

Abstract
The metabolic network in the yeast Saccharomyces cerevisiae was reconstructed using currently available genomic, biochemical, and physiological information. The metabolic reactions were compartmentalized between the cytosol and the mitochondria, and transport steps between the compartments and the environment were included. A total of 708 structural open reading frames (ORFs) were accounted for in the reconstructed network, corresponding to 1035 metabolic reactions. Further, 140 reactions were included on the basis of biochemical evidence resulting in a genome-scale reconstructed metabolic network containing 1175 metabolic reactions and 584 metabolites. The number of gene functions included in the reconstructed network corresponds to ∼16% of all characterized ORFs in S. cerevisiae. Using the reconstructed network, the metabolic capabilities of S. cerevisiae were calculated and compared with Escherichia coli. The reconstructed metabolic network is the first comprehensive network for a eukaryotic organism, and it may be used as the basis for in silico analysis of phenotypic functions.
[Supplemental material is available online at www.genome.org. The detailed genome-scale reconstructed model of Saccharomyces cerevisiae can be found at http://www.cpb.dtu.dk/models/yeastmodel.html or http://geneticcircuits.ucsd.edu/organisms/yeast.html.]
Baker's yeast, Saccharomyces cerevisiae, was the first eukaryotic genome that was fully sequenced, annotated, and made publicly available. (Goffeau 1997). Along with its industrial importance, S. cerevisiae serves as a model organism for understanding and engineering eukaryotic cell function (Dujon 1996; Botstein et al. 1997). There have been many studies aiming to unravel the function of orphan genes in the genome (Oliver 1998; Entian et al. 1999; Winzeler et al. 1999; Hughes et al. 2000), and various functional genomics techniques were first implemented in S. cerevisiae. The first genome-wide cDNA array study was designed for S. cerevisiae (DeRisi et al. 1997), which subsequently resulted in a large number of studies on expression profiling (Hughes et al. 2000). Large-scale studies have been conducted to investigate the protein–protein interactions (Uetz et al. 2000), including the use of two-hybrid systems (Ito et al. 2001). These studies and a large body of biochemical literature now enable us to functionally integrate the wealth of available genetic, molecular, and biochemical information for S. cerevisiae.
Integration of knowledge at different levels in the cascade from genes to protein and further to metabolic fluxes in a genome-scale network will be pivotal for understanding how the individual components in the system interact and influence overall cell function. The approach of analyzing a complex process at different levels was illustrated in a recent study in which expression profiles in different mutants were compared with protein levels in order to unravel the structure of the complex galactose (GAL)–regulon (Ideker et al. 2001). This coordinated and multilevel effort may have significant influence on designing metabolic engineering strategies (Østergaard et al. 2000a,b). These interactions must now be quantified through the use of a mathematical framework—something that involves a significant research effort, but which is believed to lead to fundamental new insights into cellular function (Schilling et al. 1999; Endy and Brent 2001). To gain insight into cell synthesis and the metabolic capability through mathematical modeling, a natural first step is to reconstruct the underlying metabolic network, as this is responsible for the synthesis capacity of the cell, and, as well, it allows detailed analysis of the interactions between the individual pathways functioning in the cell. Recently, genome-scale metabolic networks were reconstructed for prokaryotic cells (Edwards and Palsson 1999; Covert et al. 2001), and it was demonstrated how such reconstructed metabolic models allow direct correlation between the genomic information and metabolic activity at the flux level. In these reconstructed metabolic networks, which consist of several hundred reactions and several hundred metabolites, it was possible to simulate the phenotypic behavior under different genetic conditions and physiological environments (Edwards et al. 2001).
Here, we present the reconstruction of the metabolic network of S. cerevisiae, the first genome-scale in silico metabolic network for a eukaryotic cell. Characteristics of eukaryotic cells, such as compartmentation of reactions and involvement of transport steps across cellular membranes, were considered in the network. The structure and metabolic capabilities of the metabolic network of S. cerevisiae were compared with a genome-scale reconstructed metabolic network of Escherichia coli (Edwards and Palsson 2000).
RESULTS AND DISCUSSION
Genome-Scale Metabolic Reconstruction
A genome-scale reconstruction of a metabolic network is currently a nonautomated and iterative decision-making process that can easily require up to one man-year to assemble a satisfying reaction list specifically collected for one organism. Once a metabolic network is reconstructed, mathematical methods, such as convex analysis and linear programming, can be applied to analyze structural properties, such as connectivity, etc., and simulation of cellular behavior under different genetic and physiological conditions can be conducted. For example, results may be used for the development of metabolic engineering strategies for the construction of strains with desired and improved properties. The present work focuses on the reconstruction of the metabolic network of the yeast S. cerevisiae. Some structural properties of the network and capabilities for biomass precursor and amino acid production by the network were evaluated.
During the reconstruction process, a number of decisions on each reaction needed to be taken (Fig. 1). Is an enzyme present in the organism? Which reaction catalyzes the enzyme, and what is the stoichiometry of that reaction? If cofactors are involved, is the reaction, for example, reduced nicotinamide-adenine dinucleotide (NADH) or reduced nicotinamide-adenine dinucleotide phosphate (NADPH) dependent, or can the enzyme use both cofactors? Is the reaction reversible or irreversible? Where is the reaction localized? Furthermore, for modeling purposes, information on the biomass composition, and on growth and nongrowth-dependent adenosine-5′-diphosphate (ATP) requirements has to be available. Once the metabolic pathways have been reconstructed or a complete and satisfying reaction list is available, the model can be used to simulate metabolic behavior. However, before it can be applied, the validity of the reaction list for modeling purposes has to be tested. For model validation, we compared computed results of anaerobic and aerobic chemostat cultivation at a dilution rate of 0.1 h−1 to experimental results from Nissen et al. (1997) and Overkamp et al. (2000) (data not shown).
Figure 1.

Reconstruction of the metabolic network of S. cerevisiae. Based on the available information from the genome annotation, biochemical pathway databases, biochemistry textbooks, and recent publications, a genome-scale metabolic network of S. cerevisiae was designed. Additional physiological constraints were considered and modeled, such as growth and nongrowth-dependent ATP requirements. Compartmentation was included, and cofactor requirements of all model reactions were inspected carefully, thereby, reactions that created a net transhydrogenic effect were additionally constrained. Regulatory information was not included. The picture of the pathway map was taken from the KEGG database (www.genome.ad.jp).
A genome-scale reconstruction is based on a thorough literature examination in order to extract the current state of the art on known metabolic reactions. Here, online pathway databases, biochemistry textbooks, and the annotated genome sequence have to be consulted, and, especially, extraction of information on metabolic reactions from journal publications is essential.
In detail, the reconstruction process for S. cerevisiae was initiated by downloading a gene catalog from the KEGG metabolic pathways database (http://kegg.genome.ad.jp/dbget-bin/get_htext?S.cerevisiae.kegg+-f+T+w+C). The information contained in this gene catalog is organized into traditional pathways, such as glycolysis, pentose phosphate pathway, any amino acid biosynthetic pathway, etc., and was used throughout the reconstruction process. Details are presented for the ORF name, gene name, enzyme name, EC number (if available), and SWISS-PROT entry name, and the KEGG metabolic chart may be used to reconstruct specifically the metabolic network for S. cerevisiae. The information on EC numbers was used to search for the stoichiometry of the reactions using the Enzyme nomenclature database (http://www.expasy.ch/enzyme/). To create a comprehensive reaction list, the present information in the reconstructed network was checked for whether genes were missing using the MIPS Comprehensive Yeast Genome Database (CYGD) (http://mips.gsf.de/proj/yeast/) and Saccharomyces Genome Database (SGD) (http://genome-www.stanford.edu/Saccharomyces/). The stoichiometry of a reaction in the Enzyme nomenclature database and also in many pathway databases is often presented in a general form, such as, for example, for NADH-dependent alcohol dehydrogenase, as alcohol dehydrogenases generally accept a wide range of different aldehydes or alcohols as substrate. Hence, the stoichiometry is found as follows: An aldehyde + NADH <−> An alcohol + NAD_upper (+). For reconstruction and modeling purposes, this information is insufficient, and through an additional database and literature search, the S. cerevisiae-specific substrates and products were identified, here, acetaldehyde and ethanol. Also, a large number of reactions involve cofactors utilization, and for many of these reactions, the cofactor requirements have not yet been completely elucidated, for example, whether reactions only require either NADH or NADPH as a cofactor or whether the enzyme can use both cofactors. Some reactions are known to involve both cofactors. For example, the mitochondrial aldehyde dehydrogenase encoded by ALD4 may use both NADH and NADPH as a cofactor (Remize et al. 2000). In such cases, two reactions were included in the reconstructed metabolic network.
Concerning localization, all reactions were localized into the two main compartments, cytosol and mitochondria, as most of the common metabolic reactions in S. cerevisiae take place in these compartments. Reactions located in vivo in other compartments, or reactions for which no information is presently available on the localization, were assumed to be cytosolic. Information on localization was mainly extracted from CYGD and YPD. All corresponding metabolites were assigned appropriate localization, and a link between cytosol and mitochondria was established through either known transport and shuttle systems or through inferred reactions to meet metabolic demands. To differentiate metabolites in the mitochondria and cytosol, metabolites that were located in the mitochondria for a specific reaction end with a small m (see Web links).
Whether reactions are irreversible or reversible was extracted from pathway databases and additional literature (see Methods). When no information was available, reactions were initially defined to be reversible.
For enzyme complexes such as succinate dehydrogenase, fatty acid synthase, and complexes of the electronic transport chain, a single reaction for the corresponding genes was defined.
Further considerations were taken into account to preserve some unique features of the S. cerevisiae metabolism. S. cerevisiae lacks a gene that encodes the enzyme transhydrogenase. Insertion of a corresponding gene from Azetobacter vinelandii in S. cerevisiae has a major impact on its phenotypic behavior, especially under anaerobic conditions (Nissen et al. 2001). As a result, reactions that create a net transhydrogenic effect in the model were either constrained to zero or forced to become irreversible. For instance, the flux carried by NADH-dependent glutamate dehydrogenase (Gdh2p) was constrained to zero to avoid the appearance of a net transhydrogenase activity through coupling with the NADPH-dependent glutamate dehydrogenases (Gdh1p and Gdh3p).
Decisions also needed to be made as to whether a reaction should be present in the reconstructed metabolic model, although no corresponding confirmed gene function is available. Many reactions have shown experimentally that they must be present in S. cerevisiae or they must simply be present to allow the formation of biomass. A typical example for the former case is the oxidative branch of the pentose phosphate pathway, which is the main supplier of cytosolic NADPH. It is not currently known whether the second step in the pentose phosphate is driven nonenzymatically or enzymatically by 6-phosphogluconolactonase. Because the oxidative pathway has to be active in S. cerevisiae and because the S. cerevisiae genome contains at least four possible sites for a 6-phosphogluconolactonase (SOL1, SOL2, SOL3, SOL4), the corresponding reactions were included in the model.
The reconstruction process led to a set of biochemical reactions that might be used in constructing stoichiometric models of metabolism using metabolite balancing (Stephanopoulos et al. 1998; Edwards et al. 1999; Gombert and Nielsen 2000). These models simply rely on mass balances around metabolic intermediates and allow simulation of steady state behavior, without inclusion of information on regulatory and dynamics information. Further to the information obtained on the stoichiometry, localization, and reversibility of the reactions as described before, knowledge on the biomass composition needed to be computed as a drain of precursors or building blocks into biomass. Table 1 shows the biomass composition that was considered in the stoichiometric model and details on the calculation can be found at the mentioned Internet links. Even though the biomass composition changes under different physiological conditions, it may be assumed constant, as it has been demonstrated that a change in biomass composition merely changes the simulation results (Varma et al. 1993). Furthermore, information on the growth-associated ATP requirements (Stouthamer 1979) (maintenance of membrane potentials, turn-over of macromolecules, etc.) and on the ATP cost that is required for the polymerization of amino acids and nucleotides needed to be available.
Table 1.
Biomass Composition
| Metabolite | mmole g DW | Metabolite | mmole g DW |
| Amino acids (Oura 1972) | Mannan | 0.808 | |
| Other carbohydrates | 1.135 | ||
| Alanine | 0.459 | Ribonucleotides (Oura 1972) | |
| Arginine | 0.161 | AMP | 0.046 |
| Asparagine | 0.102 | CMP | 0.045 |
| Asparate | 0.298 | GMP | 0.046 |
| Cysteine | 0.007 | UMP | 0.060 |
| Glutamine | 0.105 | Deoxyribonucleotides(Vaughan-Martini and Martini 1993) | |
| Glutamate | 0.302 | ||
| Glycine | 0.290 | DAMP | 0.004 |
| Histidine | 0.066 | DCMP | 0.002 |
| Isoleucine | 0.193 | DGMP | 0.002 |
| Leucine | 0.296 | DTMP | 0.004 |
| Lysine | 0.286 | Lipids (Nurminen et al. 1975), Sterols (Hunter and Rose 1972), | |
| Methionine | 0.051 | Phospholipids (Kaneko et al. 1976), Fatty acids (Schulze 1995) | |
| Phenylalanine | 0.134 | ||
| Proline | 0.165 | Triacylglycerol | 0.007 |
| Serine | 0.185 | Ergosterol | 0.001 |
| Threonine | 0.191 | Zymosterol | 0.002 |
| Tryptophane | 0.028 | Phosphatidate | 0.001 |
| Tyrosine | 0.102 | Phosphatidylcholine | 0.006 |
| Valine | 0.265 | Phosphatidylethanolamine | 0.004 |
| Carbohydrates(Schulze 1995) | Phosphatidylinositol | 0.005 | |
| Phosphatidylserine | 0.002 | ||
| Glycogen | 0.519 | ||
| Trehalose | 0.023 |
(Schulze 1995) Details on the calculation of the biomass equation can be found under http://www.cpb.dtu.dk/models/yeastmodel.html and http://geneticcircuits.ucsd.edu/organisms/yeast.html.
The polymerization cost was calculated by Verduyn et al. (1991) to be 23.92 mmole ATP/g DW, and the growth-associated ATP maintenance was found by fitting the reconstructed model to the experimentally determined biomass yield of 0.51 g DW/g glucose (Verduyn 1991). Hereby, this contribution was estimated to be 35.36 mmole ATP/g DW. Thus, the sum of these two contributions is 59.28 mmole ATP/g DW, which was included in the stoichiometric model of S. cerevisiae. The ATP costs for the synthesis of building blocks, which could be derived directly from the model, was found to be 9.89 mmole ATP/g DW and, thus, the total ATP requirement for biomass growth was estimated to be 69.2 mmole ATP/g DW, which falls into the range of experimentally measured values (Verduyn et al. 1990; Verduyn 1991). Finally, a nongrowth-associated ATP requirement of 1 mmole/g DW/h was assumed to be required (Stouthamer 1979; Verduyn et al. 1990).
At this step, a first model was designed that could be applied to linear programming to simulate cellular behavior. The model was used to minimize the glucose uptake rate at a dilution rate of 0.1 h−1 for aerobic and anaerobic conditions, and results were compared with experimental results (data not shown). Initially, no agreement between computed and experimental results could be found. However, through a rigorous investigation of flux distributions and shadow price analyses, it was possible to adjust and correct the initial reaction list until simulated results were in agreement for both cases. At this step, the reconstruction process was considered to be finished.
Characteristics of the Reconstructed Network
The metabolic reconstruction process resulted in a network that consisted of 1175 metabolic reactions and 584 metabolites (Table 2). A total of 708 metabolic ORFs were included in the reconstructed network, to which 1035 reactions were assigned. Some 595 metabolic ORFs contained at least one enzyme commission (EC) number. This corresponded to ∼54% of all ORFs that were assigned an EC number in the MIPS database (595 of 1098 ORFs; Mewes et al. 1997). The remaining 46% correspond mainly to protein kinases, protein phosphates, peptidases, and proteases, which have not been included. Most of the enzymes are monofunctional, with 179 enzymes being multifunctional. Currently, the number of protein-coding genes in the S. cerevisiae genome is estimated by YPD (Costanzo et al. 2001) to be 6281, of which 4127 corresponding proteins were characterized by genetics or biochemistry, and an additional 252 proteins were assigned functions by homology searches. The total number of genes included in the reconstructed metabolic network corresponded to ∼16% of all characterized ORFs.
Table 2.
Network Characteristics of the Reconstructed Metabolic Network of Saccharomyces cerevisiae
| ORFs | 708 | ||
| Metabolites | 584 | ||
| Cytosolic metabolites | 559 | ||
| Mitochondrial metabolites | 164 | ||
| Extracellular metabolites | 121 | ||
| Reactions | 1175 | ||
| Mitochondrial reactions | 124 | ||
| Cytosolic reactions | 702 | ||
| Exchange fluxes | 349 | ||
| Cytosolic exchange fluxes | 287 | ||
| Mitochondrial exchange fluxes | 62 | ||
| Reactions with ORF assignments | 1035 | ||
| Reactions based on biochemical evidence or | |||
| Physiological considerations | 140 |
On the basis of the protein complex catalog of MIPS (Mewes et al. 1997), 26 protein complexes, which catalyzed 88 reactions, were identified in the reconstructed metabolic network. The metabolic network contained 193 ORFs coding for isoenzymes, which catalyzed 239 reactions.
A total of 140 reactions were included on the basis of biochemical evidence or physiological considerations, but currently with no annotated ORF. More than 85% of these reactions were transport reactions over the cytoplasmic or mitochondrial membrane, other reactions were mainly involved in amino acid, nucleotide, and vitamin metabolism (Table 3). A total of 349 transport reactions were included in the model, of which 287 were involved in transporting metabolites in or out of the cell, and 62 transport reactions were involved in interchanging metabolites between the cytosol and the mitochondria. Reversibility and irreversibility of reactions was carefully accounted for in the reconstruction process, so that approximately two-thirds of the reactions were assumed to be irreversible.
Table 3.
Distribution of 140 Metabolic Reactions of S. cerevisiae With Currently No Annotated ORF That Were Included in the Reconstruction Process on the Basis of Physiological and Biochemical Evidence
| Transport over Cytoplasmic Membrane | 77 |
| —Carbohydrates | 18 |
| —Nucleotides and Nucleosides | 27 |
| —Other | 32 |
| Transport over Mitochondrial Membrane | 44 |
| Amino Acid Metabolism | 2 |
| Metabolism of Cofactors, Vitamins, and Other Substances | 10 |
| Nucleotide Metabolism | 2 |
| Lipid Metabolism | 3 |
| Metabolism of Complex Lipids | 1 |
| Energy Metabolism | 1 |
A complete list of all included reactions can be downloaded at http://www.cpb.dtu.dk/models/yeastmodel.html or http://gcrg/organisms/yeast.html.
The most frequently used metabolic intermediates in the reconstructed network are presented in Table 4, showing that the most connected metabolites were involved in energy metabolism, such as ATP, etc., in redox metabolism, such as NADPH, and in nitrogen metabolism, such as glutamine and glutamate. The most frequently used metabolite was proton, due to its participation in a high number of proton-coupled transport reactions in the network. The number of reactions involving proton was much higher than in metabolic networks of prokaryotic microorganism reconstructed previously (Schilling 2000). This difference was mainly due to the larger number of proton-driven transport systems in S. cerevisiae—both in the cytosolic and in the mitochondrial membrane. For comparison, the metabolic connectivity of three prokaryotic organisms was examined (Table 4). In all 4 reconstructed networks, the 12 most connected metabolites represented the key intermediates of high-energy metabolism, redox carriers, nitrogen metabolism, and 2- and 3-carbon intermediates. Another important topological property of the reconstructed network was the number of metabolites that participate in each reaction (Fig. 2). For all four networks, the most common number was 4, representing the conversion of a substrate to a product concomitant with the conversion of a coupled cofactor from one form to another. Most frequently, this conversion involved ATP and ADP or the translocation of H+.
Table 4.
Connectivity of Metabolites in Saccharomyces cerevisiae, Escherichia coli, Haemophilus influenzae, and Helicobacter pylori
| S. cerevisiae | E. coli | H. influenzae | H. pylori | ||||||||
| metabolite | connectivity | metabolite | connectivity | metabolite | connectivity | metabolite | connectivity | ||||
| absolute | relative (%) | absolute | relative (%) | absolute | relative (%) | absolute | relative (%) | ||||
| Proton | 229 | 5.0 | ATP | 160 | 6.1 | ATP | 114 | 6.5 | ATP | 79 | 5.7 |
| ATP | 188 | 4.1 | Phosphate | 140 | 5.3 | Phosphate | 102 | 5.8 | ADP | 65 | 4.7 |
| ADP | 146 | 3.2 | ADP | 137 | 5.2 | ADP | 101 | 5.8 | Phosphate | 60 | 4.3 |
| Phosphate | 131 | 2.9 | Proton | 86 | 3.3 | Proton | 77 | 4.4 | Proton | 47 | 3.4 |
| CO2 | 90 | 2.0 | CO2 | 63 | 2.4 | CO2 | 40 | 2.3 | Diphosphate | 38 | 2.7 |
| NADP | 86 | 1.9 | Diphosphate | 56 | 2.1 | Diphosphate | 40 | 2.3 | CO2 | 36 | 2.6 |
| NADPH | 78 | 1.7 | Pyruvate | 53 | 2.0 | NADP | 31 | 1.8 | NADP | 34 | 2.4 |
| Diphosphate | 81 | 1.8 | Glutamate | 48 | 1.8 | NADPH | 30 | 1.7 | NADPH | 33 | 2.4 |
| NAD | 78 | 1.7 | NAD | 48 | 1.8 | Glutamate | 30 | 1.7 | Glutamate | 23 | 1.7 |
| NADH | 65 | 1.4 | NADH | 43 | 1.6 | NAD | 24 | 1.4 | NH3 | 19 | 1.4 |
| Glutamate | 68 | 1.5 | NADP | 41 | 1.6 | Pyruvate | 22 | 1.3 | Pyruvate | 18 | 1.3 |
| NH3 | 56 | 1.2 | NH3 | 41 | 1.6 | NH3 | 22 | 1.3 | COA | 18 | 1.3 |
(Absolute) Connectivity is defined as the number of occurrences of a metabolite in any of the reactions of the reconstructed metabolic networks. The relative connectivity has been calculated on the basis of the summed frequency of all metabolites in the reconstructed metabolic networks, which is 4551, 2637, 1753, and 1389 in S. cerevisiae, E. coli, H. influenzae, and H. pylori, respectively.
Figure 2.

Reaction classification by number of participating metabolites.
A total of 184 metabolites were not connected to the overall metabolic network, showing that either reactions linking these metabolites to the overall metabolic network have not been identified yet, proteins may have been assigned wrong functions in the annotation process, or S. cerevisiae has lost some of its metabolic functions during evolution. This result shows that the information on the metabolic network in S. cerevisiae is currently still incomplete, however, the presented reconstructed metabolic network may be useful in guiding the assignment of orphan ORFs (Förster et al. 2002) or the identification of erroneous assignments. An example of an unlinked metabolite was formaldehyde. It appeared only in one reaction in the metabolic network through the inclusion of the gene SFA1, which codes for a formaldehyde dehydrogenase. It is as yet unknown which role formaldehyde plays in the natural environment of S. cerevisiae. From the observation that S. cerevisiae contains a formaldehyde as well as formate dehydrogenases, it may be concluded that it either encounters these C1 compounds in its natural environment or generates them in its metabolic network (J. Pronk, pers. comm.).
The Reconstructed Metabolic Network of S. cerevisiae Versus MIPS Entries and Enzyme Commission Assignments
Throughout the reconstruction process, 595 ORFs have been assigned an EC number, corresponding to 850 reactions. The most abundant enzyme class is transferases followed by oxidoreductases, hydrolases, lyases, ligases, and isomerases (Fig. 2.) A similar tendency was found for a reconstructed metabolic network of E. coli (Edwards and Palsson 2000), but this network contains more lyases than hydrolases. The comparison of the number of ORFs with the number of reactions in each enzyme category suggests that in S. cerevisiae, isomerases, and transferases are less substrate specific (ratio of number of reactions to number of ORFs) than any of the other enzyme classes, whereas in E. coli transferases and hydrolases are the least substrate-specific enzyme classes (Fig. 3).
Figure 3.

Number of reactions and ORFs included in the S. cerevisiae metabolic network arranged by enzyme categories. For comparison, data for an E. coli (Edwards and Palsson 2000) metabolic network is shown. (EC 1) Oxidoreductases; (EC 2) transferases; (EC 3) hydrolases; (EC 4) lyases; (EC 5) isomerases; (EC 6) ligases.
The number of ORFs included in the metabolic reconstruction of S. cerevisiae was also compared with the functional categories as defined by MIPS. Not surprisingly, most of the ORFs fall into two main classes, such as metabolism and energy, followed by the classes of transport facilitation and cellular transport and transport mechanism. Furthermore, for ∼430 ORFs, information about localization is available as characterized by the functional category, cellular organization (Table 5).
Table 5.
ORFs in the Saccharomyces cerevisiae Metabolic Network Sorted Into Functional Categories According to the Munich Information Center for Protein Sequences (MIPS)
| Functional Category | No. of ORFs |
| Metabolism | 646 |
| Energy | 158 |
| Cell Growth, Cell Division and DNA Synthesis | 30 |
| Transcription | 2 |
| Protein Synthesis | 15 |
| Protein Destination | 43 |
| Transport Facilitation | 104 |
| Cellular and Transport Mechanism | 97 |
| Cellular Biogenesis | 23 |
| Cellular Communication/Signal Transduction | 10 |
| Cell Rescue, Defense, Cell Death and Ageing | 32 |
| Ionic Homeostasis | 42 |
| Cellular Organization | 434 |
| Classification not yet Clear-cut | 3 |
| Unclassified Proteins | 14 |
The large functional classes of metabolism and energy were investigated in more detail (Fig. 4). Analyzing the functional category metabolism (Fig. 4A) revealed that most ORFs are involved in C-compound and carbohydrate metabolism, followed by amino acid metabolism, lipid, fatty acid and isoprenoid metabolism, and nucleotide metabolism. Comparison with MIPS entries showed that the number of ORFs included in the reconstructed network is different from the MIPS database. This difference is either due to exclusion of ORFs, which are involved in regulation, such as ORFs encoding activators or negative regulators, or exclusion of ORFs, which have assigned function based on similarity searches. The second largest functional category classified by the MIPS database is that of the lipid, fatty acid, and isopreniod metabolism. However, during the metabolic reconstruction process, more ORFs were included in the functional category, amino-acid metabolism, than in the functional category, lipid, fatty acid, and isoprenoid metabolism, based on a high number of ORFs that have been assigned function using similarity searches. This result is consistent with the fact that the amino acid metabolism is currently still better understood than the much more complex lipid metabolism.
Figure 4.

ORFs sorted into the functional categories of MIPS, metabolism (A) and energy (B). (AA) Amino-Acid Metabolism; (N2/S) nitrogen and sulphur metabolism; (N) nucleotide metabolism; (P) phosphate metabolism; (C) C-compound and carbohydrate metabolism; (L) lipid, fatty-acid and isoprenoid metabolism; (V) metabolism of vitamins, cofactors, and prosthetic groups; (S) secondary metabolism; (EMP) glycolysis and gluconeogenesis; (PPP) pentose-phosphate pathway; (TCA) tricarboxylic-acid pathway; (RES) respiration; (FER) fermentation; (ER) metabolism of energy reserves (glycogen, trehalose); (GLYC) glyoxylate cycle; (OX) oxidation of fatty acids; (O) other energy generation activities.
Details of the functional class energy metabolism are shown in Figure 4B, elucidating the fact that traditional pathways of the primary metabolism, such as glycolysis, gluconeogenesis, pentose-phosphate pathway, TCA cycle, and glyoxylate cycle are very well described. The functional classes of respiration, fermentation, etc., contain a higher number of proteins that are involved in regulation and transport. In addition, these categories contain a high number of ORFs that have been assigned function by similarity searches and for many cases, the function has not been fully elucidated.
Biosynthesis of Amino Acid and Precursor Metabolites—Metabolic Capabilities of S. cerevisiae
All building blocks needed for synthesis of macromolecules constituting cell mass can be generated from a set of 12 precursor metabolites (Stephanopoulos et al. 1998). The capability of the reconstructed genome-based S. cerevisiae and E. coli networks to produce these precursor metabolites using glucose as the sole carbon source was computed by use of linear optimization (Fell and Small 1986; Varma and Palsson 1993). Similarly, the maximum production of the 20 common amino acids was calculated for both organisms. In both cases, S. cerevisiae was found to be more efficient in producing precursor metabolites and amino acids (Fig. 5A,B). This result is somewhat surprising, as E. coli has been recognized and is widely used as a host for industrial amino acid production. Investigation of the corresponding flux distributions shows that the difference is caused by the higher ATP maintenance requirements in E. coli. If ATP maintenance requirements are not considered, the S. cerevisiae and E. coli networks generate similar systemic yields, except for acetyl-CoA, glutamate, glutamine, and glycine. Thus, the analysis shows that S. cerevisiae may be a suitable host for industrial amino acid production.
Figure 5.

Maximum precursor and amino acid production in S. cerevisiae and E. coli including nongrowth-specific ATP maintenance (A,B) (in mole/mole glucose). (3PG) 3-Phospho glycerate; (ACCOA) acetyl-CoA; (AKG) 2-Oxoglutarate; (ALA) alanine; (ARG) arginine; (ASN) asparagine; (ASP) aspartate; (CYS) cysteine; (E4P) eErythrose 4 -phosphate; (F6P) fructose 6-phosphate; (G6P) glucose 6-phosphate; (GLN) glutamine; (GLU) glutamate; (GLY) glycine; (HIS) histidine; (ILE) isoleucine; (LEU) leucine; (LYS) lysine; (MET) methionine; (OA) oxaloacetate; (PEP) phosphoenolpyruvate; (PHE) phenylalanine; (PRO) pProline; (PYR) pyruvate; (R5P) ribose 5-phosphate; (SER) serine; (SUCCOA) succinyl-CoA; (T3P1) glyceraldehyde 3-phosphate; (THR) threonine; (TRP) tryptophane; (TYR) tyrosine; (VAL) valine.
In conclusion, the metabolic network of S. cerevisiae was reconstructed using a procedure based on information from genomic databases, reaction databases, and a comprehensive literature search on S. cerevisiae. Although it is incomplete, given the number of orphan ORFs, it is a first step toward cataloging and characterizing the entire metabolic portfolio of a eukaryotic organism. This conclusion is supported by the myriad of specific and insightful information derived from the list of metabolic reactions. The potential of the reconstructed model may further be used for the analysis of phenotypic behavior under different genetic and physiological conditions (I. Famili, J. Förster, J. Nielsen, and B.Ø. Palsson, in prep.) The reconstructed metabolic network of S. cerevisiae represents a strong platform for reconstruction of metabolic networks of higher organisms, such as plants, animal, and human. Such reconstructed metabolic networks will serve an important role in systems biology, as the analysis of reconstructed metabolic networks will facilitate the exploration of metabolism for drug targets (Schuster et al. 1999), enable the design of microbial strains with improved characteristics through metabolic engineering (Nielsen 2001), and serve as a tool in functional annotation (Selkov et al. 2000).
METHODS
Metabolic Reconstruction Process
The reconstruction process is shown in Figure 1 and is described in detail in the main text. In brief, the reconstruction process involves the collection of all known enzymatic reactions in the metabolic pathways of S. cerevisiae. Tables 6 and 7 contain information on the online genome and pathway databases and key references used for the reconstruction process. Furthermore, journal publications were used to identify specific information on the reactions.
Table 6.
Online Resources for the Reconstruction of the Metabolic Network of Saccharomyces cerevisiae
| Database | Link |
| Genome Databases | |
| Munich Information Center for Protein Sequences Database (MIPS) | http://mips.gsf.de/proj/yeast/ |
| Saccharomyces Genome Database (SGD) | http://genome-www.stanford.edu/Saccharomyces/ |
| Yeast Proteome Database | http://www.proteome.com/databases/YPD/YPDsearch-quick.html |
| Pathway and other databases | |
| KEGG Database | http://kegg.genome.ad.jp/kegg/kegg2.html |
| ExPASy Biochemical Pathways | http://www.expasy.ch/cgi-bin/search-biochem-index |
| ExPASy Enzyme Database | http://www.expasy.ch/enzyme/ |
| ERGO | http://www.integratedgenomics.com/ |
| Swiss-Prot | http://www.expasy.ch/sprot |
Table 7.
Key references consulted additionally to online resources for the reconstruction of the metabolic network of Saccharomyces cerevisiae
| Metabolism | Reference |
| Amino acid biosynthesis | (Strathern et al. 1982) |
| Lipid synthesis | (Daum et al. 1998) (Parks 1978; Dickinson and Schweizer 1998; Dickson 1998; Dickson and Lester 2000) |
| Nucleotide Metabolism | (Strathern et al. 1982) (Michal 1999) |
| Oxidative phosphorylation and electron transport | (Verduyn et al. 1991) (Overkamp et al. 2000) |
| Primary Metabolism | (Zimmerman and Entian 1997) (Strathern et al. 1982; Dickinson and Schweizer 1998) |
| Transport over cytoplasmic membrane | (Paulsen et al. 1998) (André 1995; Regenberg 1999; Wieczorke et al. 1999) |
| Transport over mitochondrial membrane |
(Palmieri et al. 2000a,b,c,d) (Tzagoloff 1982; André 1995; Pallotta et al. 1998; Paulsen et al. 1998) |
Linear Programming
The reactions of the reconstructed metabolic model were formulated as a stoichiometric model S · v = 0, as, for example, described in Edwards et al. (1999) or Stephanopoulos et al. (1998). This model describes cellular behavior under pseudo steady-state conditions, and S is defined as the stoichiometric matrix that contains the stoichiometric coefficients of internal (balanced) metabolite i in the jth reactions and v is the flux vector that corresponds to the flux of the jth reaction. The stoichiometric model was solved using linear programming, an approach often referred to as flux balance analysis (Edwards et al. 1999).
The linear programming problem was formulated by defining an objective function Z:
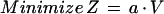 |
 |
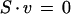 |
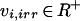 |
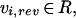 |
in which a was a row vector containing weights of the individual variables specifying the influence of the individual fluxes on the objective function Z. The elements of the flux vector v were constrained for the definition of reversible and irreversible reactions, vi,rev and vi,irr, respectively. Uptake was defined for glucose, sulfate, ammonia, phosphate, oxygen (for aerobic growth), and ergosterol and zymosterol (for anaerobic growth). Secretion was defined for all major metabolic products, such as ethanol, glycerol, succinate, acetate, pyruvate, and for all amino acid, organic acids (see supplementary material).
The consistency of the model was checked at anaerobic and aerobic conditions at a dilution rate of 0.1 h−1 (objective function Z = μ) and compared with experimental results from Nissen et al. (1997) and Overkamp et al. (2000), respectively.
For the maximization of precursors or building blocks of biomass, an additional reaction was defined in the model and maximized for. The general format of the additional reaction was as follows: precursor → precursorOut, and the objective was the maximization of that particular reaction.
All calculations were carried out using the commercially available software Lindo (Lindo Systems Inc.).
Shadow Prices
Shadow prices are derived from the dual variable of a linear programming problem (see, for example, Bertsimas and Tsitsiklis 1997). Its' definition is:
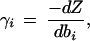 |
in which bi corresponds to a potential uptake or secretion rate of metabolite i. Negative shadow prices describe metabolites that are demanded by the metabolic network and positive shadow prices identify metabolites that the metabolic network would like to excrete in order to improve the objective value Z.
Preliminate versions of the reconstructed models were unable to model cellular behavior; either the model did not allow growth or growth reached infinity. In such cases, three strategies were considered for identifying the missing or incorrect information in the model during the reconstruction process. First, investigation of the flux distribution, second, investigation of shadow prices, and third, definition of new linear programming problems, such as maximization of precursors or building blocks that are necessary to synthesize biomass.
Acknowledgments
Research activities in the field of functional genomics at the Center for Process Biotechnology are financially supported by the Danish Biotechnology Instrument Center (DABIC). We thank the Whitaker Foundation for their support through the Graduate Fellowship in Biomedical Engineering to I.F., the National Science Foundation through grant nos. 9873384 and 9814092, and the National Institutes of Health through grant no. GM57089.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL jn@biocentrum.dtu.dk; FAX 45-45-88-41-48.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.234503.
REFERENCES
- 1.André B. 1995. An overview of membrane transport proteins in Saccharomyces cerevisiae. Yeast 11: 1575-1611. [DOI] [PubMed] [Google Scholar]
- 2.Bertsimas D. and Tsitsiklis, J.N., 1997. Linear optimization. Athena Scientific, Belmont, MA.
- 3.Botstein D., Chervitz, S.A., and Cherry, J.M. 1997. Yeast as a model organism. Science 277: 1259-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costanzo M.C., Crawford, M.E., Hirschman, J.E., Kranz, J.E., Olsen, P., Robertson, L.S., Skrzypek, M.S., Braun, B.R., Hopkins, K.L., Kondu, P., et al. 2001. YPD, PombePD and WormPD: Model organism volumes of the BioKnowledge library, an integrated resource for protein information. Nucleic Acids Res. 29: 75-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Covert M.W., Schilling, C.H., Famili, I., Edwards, J.S., Goryanin, I.I., Selkov, E., and Palsson, B.Ø. 2001. Metabolic modeling of microbial strains in silico. Trends Biochem. Sci. 26: 179-186. [DOI] [PubMed] [Google Scholar]
- 6.Daum G., Lees, N.D., Bard, M., and Dickson, R. 1998. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 14: 1471-1510. [DOI] [PubMed] [Google Scholar]
- 7.DeRisi J.L., Iyer, V.R., and Brown, P.O. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278: 680-686. [DOI] [PubMed] [Google Scholar]
- 8.Dickinson J.R. and Schweizer, M., 1998. The metabolism and molecular physiology of Saccharomyces cerevisiae, pp. 1798–1998. Taylor & Francis, London, UK.
- 9.Dickson R. 1998. Sphingolipid functions in Saccharomyces cerevisiae: Comparison to mammals. Annu. Rev. Biochem. 67: 27-48. [DOI] [PubMed] [Google Scholar]
- 10.Dickson R. and Lester, R.L. 2000. Metabolism and selected functions of sphingolipids in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1438: 305-321. [DOI] [PubMed] [Google Scholar]
- 11.Dujon B. 1996. The yeast genome project: What did we learn? Trends Genet. 12: 263-270. [DOI] [PubMed] [Google Scholar]
- 12.Edwards J.S. and Palsson, B.Ø. 1999. Systems Properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 274: 17410-17416. [DOI] [PubMed] [Google Scholar]
- 13.___, 2000. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci.. 97: 5528-5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards J.S., Ramakrishna, R., Schilling, C.H., and Palsson, B.Ø. 1999. Metabolic flux balance analysis. In Metabolic engineering (eds. S.Y. Lee & E.T. Papoutsakis), pp. 13–57. Marcel Dekker Inc., New York, NY.
- 15.Edwards J.S., Ibarra, R.U., and Palsson, B.Ø. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19: 125-130. [DOI] [PubMed] [Google Scholar]
- 16.Endy D. and Brent, R. 2001. Modelling cellular behaviour. Nature 409: 391-395. [DOI] [PubMed] [Google Scholar]
- 17.Entian K.-D., Schuster, T., Hegemann, J.H., Becher, D., Feldmann, H., Güldener, U., Götz, R., Hansen, M., Hollenberg, C.P., Jansen, G., et al. 1999. Functional analysis of 150 deletion mutants in Saccharomyces cerevisiae by a systematic approach. Mol. Gen. Genet. 262: 683-702. [DOI] [PubMed] [Google Scholar]
- 18.Fell D.A. and Small, J. 1986. Fat synthesis in adipose tissue. An examination of stoichiometric constraints. Biochem. J. 238: 781-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Förster J., Gombert, A.K., and Nielsen, J. 2002. A functional genomics approach using metabolomics and in silico pathway analysis. Biotechnol. Bioeng. 79: 703-712. [DOI] [PubMed] [Google Scholar]
- 20.Goffeau A. 1997. The yeast genome directory. Nature 387: 5-6. [PubMed] [Google Scholar]
- 21.Gombert A.K. and Nielsen, J. 2000. Mathematical modelling of metabolism. Curr. Opin. Biotechnol. 11: 180-186. [DOI] [PubMed] [Google Scholar]
- 22.Hughes T.R., Marton, M.J., Jones, A.R., Roberts, C.J., Stoughton, R., Armour, C.D., Bennett, H.A., Coffey, E., Dai, H., He, Y.D., et al. 2000. Functional discovery via a compendium of expression profiles. Cell 102: 109-126. [DOI] [PubMed] [Google Scholar]
- 23.Hunter K. and Rose, A.H. 1972. Lipid composition of Saccharomyces cerevisiae as influenced by growth temperature. Biochim. Biophys. Acta 260: 639-653. [DOI] [PubMed] [Google Scholar]
- 24.Ideker T., Thorsson, V., Ranish, J.A., Christmas, R., Buhler, J., Eng, J.K., Bumgarner, R., Goodlett, D.R., Aebersold, R., and Hood, L. 2001. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929-934. [DOI] [PubMed] [Google Scholar]
- 25.Ito T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., and Sakaki, Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci.. 98: 4569-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneko H., Hosohara, M., Tanaka, M., and Itoh, T. 1976. Lipid composition of 30 species of yeast. LIPIDS 11: 837-844. [DOI] [PubMed] [Google Scholar]
- 27.Mewes H.W., Albermann, K., Heumann, K., Liebl, S., and Pfeiffer, F. 1997. MIPS: A database for protein sequences, homology data and yeast genome information. Nucleic Acids Res. 25: 28-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michal G., 1999. Biochemical pathways. Spektrum, Akad. Verl., Heidelberg, Berlin, Germany.
- 29.Nielsen J. 2001. Metabolic engineering. Appl. Microbiol. Biotechnol. 55: 263-283. [DOI] [PubMed] [Google Scholar]
- 30.Nissen T.L., Schulze, U., Nielsen, J., and Villadsen, J. 1997. Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology 143: 203-218. [DOI] [PubMed] [Google Scholar]
- 31.Nissen T.L., Anderlund, M., Nielsen, J., Villadsen, J., and Kielland-Brandt, M.C. 2001. Expression of a cytoplasmic transhydrogenase in Saccharomyces cerevisiae results in formation of 2-oxoglutarate due to depletion of the NADPH pool. Yeast 18: 19-32. [DOI] [PubMed] [Google Scholar]
- 32.Nurminen T., Konttinen, K., and Suomalatnen, H. 1975. Neutral lipids in the cells and cell envelope fractions of aerobic baker's yeast and anaerobic brewer's yeast. Chem. Phys. Lipids 14: 15-32. [DOI] [PubMed] [Google Scholar]
- 33.Oliver S.G. 1998. Introduction to functional analysis of the yeast genome. In Methods in microbiology (eds. A.J.P. Brown & M. Tuite), pp. 1–13. Academic Press, San Diego, CA.
- 34.Østergaard S., Olsson, L., Johnston, M., and Nielsen, J. 2000a. Increasing galactose consumption by Saccharomyces cerevisiae through metabolic engineering of the GAL gene regulatory network. Nat. Biotechnol. 18: 1283-1286. [DOI] [PubMed] [Google Scholar]
- 35.Østergaard S., Olsson, L., and Nielsen, J. 2000b. Metabolic engineering of Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 64: 34-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oura E., 1972. The effect of aeration on the growth energetics and biochemical composition of baker's yeast, with an appendix: Reactions leading to the formation of yeast cell material from glucose and ethanol. Helsinki University, Helsinki, Finland.
- 37.Overkamp K.M., Bakker, B.M., Kotter, P., van Tuijl, A., de Vries, S., van Dijken, J.P., and Pronk, J.T. 2000. In vivo analysis of the mechanisms for oxidation of cytosolic NADH by Saccharomyces cerevisiae mitochondria. J. Bacteriol. 182: 2823-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pallotta M.L., Brizio, C., Fratinni, A., de Virgilio, C., Barile, M., and Passarella, S. 1998. Saccharomyces cerevisiae mitochondria can synthesise FMN and FAD from externally added riboflavin and export them to the extramitochondrial phase. FEBS Lett. 428: 245-249. [DOI] [PubMed] [Google Scholar]
- 39.Palmieri L., Lasorsa, F.M., de Palma, A., Palmieri, F., Runswick, M.J., and Walker, J.E. 2000a. Identification of the yeast ACR1 gene product as a succinate-fumarate transporter essential for growth on ethanol and acetate. FEBS Lett. 417: 114-118. [DOI] [PubMed] [Google Scholar]
- 40.Palmieri L., Lasorsa, F.M., Vozza, A., Agrimi, G., Fiermonte, G., Runswick, M.J., Walker, J.E., and Palmieri, F. 2000b. Identification and functions of new transporters in yeast mitochondria. Biochim. Biophys. Acta 1459: 363-369. [DOI] [PubMed] [Google Scholar]
- 41.Palmieri L., Runswick, M.J., Fiermonte, G., Walker, J.E., and Palmieri, F. 2000c. Yeast mitochondrial carriers: Bacterial expression, biochemical identification and metabolic significance. J. Bioenerg. Biomembr. 32: 67-77. [DOI] [PubMed] [Google Scholar]
- 42.Palmieri L., Vozza, A., Agrimi, G., De Marco, V., Runswick, M.J., Palmieri, F., and Walker, J.E. 2000d. Identification of the yeast mitochondrial transporter for oxaloacetate and sulfate. J. Biol. Chem. 32: 22184-22190. [DOI] [PubMed] [Google Scholar]
- 43.Parks L.W. 1978. Metabolism of sterols in yeast. Crit. Rev. Microbiol. 6: 301-341. [DOI] [PubMed] [Google Scholar]
- 44.Paulsen I.T., Sliwinski, M.K., Nelissen, B., Goffeau, A., and Saier, M.H., Jr. 1998. Unified inventory of established and putative transporters encoded within the complete genome of Saccharomyces cerevisiae. FEBS Lett. 430: 116-125. [DOI] [PubMed] [Google Scholar]
- 45.Regenberg B., 1999. Amino acid uptake in Saccharomyces cerevisiae, substrate specificity and regulation of the permeases. The Royal Veterinary and Agricultural University, Frederiksberg C, Denmark.
- 46.Remize F., Andrieu, E., and Dequin, S. 2000. Engineering of the pyruvate dehydrogenase bypass in Saccharomyces cerevisiae: Role of the cytosolic Mg(2+) and mitochondrial K(+) acetaldehyde dehydrogenases Ald6p and Ald4p in acetate formation during alcoholic fermentation. Appl. Environ. Microbiol. 66: 3151-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schilling C.H., 2000. On systems biology and the pathway analysis of metabolic networks. University of California, San Diego, CA.
- 48.Schilling C.H., Edwards, J.S., and Palsson, B.Ø. 1999. Toward metabolic phenomics: Analysis of genomic data using flux balances. Biotechnol. Prog. 15: 288-295. [DOI] [PubMed] [Google Scholar]
- 49.Schulze U., 1995. Anaerobic physiology of Saccharomyces cerevisiae. Department of Biotechnology, Technical University of Denmark, Kgs. Lyngby, Denmark.
- 50.Schuster S., Dandekar, T., and Fell, D.A. 1999. Detection of elementary flux modes in biochemical networks: A promising tool for pathway analysis and metabolic engineering. Trends Biochem. Sci. 17: 53-60. [DOI] [PubMed] [Google Scholar]
- 51.Selkov E., Overbeek, R., Kogan, Y., Chu, L., Vonstein, V., Holmes, D., Silver, S., Haselkorn, R., and Fonstein, M. 2000. Functional analysis of gapped microbial genomes: Amino acid metabolism of Thiobacillus ferrooxidans. Proc. Natl. Acad. Sci. 97: 3509-3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephanopoulos G., Aristidou, A.A., and Nielsen, J., 1998. Metabolic engineering—Principles and methodologies. Academic Press, San Diego, CA.
- 53.Stouthamer A.H. 1979. The search for correlation between theoretical and experimental growth yields. Microb. Biochem. 21: 1-48. [Google Scholar]
- 54.Strathern J.N., Jones, E.W., and Broach, J.R., 1982. The molecular biology of the yeast Saccharomyces—Metabolism and gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 55.Tzagoloff A., 1982. Mitochondria. Plenum Pub. Corp., New York, NY.
- 56.Uetz P., Giot, L., Cagney, G., Mansfield, T.A., Judson, R.S., Knight, J.R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403: 623-631. [DOI] [PubMed] [Google Scholar]
- 57.Varma A. and Palsson, B.Ø. 1993. Metabolic capabilities of Escherichia coli: I. Synthesis of biosynthetic precursors and cofactors. J. Theor. Biol. 165: 477-502. [DOI] [PubMed] [Google Scholar]
- 58.Varma A., Boesch, B.W., and Palsson, B.Ø. 1993. Stoichiometric interpretation of Escherichia coli glucose catabolism under various oxygenation rates. Appl. Environ. Microbiol. 59: 2465-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vaughan-Martini A. and Martini, A. 1993. A taxonomic key for the genus Saccharomyces. System. Appl. Microbiol. 16: 113-119. [DOI] [PubMed] [Google Scholar]
- 60.Verduyn C. 1991. Physiology of yeasts in relation to biomass yields. Antonie van Leeuwenhoek 60: 325-353. [DOI] [PubMed] [Google Scholar]
- 61.Verduyn C., Postma, E., Scheffers, W.A., and van Dijken, J.P. 1990. Physiology of Sacchomyces cerevisiae in anaerobic glucose- limited chemostate cultures. J. Gen. Microbiol. 136: 395-403. [DOI] [PubMed] [Google Scholar]
- 62.Verduyn C., Stouthamer, A.H., Scheffers, W.A., and van Dijken, J.P. 1991. A theoretical evaluation of growth yields of yeasts. Antonie van Leeuwenhoek 59: 49-63. [DOI] [PubMed] [Google Scholar]
- 63.Wieczorke R., Krampe, S., Weierstall, T., Freidel, K., Hollenberg, C.P., and Boles, E. 1999. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett. 464: 123-128. [DOI] [PubMed] [Google Scholar]
- 64.Winzeler E.A., Shoemaker, D.D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J.D., Bussey, H., et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901-906. [DOI] [PubMed] [Google Scholar]
- 65.Zimmerman F.K. and Entian, K.-D. 1997. Yeast sugar metabolism (eds. F.K. Zimmerman & K.-D. Entian) Technomic Publishing Co., Inc., Lancaster, PA.