17460–7. doi: 10.1074/jbc.M701043200. [DOI] [PubMed] [Google Scholar]
- 86.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 87.Remillard CV, Lupien MA, Crépeau V, Leblanc N. Role of Ca2+- and swelling-activated Cl- channels in alpha1-adrenoceptor-mediated tone in pressurized rabbit mesenteric arterioles. Cardiovasc Res. 2000;46:557–68. doi: 10.1016/S0008-6363(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 88.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–29. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shmygol A, Noble K, Wray S. Depletion of membrane cholesterol eliminates the Ca2+-activated component of outward potassium current and decreases membrane capacitance in rat uterine myocytes. J Physiol. 2007;581:445–56. doi: 10.1113/jphysiol.2007.129452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sones WR, Davis AJ, Leblanc N, Greenwood IA. Cholesterol depletion alters amplitude and pharmacology of vascular calcium-activated chloride channels. Cardiovasc Res. 2010;87:476–84. doi: 10.1093/cvr/cvq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Srivastava A, Romanenko VG, Gonzalez-Begne M, Catalán MA, Melvin JE. A variant of the Ca2+-activated Cl channel Best3 is expressed in mouse exocrine glands. J Membr Biol. 2008;222:43–54. doi: 10.1007/s00232-008-9098-4. [DOI] [PubMed] [Google Scholar]
- 92.Sun H, Xia Y, Paudel O, Yang XR, Sham JS. Chronic hypoxia-induced upregulation of Ca2+-activated Cl- channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol. 2012;590:3507–21. doi: 10.1113/jphysiol.2012.232520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thomas-Gatewood C, Neeb ZP, Bulley S, Adebiyi A, Bannister JP, Leo MD, Jaggar JH. TMEM16A channels generate Ca²⁺-activated Cl⁻ currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2011;301:H1819–27. doi: 10.1152/ajpheart.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tien J, Lee HY, Minor DL, Jr., Jan YN, Jan LY. Identification of a dimerization domain in the TMEM16A calcium-activated chloride channel (CaCC) Proc Natl Acad Sci U S A. 2013;110:6352–7. doi: 10.1073/pnas.1303672110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Toma C, Greenwood IA, Helliwell RM, Large WA. Activation of potassium currents by inhibitors of calcium-activated chloride conductance in rabbit portal vein smooth muscle cells. Br J Pharmacol. 1996;118:513–20. doi: 10.1111/j.1476-5381.1996.tb15432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsunenari T, Nathans J, Yau KW. Ca2+-activated Cl- current from human bestrophin-4 in excised membrane patches. J Gen Physiol. 2006;127:749–54. doi: 10.1085/jgp.200609527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW, Nathans J. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem. 2003;278:41114–25. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–71. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang M, Yang H, Zheng LY, Zhang Z, Tang YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG, et al. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. 2012;125:697–707. doi: 10.1161/CIRCULATIONAHA.111.041806. [DOI] [PubMed] [Google Scholar]
- 100.Ward SM, Kenyon JL. The spatial relationship between Ca2+ channels and Ca2+-activated channels and the function of Ca2+-buffering in avian sensory neurons. Cell Calcium. 2000;28:233–46. doi: 10.1054/ceca.2000.0151. [DOI] [PubMed] [Google Scholar]
- 101.Wu MM, Lou J, Song BL, Gong YF, Li YC, Yu CJ, Wang QS, Ma TX, Ma K, Hartzell HC, et al. Hypoxia augments anoctamin 1-encoded calcium-activated chloride current in cardiac vascular endothelial cells of neonatal mouse. Br J Pharmacol. 2014 doi: 10.1111/bph.12730. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiao Q, Prussia A, Yu K, Cui YY, Hartzell HC. Regulation of bestrophin Cl channels by calcium: role of the C terminus. J Gen Physiol. 2008;132:681–92. doi: 10.1085/jgp.200810056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–5. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]