

Abstract
The voltage-gated sodium channel NaV1.8 (encoded by SCN10A) is predominantly expressed in dorsal root ganglia (DRG) and plays a critical role in pain perception. We analyzed SCN10A transcripts isolated from human DRGs using deep sequencing and found a novel splice variant lacking exon 11, which codes for 98 amino acids of the domain I/II linker. Quantitative PCR analysis revealed an abundance of this variant of up to 5–10% in human, while no such variants were detected in mouse or rat. Since no obvious functional differences between channels with and without the exon-11 sequence were detected, it is suggested that SCN10A exon 11 skipping in humans is a tolerated event.
Keywords: sodium channel, patch clamp, exon skipping, protein kinase A, pain
Introduction
Voltage-gated sodium (NaV) channels are important constituents in the membranes of excitable cells because they mediate the fast rising phase of action potentials and thus enable cellular excitation. The family of mammalian NaV channels comprises nine large α-subunit isoforms (NaV1.1–1.9) with different functional characteristics and four β-subunits (β1–4), which associate with an α-subunit and modulate its functions (for review see ref. 1). The α-subunit of NaV channels has a pseudotetrameric structure; a central pore is surrounded by four homologous domains that are linked by large cytoplasmic loops (Fig. 1A). Together with the N- and C-termini, these loops are involved in the regulation of channel functions by means of posttranslational modification and interaction with various intracellular signaling molecules.2
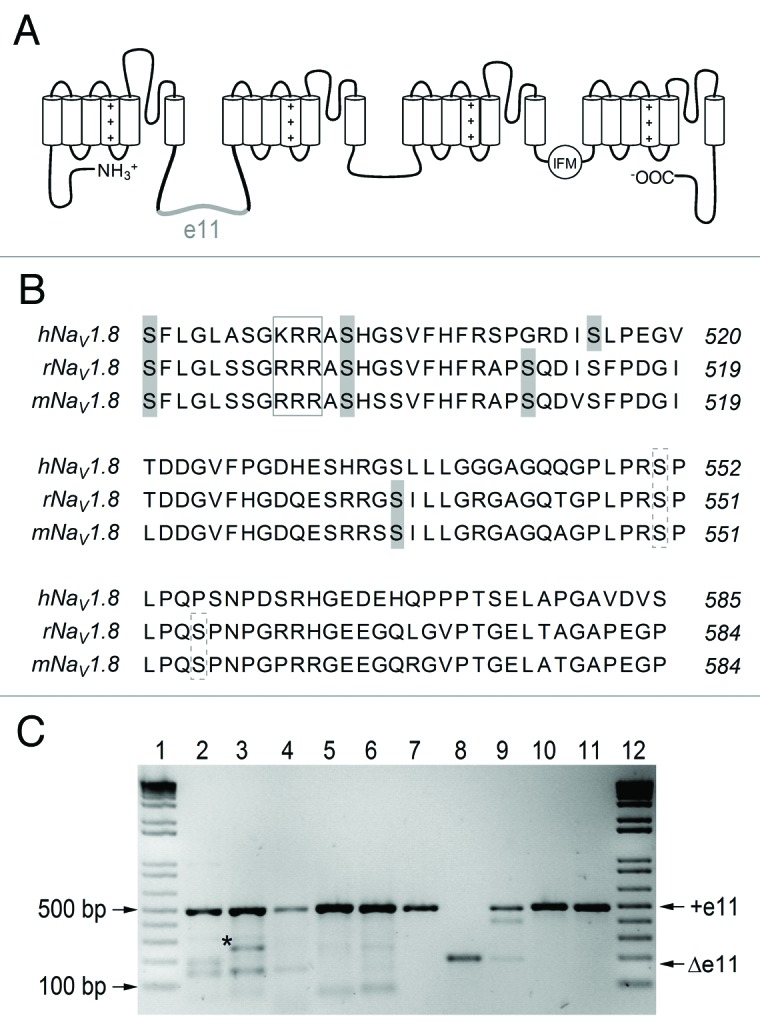
Figure 1. Skipping of exon 11 in SCN10A. (A) Topological model of human NaV1.8 highlighting the position of amino acid residues encoded by exon 11 (e11, gray) in the cytosolic linker connecting domains I and II. (B) Multiple sequence alignment of the region coding for exon 11 of SCN10A for human, rat, and mouse. The exon 11-encoded sequence includes three putative PKA consensus sites (RXXS) for human NaV1.8 and four in rat and mouse (gray shaded serines). Furthermore, there is a putative p38 site (broken boxes, consensus SP) and an ER retention motif (box, RRR) in the rat and mouse sequence that is not conserved in human. (C) Evidence of the existence of the Δe11 isoform in three human individuals (lane 2–4), but not in rat (lane 5) or mouse (lane 6). Positive controls were performed on the expression constructs used in this study and show hNaV1.8 (lane 7), hNaV1.8-Δe11 (lane 8), a 9:1 mixture of hNaV1.8 and hNaV1.8-Δe11 (lane 9), mNaV1.8 (lane 10), and rNaV1.8 (lane 10). Reference bands of a size marker are indicated in lanes 1 and 12. +e11: full-length isoform (note, the species-specific PCR amplicons are very similar in size: 493, 501 and 493 bp for the three species, respectively); Δe11: short isoform with skipped exon 11; asterisk: unspecific PCR product (lane 3), as revealed by cloning and Sanger sequencing.
NaV1.8 is primarily expressed in afferent neurons of dorsal root ganglia (DRG);3,4 it is involved in pain signaling5,6 and its activity is highly regulated. For example, NaVβ3 interacts with the rat NaV1.8 channel and masks a putative ER retention motif (sequence RRR) in the domain I/II linker, thus leading to enhanced surface expression of NaV1.8.7 Furthermore, phosphorylation of rat NaV1.8 by p38 mitogen-activated protein kinase (MAPK) at two serine residues located in the exon 11-encoded fragment of the domain I/II linker (Fig. 1B, S551 and S556) leads to enhanced current in DRG neurons.8 Moreover, rat NaV1.8 has been shown to be regulated by protein kinase A (PKA) phosphorylation at five consensus sites located in the domain I/II linker.9 Stimulation of PKA by the adenylyl cyclase activator forskolin (FSK) resulted in an increase in NaV1.8 peak current.9 However, these functional regulations were investigated using rat NaV1.8; a sequence alignment with the human protein reveals fewer putative phosphorylation sites and the sequence KRR instead of the putative ER retention signal sequence RRR (Fig. 1B).
Alternative mRNA splicing is a cellular mechanism utilized to adjust channel functions as was abundantly shown for NaV1.3,10 NaV1.511,12 and NaV1.7.13 Much less is known about the alternative splicing of SCN10A, the gene encoding NaV1.8. The only reported regulation of SCN10A on the level of mRNA is the alternative usage of NAGNAG tandem acceptors at the end of intron 16 which results in the inclusion or exclusion of glutamine at position 1030 in the domain II/III linker.14,15
In this study we describe a splice variant unique to human SCN10A mRNA resulting from skipping of exon 11.
Results and Discussion
Roche 454 sequencing of cDNA obtained from human DRG neurons revealed a novel splice variant of SCN10A, explained by skipping of exon 11, which has a length of 294 nt. As a result, there exist two isoforms of the human NaV1.8 protein: the shortened NaV1.8−Δe11 and the canonical NaV1.8. The abundance of the Δe11 mRNA isoform was 31% (206 of 669 reads and 233 of 741 reads, for two sequencing directions) in the 454 sequence data. The occurrence of the Δe11 isoform was independently analyzed in DRGs of three additional human individuals (Fig. 1C), and the fraction of Δe11 mRNA was approximately 5 to 10% in these samples. High-precision quantitative measurements using PCR-based techniques were precluded by the relatively large length difference of the isoforms, which predict an isoform-specific bias in PCR efficiency.16 For the same reason, our PCR-based estimate of Δe11 isoform represents an upper limit of the natural amount. The screening of mouse and rat DRG revealed no Δe11 or other variants (Fig. 1C). Thus, the splice event appears to be human specific.
The conserved genomic region of exon 11 (17 nt intron, 294 nt exon and 10 nt intron) was analyzed based on an alignment of the homologous sequence from 29 mammalian organisms. The sequence is 72.8% identical between human and mouse, clearly below the average human-mouse coding sequence identity of 85%, which indicates that the exon has low constraint under purifying selection. Consistently, predictions of splicing regulatory motifs (ESEfinder 3.0 and RegRNA) reveal very different motif patterns (data not shown), giving no indication of what the specific mechanisms of differential splicing regulation might be.
Skipping of exon 11 reduces the length of the 260 amino acid-long linker connecting domains I and II by 98 residues (S488 to S585, Figure 1A). To characterize the human NaV1.8 channel lacking exon 11, a construct representing the short isoform NaV1.8−Δe11 was compared with NaV1.8 upon heterologous expression in the mouse neuroblastoma cell line Neuro-2A and analysis with the whole-cell patch-clamp method. NaV-mediated currents endogenous to Neuro-2A were completely blocked with 300 nM TTX, which was used throughout this study (peak current at 0 mV was 3.72 ± 1.64 nA before, and 0.03 ± 0.01 nA after TTX application, both n = 6).
Both hNaV1.8 isoforms produced robust Na+ currents in this cell line with no apparent difference in the peak inward-current densities (98.9 ± 17.4 pA/pF, n = 36 for hNaV1.8 and 91.9 ± 14.0 pA/pF, n = 25 for hNaV1.8−Δe11 at 10 mV, P > 0.7) (Fig. 2A, 2G). Deletion of about half the domain-I/II linker sequence thus has no effect on the production of functional NaV1.8 channel protein and its trafficking to the plasma membrane. Representative current traces are shown in Figure 2A. Parameters characterizing the voltage dependence of channel activation (Fig. 2B) were almost identical for the two channel types with Vm values of –16.2 ± 1.4 mV and –15.3 ± 1.3 mV and km values of 18.4 ± 0.5 mV and 19.0 ± 0.3 mV for hNaV1.8 (n = 30) and hNaV1.8−Δe11 (n = 23), respectively.
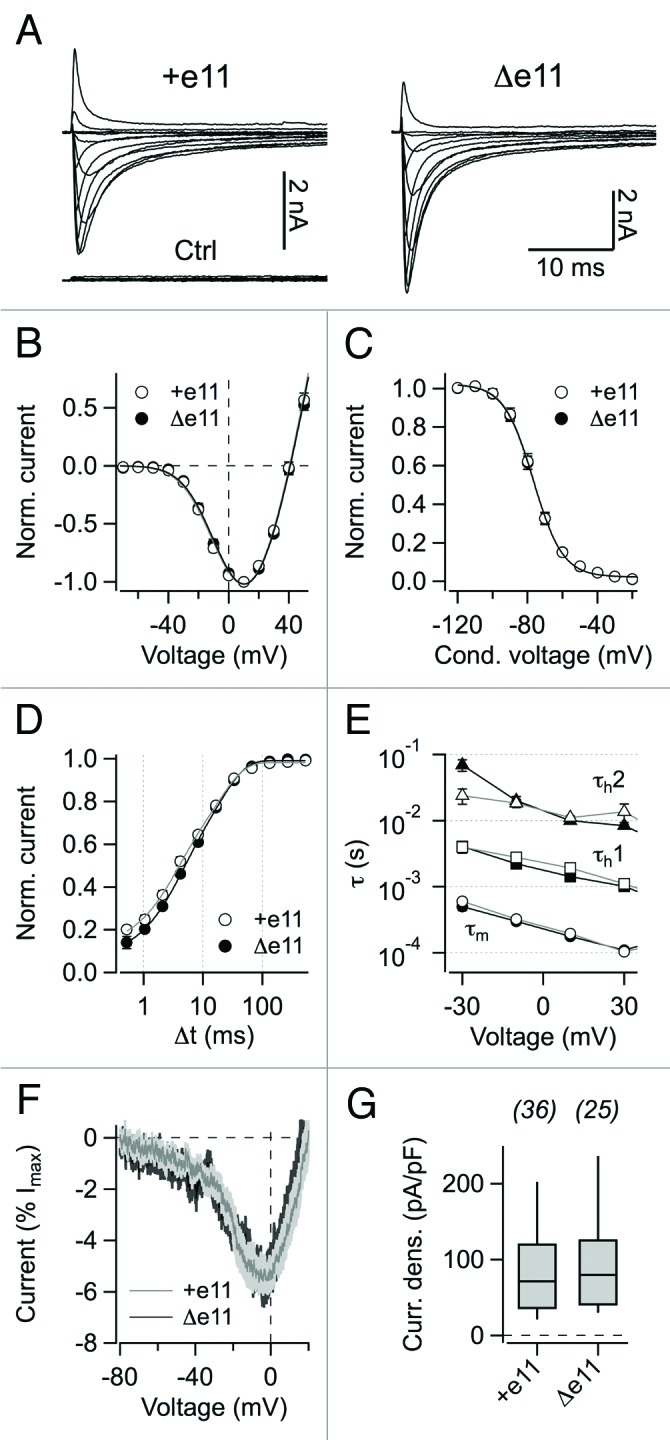
Figure 2. Electrophysiological properties. (A) Current traces of hNaV1.8 and hNaV1.8−Δe11 channels recorded in whole-cell patch-clamp configuration in the presence of 300 nM TTX after heterologous expression in Neuro-2A cells. Cells were held at –120 mV and current traces resulting from depolarizing pulse steps from -70 to 50 mV were recorded in steps of 10 mV. “Ctrl” refers to a nontranfected Neuro-2A cell. (B) Current-voltage relationships, normalized to the peak inward current, with superimposed fits according to Eq. (1). Error bars indicate s.e.m. values for n = 30 (hNaV1.8) and n = 23 (hNaV1.8−Δe11). (C) Voltage dependence of steady-state inactivation averaged for both channel variants. Superimposed curves are Boltzmann fits (Eq. (3)). Error bars indicate s.e.m. values for n = 27 (hNaV1.8) and n = 16 (hNaV1.8−Δe11). (D) Recovery from inactivation at –120 mV with superimposed double-exponential functions (Eq. (4)). Error bars indicate s.e.m. values for n = 15 independent experiments for both channel isoforms. (E) Kinetics of channel activation (τm, circles) and inactivation (τh1, squares and τh2, triangles) were determined by applying a Hodgkin-Huxley fit function (Eq. (2)) to current traces recorded from the indicated depolarizing steps using m = 3 and h = 1 gate. Straight lines connect the data points. (F) Superposition of mean currents stimulated with ramp voltage protocols (200 mV/s); the light gray area denotes the s.e.m value; n = 9 for hNaV1.8 and 12 for hNaV1.8−Δe11. (G) Peak current densities at 10 mV for hNaV1.8 and hNaV1.8−Δe11 channels expressed in Neuro-2A cells. Cell numbers are listed on top.
Steady-state inactivation (Fig. 2C) for both channel isoforms showed that Vh was –76.1 ± 1.3 mV and –77.0 ± 1.8 mV and kh was 8.0 ± 0.3 mV and 7.7 ± 0.3 mV for hNaV1.8 (n = 27) and hNaV1.8−Δe11 (n = 16), respectively. Recovery from fast inactivation (Fig. 2D) occurred with time constants of τfast 3.5 ± 0.5 ms and 4.2 ± 0.5 ms, τslow 23.8 ± 3.0 ms and 25.9 ± 3.6 ms for hNaV1.8 (n = 15) and hNaV1.8−Δe11 (n = 15), respectively. The fraction of the fast recovering current amplitude was 0.49 ± 0.05 for hNaV1.8 and 0.50 ± 0.05 for hNaV1.8−Δe11 (all P > 0.2). Thus, voltage dependence of fast inactivation and recovery from fast inactivation for hNaV1.8 did not differ from hNaV1.8−Δe11 (Fig. 2). Time courses of channel activation and inactivation were determined applying a Hodgkin-Huxley fit function with m = 3 and h = 1 gates to current responses resulting from 40 ms test pulses to –30, –10, 10, and 30 mV from a holding potential of –120 mV. No differences between hNaV1.8 and hNaV1.8−Δe11 were observed (Fig. 2E). Ramp currents evoked by slow (200 mV/s) voltage-ramp protocols revealed no difference between NaV1.8 and NaV1.8−Δe11 in ramp current amplitude either (all P > 0.05, Figure 2F).
As reported previously,7 heterologous expression of rat NaV1.8 in non-neuronal cells is restricted because of an ER-retention motif (sequence RRR) in a region of the domain-I/II linker that is part of exon 11. This motif is not conserved between rodents and human since human NaV1.8 harbors a KRR motif. To check for an enhanced membrane trafficking of hNaV1.8−Δe11, we transfected HEK 293 cells, which normally allow only very poor expression of NaV1.8. For both channel types only very small current densities (2.8 ± 2.3 pA/pF, n = 31 for hNaV1.8 and 4.2 ± 2.0 pA/pF, n = 37 for hNaV1.8−Δe11 at 10 mV, P > 0.6) were observed; the current density values were in the range of non-transfected HEK 293 cells (5.1 ± 0.7 pA/pF, n = 5).
ND7/23 cells – a hybridoma cell line of mouse neuroblastoma and rat DRG neurons –, transfected with either NaV1.8 or NaV1.8−Δe11, were assayed in the whole-cell patch-clamp mode using physiological solutions to retain activity of intracellular enzymes. Under control conditions, the two channel types did not show differences regarding parameters of voltage-dependence of channel activation. Vm was –15.9 ± 1.2 mV and km was 16.7 ± 1.0 mV (n = 7) for hNaV1.8 and –15.9 ± 1.5 mV and 16.3 ± 0.8 mV (n = 8) for hNaV1.8−Δe11, respectively. As a positive control, forskolin stimulation of rat NaV1.8 was monitored according to Fitzgerald et al.9 rNaV1.8 showed a strong response to forskolin stimulation regarding an increase in the peak current amplitude by a factor of 1.48 ± 0.07 (P < 0.002) with a single-exponential time constant of 36.3 ± 4.9 s at 0 mV (n = 11). Voltage of half-maximal activation per gate was shifted to the left by about 6.8 ± 0.7 mV (n = 9; P < 0.001) compared with control. However, hNaV1.8 did not respond to forskolin application. Peak current amplitude was only insignificantly altered by a factor of 1.03 ± 0.08 (n = 7; P = 0.76). The current-voltage relationship was also not affected (n = 6; P = 0.98). hNaV1.8−Δe11 behaved like the long channel isoform (IFSK/ICtrl = 1.02 ± 0.10, n = 8, P = 0.59; IV: n = 5, P = 0.42; Figure 3).
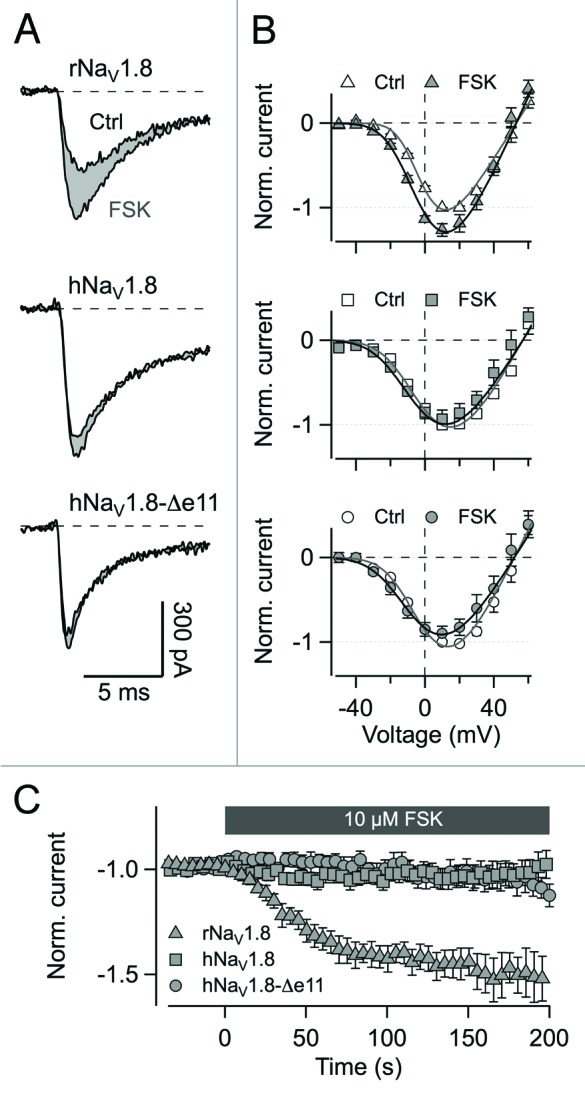
Figure 3. Stimulation of protein kinase A phosphorylation by the adenylyl cyclase activator forskolin. (A) Current traces evoked by depolarization from −80 mV to 0 mV before and after extracellular application of 10 µM forskolin (FSK) for NaV1.8 from rat, hNaV1.8, and hNaV1.8−Δe11 in ND7/23 cells. (B) Current-voltage relationships for the channels in (A) before and after FSK application normalized to the peak inward current and averaged for 6–8 cells ± s.e.m. (C) Time course of peak current changes upon FSK stimulation normalized to control level for NaV1.8 from rat, hNaV1.8, and hNaV1.8−Δe11. Application of FSK starts at t = 0 s. Data points are mean values for 7–11 cells each ± s.e.m.
A substantial deletion of the domain I/II linker apparently has almost no immediate functional consequences for the channel protein. This finding is compatible with an observation by Faber et al.17 who described the mutation L554P in human NaV1.8, resulting in the formation of a PPP motif within the sequence encoded by exon 11. Upon expression in mouse DRG neurons, NaV1.8-L554P channels revealed no differences compared with wild-type channels regarding their voltage dependence of activation and steady-state inactivation.17 Only recovery from inactivation seemed to be slightly faster for the mutant, and increased ramp current amplitudes were reported.17 No such effect was observed for hNaV1.8 and hNaV1.8−Δe11, but the inactivation properties of hNaV1.8 measured in Neuro-2A cells are strongly different from those described for transfected DRG neurons.17
The domain I/II linker contains several putative protein interaction sites presumably contributing to the physiological regulation of NaV channels. It was shown that rat NaV1.8 possesses an ER retention/retrieval motif in the region encoded by exon 11; this motif can be masked by NaVβ3 to allow for membrane trafficking.7 Furthermore, it is commonly observed that NaV1.8 channels can be expressed in neuronal cells while heterologous expression in HEK 293 cells is very poor. A likely explanation could be the endogenous expression of NaVβ1 and NaVβ3 in neuronal cells, such as in ND7/23,18 affecting the retention mechanism. However, here we did not observe any difference in functional channel expression of both isoforms, neither in neuronal nor in HEK 293 cells. Since human NaV1.8 channels contain a KRR motif, this might be a weaker ER retention signal or a motif that does not interact with the NaVβ3 subunit. Thus, the KRR retention motif can be excluded as a molecular reason for the weak expression of NaV1.8 channels in HEK 293 cells. Since ND7/23 and Neuro-2A cells used in this study are derived from rodents – where the splice variant does not exist − such cells may lack another as yet unidentified interaction protein who’s interaction with the domain I/II linker is possibly affected once the exon 11-encoded sequence is absent.
Apparently, skipping of exon 11 has no impact on the functionality of human NaV1.8 channels expressed in the neuronal cell lines Neuro-2A and ND7/23. The sequence of exon 11 encodes 98 amino acids whereof 30 differ between the rat, mouse, and human orthologs. This affects the PKA consensus sequences (RXXS) resulting in three such sites in the human but four in the rat and mouse channel isoform, whereby one of the human sites is not conserved on the rodent forms. The putative ER retention signal (sequence RRR vs. KRR) and one of the two interaction sites (consensus SP) of p38 kinase are affected as well. Regulatory impact of such sites has been reported for rat NaV1.8 only,7-9,19 and in this study no effect of forskolin stimulation was detectable for hNaV1.8, while currents mediated by rat NaV1.8 was augmented. Based on a protein sequence comparison with NaV1.8 from other species (P. troglodytes, M. mulatta, C. lupus and B. taurus) an evolutionary loss of function in exon 11 can be hypothesized. It appears that human NaV1.8 channels are less subject to cellular regulation owing to their diverging sequence of the domain I/II linker and, hence, skipping of exon 11 is tolerated in humans due to a lack of phenotypic consequences.
Materials and Methods
Tissues, RNA extraction, and reverse transcription
Human DRG tissue was sampled during routine autopsy for neuropathological examinations in accordance with ethical regulations of the state of Saxony-Anhalt and the European Communities Council Directives, and approved by the Institutional Review Board of the Otto-von-Guericke University, Magdeburg, Germany. Samples were frozen on dry ice immediately after sampling, anonymized, and stored at –80 °C. Neuropathological examinations revealed no pathological alterations in the human DRGs analyzed. Rats and mice were fed standard chow and water ad libitum, and animal care procedures were performed according to local guidelines of animal protection, approved by the Thuringian Animal Care Council (registration number: 02–101/13). Tissues from adult C57/B6-J mice and Wistar rats, killed by cervical dislocation, were frozen on dry ice and stored at –80 °C. DRGs were pooled from one specimen. Total RNA from DRG was purified using an RNeasy Mini Kit (Qiagen) and subsequently reversed transcribed using the SuperScriptIIITM First-Strand Synthesis System (Invitrogen).
Isoform identification
An RT-PCR amplicon spanning exon 11 was included in an amplicon tiling path across human SCN10A mRNA. RT-PCR was performed using Taq polymerase (BioLine BioMix Red, Luckenwalde, Germany), 1 rxn unit cDNA from human Medulla oblongata (BioChain, Newark, CA), and primers (Metabion, Martinsried, Germany) 5′-CTCTGTGGGA ACAGCAGTGAT-3′ with 5′-CTTCTCCCCT TGCGATGTGT-3′. Concentrations of the PCR products were measured on a UV/VIS spectrophotometer ND-1000 (Nanodrop, Germany), and the products were mixed in an equimolar ratio. Ligation of 454 adaptors A and B, the emulsion PCR protocol (FLX emPCR amplicon), and sequencing on a 454 GS FLX was performed according to standard protocols (Roche Diagnostics, Basel, Switzerland). The obtained reads were aligned to a reference sequence based on the RefSeq transcript (NM_006514.2), and variants were identified and quantified using in-house software.
Isoform quantification
Amplicons spanning the splice-variable SCN10A region were obtained using Taq polymerase, 1 µl cDNA (corresponding to 40 ng isolated total RNA), and primers 5′-CACCCCGCTC TGATCCTTAC-3′ with 5′-TGAGACAAGC TGGTCAAGCA-3′ (human SCN10A), 5′-TCAGAAGGCT CGACAGATGAC-3′ with 5′-GGGTGGGCAC TTCAGCTTAG-3′ (mouse Scn10a), 5′-CTCCACGGAT GACAACAGGT-3′ with 5′-GGGTGGGCAC TTCAGCTTAG-3′ (rat Scn10a). The thermocycle protocol was 2 min initial denaturation at 94 °C, followed by 40 cycles of 45 s denaturation at 94 °C, 50 s annealing at 56 °C, 1 min extension at 72 °C, and a final 10 min extension step at 72 °C. PCR products were separated on a 1.5% agarose gel and visualized using ethidium bromide.
Channel constructs
The pCMV6-XL5 expression plasmid coding for human wild-type NaV1.8 (SCN10A, NM_006514.2) was purchased from OriGene Technologies Inc. (Rockville, Maryland, USA). A variant lacking the region coded by exon 11 (deleted residues S488 to S585; NaV1.8−Δe11) was constructed using a PCR-based strategy and verified by sequencing. A pcDNA3 plasmid was used for expression of rat NaV1.8 (AAC52619, ref. 20).
Cell culture and transfection
Neuro-2A (DSMZ, Braunschweig, Germany) and ND7/23 (provided by Dr. C. Nau, Lübeck) cells were maintained in 90% Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal calf serum in a 10% CO2 incubator at 37 °C. HEK 293 cells (CAMR, Porton Down, Salisbury, UK) were maintained in 45% Dulbecco’s Modified Eagles Medium (DMEM) and 45% Ham’s F12 Medium supplemented with 10% fetal calf serum in a 5% CO2 incubator at 37 °C. Cells were trypsinized, diluted with culture medium, and grown in 35-mm dishes. When grown to 30–50% confluence, cells were transfected with a 5:1 ratio of the NaV channel expression plasmids and a vector encoding the CD8 antigen using the Superfect transfection kit (Qiagen). Dynabeads (Deutsche Dynal GmbH, Hamburg, Germany) were used for visual identification of individual transfected cells. Electrophysiological recordings were performed 1–2 d after transfection.
Electrophysiology
Whole-cell voltage-clamp experiments were performed as previously described.21 Briefly, borosilicate glass patch pipettes with resistances of 0.9–1.5 MΩ were used. The series resistance was compensated for about 80% in order to minimize voltage errors. An EPC-10 patch-clamp amplifier was operated by PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Leak and capacitive currents were corrected with a p/6 method. Currents were low-pass filtered at 5 kHz and sampled at a rate of 25 kHz. All experiments were performed at constant temperature (19–21 °C). Data analysis was performed using FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA) software. Data are presented as mean ± standard error of the mean (n = number of independent experiments). Data sets were tested for statistical significance using a two-sided Student’s t test with unequal variances when appropriate.
Current-voltage relationships
From a holding potential of –120 mV, test depolarizations in the range from –70 to 50 mV in steps of 10 mV were applied at an interval of 5 s. The peak currents were fit with a Hodgkin-Huxley activation formalism involving m = 3 activation gates and a linear single-channel characteristic:
(Eq. 1)
where I is the peak current and V is the test pulse voltage. Vm is the voltage of half-maximal gate activation, km is the corresponding slope factor, Gmax is the maximal conductance of all channels and Erev the reversal potential.
Voltage ramp recordings
Voltage ramps from –80 to 20 mV in 500 ms were applied. Ramp currents were normalized to the maximal transient inward currents recorded in the IV and plotted against the ramp voltage.
Inactivation kinetics
The kinetics of activation and fast channel inactivation were analyzed using a Hodgkin-Huxley function to fit the current decay during depolarizing pulses to –30, –10, 10, and 30 mV from a holding potential of –120 mV:
(Eq. 2)
where I is the current, τh1 and τh2 are the fast and slow time constant describing the current decay, r12 is the amplitude ratio of the fast and the slow component, and hinf is the steady-state inactivation, τm is the time constant describing activation kinetics.
Steady-state inactivation
From a holding potential of –120 mV cells were conditioned for 500 ms at voltages ranging from –120 to –20 mV in steps of 10 mV. Subsequently, peak current was determined at 0 mV. The repetition interval was 20 s. The peak current plotted vs. the conditioning voltage was described with a Boltzmann function:
(Eq. 3)
with the half-maximal inactivation voltage Vh and the corresponding slope factor kh.
Recovery from fast inactivation
From a holding potential of −120 mV channels were inactivated with a 10-ms pulse to 0 mV. A second pulse to 0 mV, applied after a variable time interval at −120 mV, was used to assay the recovery of the channels from fast inactivation:
(Eq. 4)
with time t, the current amplitudes I1 (first pulse) and I2 (second pulse) and the time constants for the fast and slow components of the recovery from inactivation τfast and τslow, respectively.
Solutions and chemicals
In most experiments the bath solution contained (in mM): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES (pH 7.4 with NaOH) and the patch pipettes contained (in mM): 35 NaCl, 105 CsF, 10 EGTA, 10 HEPES (pH 7.4 with CsOH). Forskolin (Sigma-Aldrich, Steinheim, Germany) experiments were performed in bath solution containing (in mM): 100 NaCl, 5 KCl, 2.5 MgCl2, 40 TEA, 0.01 CdCl2, 10 glucose, 2 Na2HPO4, 5 HEPES (pH 7.4 with NaOH); the corresponding pipette solution contained (in mM): 120 CsCl, 10 NaCl, 2.5 KCl, 10 MgCl2, 0.5 Na2ATP, 5 EGTA, 5 HEPES (pH 7.4 with CsOH). Forskolin was dissolved in bath solution and applied focally to the cells at a concentration of 10 µM. To completely block Na+ channels endogenous to Neuro-2A and ND7/23 cells, external solutions were supplemented with 300 nM tetrodotoxin. In addition, the holding potential was set to −80 mV in all forskolin experiments to optimize cell viability.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (SFB 604, S.H.H., M.P.) and Landesgraduiertenprogramm Thüringen (J.S.).
References
- 1.Goldin AL. Evolution of voltage-gated Na(+) channels. J Exp Biol. 2002;205:575–84. doi: 10.1242/jeb.205.5.575. [DOI] [PubMed] [Google Scholar]
- 2.Malik-Hall M, Poon WY, Baker MD, Wood JN, Okuse K. Sensory neuron proteins interact with the intracellular domains of sodium channel NaV1.8. Brain Res Mol Brain Res. 2003;110:298–304. doi: 10.1016/S0169-328X(02)00661-7. [DOI] [PubMed] [Google Scholar]
- 3.Renganathan M, Cummins TR, Hormuzdiar WN, Waxman SG. α-SNS produces the slow TTX-resistant sodium current in large cutaneous afferent DRG neurons. J Neurophysiol. 2000;84:710–8. doi: 10.1152/jn.2000.84.2.710. [DOI] [PubMed] [Google Scholar]
- 4.Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–40. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- 5.Joshi SK, Mikusa JP, Hernandez G, Baker S, Shieh CC, Neelands T, Zhang XF, Niforatos W, Kage K, Han P, et al. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain. 2006;123:75–82. doi: 10.1016/j.pain.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Zimmermann K, Leffler A, Babes A, Cendan CM, Carr RW, Kobayashi J, Nau C, Wood JN, Reeh PW. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447:855–8. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]
- 7.Zhang ZN, Li Q, Liu C, Wang HB, Wang Q, Bao L. The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the β3 subunit. J Cell Sci. 2008;121:3243–52. doi: 10.1242/jcs.026856. [DOI] [PubMed] [Google Scholar]
- 8.Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol. 1999;516:433–46. doi: 10.1111/j.1469-7793.1999.0433v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thimmapaya R, Neelands T, Niforatos W, Davis-Taber RA, Choi W, Putman CB, Kroeger PE, Packer J, Gopalakrishnan M, Faltynek CR, et al. Distribution and functional characterization of human Nav1.3 splice variants. Eur J Neurosci. 2005;22:1–9. doi: 10.1111/j.1460-9568.2005.04155.x. [DOI] [PubMed] [Google Scholar]
- 11.Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–8. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- 12.Walzik S, Schroeter A, Benndorf K, Zimmer T. Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS One. 2011;6:e19188. doi: 10.1371/journal.pone.0019188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatelier A, Dahllund L, Eriksson A, Krupp J, Chahine M. Biophysical properties of human Na v1.7 splice variants and their regulation by protein kinase A. J Neurophysiol. 2008;99:2241–50. doi: 10.1152/jn.01350.2007. [DOI] [PubMed] [Google Scholar]
- 14.Kerr NC, Holmes FE, Wynick D. Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J Biol Chem. 2004;279:24826–33. doi: 10.1074/jbc.M401281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schirmeyer J, Szafranski K, Leipold E, Mawrin C, Platzer M, Heinemann SH. A subtle alternative splicing event of the Na(V)1.8 voltage-gated sodium channel is conserved in human, rat, and mouse. J Mol Neurosci. 2010;41:310–4. doi: 10.1007/s12031-009-9315-3. [DOI] [PubMed] [Google Scholar]
- 16.Schindler S, Heiner M, Platzer M, Szafranski K. Comparison of methods for quantification of subtle splice variants. Electrophoresis. 2009;30:3674–81. doi: 10.1002/elps.200900292. [DOI] [PubMed] [Google Scholar]
- 17.Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, Persson AK, Hoeijmakers JG, Gerrits MM, Pierro T, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A. 2012;109:19444–9. doi: 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.John VH, Main MJ, Powell AJ, Gladwell ZM, Hick C, Sidhu HS, Clare JJ, Tate S, Trezise DJ. Heterologous expression and functional analysis of rat Nav1.8 (SNS) voltage-gated sodium channels in the dorsal root ganglion neuroblastoma cell line ND7-23. Neuropharmacology. 2004;46:425–38. doi: 10.1016/j.neuropharm.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Liu C, Li Q, Su Y, Bao L. Prostaglandin E2 promotes Na1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic. 2010;11:405–17. doi: 10.1111/j.1600-0854.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- 20.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–62. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Gordon D, Heinemann SH. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and Lqh alphaIT. Pflugers Arch. 2000;439:423–32. doi: 10.1007/s004240050959. [DOI] [PubMed] [Google Scholar]