

Abstract
Cannabis sativa L. is an important economic plant for the production of food, fiber, oils, and intoxicants. However, lack of sufficient simple sequence repeat (SSR) markers has limited the development of cannabis genetic research. Here, large-scale development of expressed sequence tag simple sequence repeat (EST-SSR) markers was performed to obtain more informative genetic markers, and to assess genetic diversity in cannabis (Cannabis sativa L.). Based on the cannabis transcriptome, 4,577 SSRs were identified from 3,624 ESTs. From there, a total of 3,442 complementary primer pairs were designed as SSR markers. Among these markers, trinucleotide repeat motifs (50.99%) were the most abundant, followed by hexanucleotide (25.13%), dinucleotide (16.34%), tetranucloetide (3.8%), and pentanucleotide (3.74%) repeat motifs, respectively. The AAG/CTT trinucleotide repeat (17.96%) was the most abundant motif detected in the SSRs. One hundred and seventeen EST-SSR markers were randomly selected to evaluate primer quality in 24 cannabis varieties. Among these 117 markers, 108 (92.31%) were successfully amplified and 87 (74.36%) were polymorphic. Forty-five polymorphic primer pairs were selected to evaluate genetic diversity and relatedness among the 115 cannabis genotypes. The results showed that 115 varieties could be divided into 4 groups primarily based on geography: Northern China, Europe, Central China, and Southern China. Moreover, the coefficient of similarity when comparing cannabis from Northern China with the European group cannabis was higher than that when comparing with cannabis from the other two groups, owing to a similar climate. This study outlines the first large-scale development of SSR markers for cannabis. These data may serve as a foundation for the development of genetic linkage, quantitative trait loci mapping, and marker-assisted breeding of cannabis.
Introduction
Cannabis sativa L., a member of the Cannabinaceae, is a diploid (2n = 20) monocotyledon, and one of the oldest cultivated plants. It has been cultivated or grows wild around the world, and is used in diverse applications [1]. Cannabis is a botanical genus of flowering plants that is divided into two species. Hemp, which has low tetrahydrocannabinol (THC) content, is mainly used to cultivate for fiber and seed production, whereas marijuana, with its relatively higher THC content, is used for its psychoactive potency [2]. Although cannabis cultivation is being restricted in many countries due to its potential use as a recreational drug, there has been a resurgence of interest in cannabis for its agronomic potential. In fact, hemp was used for textile production in China more than 6,000 years ago, and is still widely grown in China now.
Identification of variability among its genotypes is the key factor in any crop's improvement. Molecular markers play an important role in selecting these diverse genotypes. During the initial stages of development of molecular markers, some types of markers were used to analyse the genetic diversity in cannabis. These included random amplified polymorphic DNA (RAPD) [3]–[5], amplified fragment length polymorphisms (AFLP) [6]–[8], and inter simple sequence repeat amplification (ISSR) [9]. However, these markers have the common shortcoming of poor repeatability and dominance. Single nucleotide polymorphism (SNP) was mainly used for studies of specific genes in cannabis [10]. More recently, microsatellite markers were proven highly useful in applied breeding programs [11]. Microsatellites, known as simple sequence repeats (SSRs), are tandem repeats of short (1 to 6 bp) DNA sequences that exist throughout the entire genome of eukaryotic organisms, including both non-coding and coding regions. SSRs are among the most useful genetic markers in biology. SSR loci have many distinguishing features, such as high information content, co-dominant inheritance patterns, consistent distribution along chromosomes, reproducibility, and locus specificity [12]–[14]. Furthermore, SSRs demonstrate an impressive ability to be transferred among related species, making them excellent markers for comparative genetic and genomic analyses [15]. Therefore, SSRs are widely used for genetic studies in recent years. However, only small-scale SSR markers have been reported in cannabis [1], [2], [16], which is far from sufficient for effective genetic mapping and marker-assisted breeding. The complete genome and transcriptome sequence of cannabis was published in 2011 [17], providing a foundation for cannabis research. Nowadays, identification of SSRs from expressed sequence tags (ESTs) is the preferred method for rapid and inexpensive marker development. Furthermore, these EST-SSRs can then be directly linked to the genes that confer important agronomic traits. Thus, SSRs based on the ESTs have become one of the most important methods in agriscience for the analysis of genetic diversity, genotype identification, high density genetic mapping, molecular tagging of genes, and marker assisted selection (MAS) breeding [18]. There are many reports about the application of EST-SSR markers in various plants at present, including sugar cane [19], barley [20], grapes [21], Chinese cabbage [22], and citrus [23]. Therefore, developing EST-SSR in cannabis on a large scale is important and urgent.
In the present study, potential SSR loci were characterized from the cannabis ESTs [17] and large-scale development of EST-SSRs in the genus was performed for the first time. This was followed with evaluation of the quality of the novel SSR markers and subsequent selection of a group of them for genetic diversity analysis. These results will provide a valuable resource for genetic and genomic studies of cannabis for genetic studies.
Materials and Methods
Plant materials
One hundred and fifteen cannabis varieties were used for the polymorphic analysis of SSR markers. Among them, 100 varieties were from 18 provinces of China, Longdama 1 (LDM), Jinma 1 (JM1), Wandama 1 (WDM), Yunma 1 (YM1), Yunma 4 (YM4), Yunma 5 (YM5) and Yunwan 6 (YW6) were cultivated varieties and the others were all wild varieties in China. The other 15 varieties were from Ukraine, Poland, and France, respectively (Table S1). Seeds for the study were collected from the Institute of Bast Fiber Crops, China Academy of Agriculture Science, Changsha, China.
DNA extraction and quantification
The cannabis seeds were grown in pots under natural conditions. DNA was extracted from seedlings (100 mg) by the cetyltrimethyl ammonium bromide (CTAB) method [24]. After extraction, 3 µL of the DNA sample (for all varieties) was loaded in 1.0% agarose gels. A DNA marker was loaded as a control to assess the quality and the quantity of DNA. Based on the marker standards, DNA samples were normalized to a uniform concentration (approximately 10 ng/µL) and used for SSR genotyping.
SSRs sources and primer development
Potential SSR markers were detected among the 32,324 sequences using the AutoSSR software [17], [25]. The parameters were adjusted for identification of perfect di-, tri-, tetra-, penta-, and hexanucleotide motifs with a minimum of 9, 6, 5, 4, and 3 repeats, respectively. The ESTs sequences were used to design primers flanking the potential SSRs. Input criteria for the Primer 3.0 software for designing primers were as follows: length, 17–23 bp; GC content, 40–60%; and estimated amplicon size, 100–400 bp [26]. Primers were synthesized by Sangon Biotech (Shanghai) Co., Ltd.
SSRs amplification and detection of polymorphisms
Approximately, 10 ng of template DNA was added to a 10-µL PCR mix containing 1×PCR buffer with MgCl2 (Perkin-Elmer), 250 µM dNTPs, 0.2 µM primers (forward and reverse), and 0.5 units of TaqGold DNA Polymerase (Perkin-Elmer). The PCR reaction profile comprised a 10-min incubation at 94°C, then a cycle of 94°C for 30 s, 50–60°C for 30 s and 72°C for 40 s, repeated 35 times. Following cycling, the reaction was held at 72°C for 10 min, before a final 4°C hold. The entire reaction is carried out in the BioRAD S1000 Thermal cycler. The PCR product quality was checked in a 1.0% agarose gel, using 3 µL of the PCR reaction and the remaining reaction was then subjected to electrophoresis on a 8% polyacrylamide gel, consisting of 30% PA (acrylamide+N, N-methylene bisacrylamide, Biosharp) 12 mL, 10×TBE 4 mL, TEMED 50 µL, 10% APS (ammonium peroxydisulfate, Biosharp) 950 µL, and ddH2O to a total volume of 45 mL. Electrophoresis was carried out in 1×TBE buffer at 220 U for 90 min. Gels were stained with 0.1% silver nitrate following a chromogenic reaction with 1.5% NaOH (including 1% formaldehyde), and finally photographed in white light. We screened 24 typical cannabis varieties to assess the usefulness of the SSR primer pairs developed in this study.
Determination of genetic diversity among 115 cannabis varieties
Forty-five markers (Table S2) were selected from the 117 EST-SSR primers developed to analyse the relationship of cannabis varieties. The allelic data were converted into a binary matrix using the score 1, 0 for presence and absence of the allele. The binary data were analysed using the Numerical Taxonomy Multivariate Analysis System (NTSYS-pc) version 2.10 software [27]. Genetic similarity (GS) coefficients were calculated based on the coefficient for similarity matching by using the SIMQUAL module of the software. Using the GS matrix, we constructed a dendrogram using the unweighted pair group method with arithmetic average (UPGMA) to determine genetic relationships among the 115 genotypes. Principal Coordinate Analysis (PCoA) was also performed using NTSYS-pc software to resolve the patterns of clustering among genotypes. The effective allele number (Ne), Shannon's informative index (I), expected heterozygosity (HE), and the percentage of polymorphic loci (PP) were calculated by Popgen Ver.132 [28].
Results
Development and characterization of SSR markers
In this study, EST-SSR loci were detected from the 32,324 EST sequences of the cannabis transcriptome using AutoSSR software [17], [25]. A total of 4,577 potential SSR loci were identified from 3,624 EST sequences (Table 1). The frequency of occurrence for SSRs was 1 SSR per 8.49 kb, and 11.21% of the EST sequences contained SSR loci. Among the 3,624 EST sequences, 505 sequences contained 2 SSR loci, 114 sequences contained 3 SSR loci, 33 sequences contained 4 SSR loci, and 16 sequences contained more than 4 SSR loci. Moreover, 121 sequences contained several SSR motifs that were present in a compound formation. Finally, a total of 3,442 SSR markers were developed and made publicly available (Table 1, Table S3).
Table 1. Results of searches for EST-SSRs in cannabis.
| Search item | Numbers |
| Total number of sequences searched | 32324 |
| Total size of sequences searched (bp) | 39038131 |
| Total number of SSRs identified | 4577 |
| Total number of SSR-containing sequences | 3624 |
| Number of sequences containing more than 1 SSR | 668 |
| Dinucleotides | 748 |
| Trinucleotides | 2334 |
| Tetranucleotides | 174 |
| Pentanucleotides | 171 |
| Hexanucleotides | 1150 |
| Marker number of SSRs present in compound formation | 121 |
| Total number of SSR markers developed | 3442 |
In order to further characterize the potential SSR loci, the types, frequencies, and distributions of the 4,577 SSRs were analysed. The result indicated that trinucleotide was the most abundant (2,334, 50.99%) type of repeat. The number of hexanucleotide, dinucleotide, tetranucleotide, and pentanucleotide markers were 1150 (25.13%), 748 (16.34%), 174 (3.8%), and 171 (3.74%), respectively (Table 1). Table 2 presents the lengths of the EST-SSRs based on the number of repeat units. The results showed that 3,385 SSRs (73.96%) were calculated to range from 18 to 22 bp. Only 90 SSRs (1.97%) were longer than 40 bp (Table 2).
Table 2. Frequency of EST-SSRs in cannabis.
| Motiflength | Repeat numbers | ||||||||
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ≥11 | |
| Di | 265 | 142 | 341 | ||||||
| Tri | 1262 | 557 | 249 | 158 | 51 | 57 | |||
| Tetra | 126 | 42 | 6 | ||||||
| Penta | 137 | 25 | 9 | ||||||
| Hexa | 931 | 177 | 30 | 8 | 3 | 1 | |||
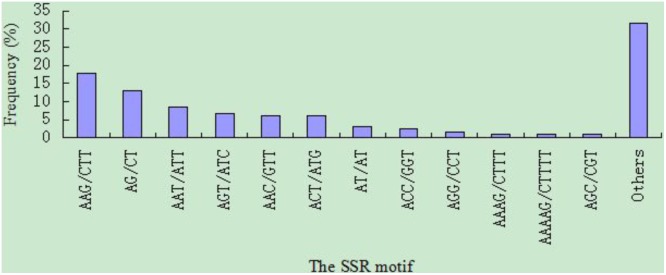
Among the 3,442 EST-SSRs a total of 318 motif sequence types were identified. The abundance of each motif was 3, 10, 19, 51, and 235 of dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeats, respectively. The most abundant type of repeat motif was the AAG/CTT trinucleotide repeat (822, 17.96%), followed by AG/CT (590, 12.89%), AAT/ATT (395, 8.63%), AGT/ATC (303, 6.62%), AAC/GTT (278, 6.07%), ACT/ATG(276, 6.03%), AT/AT (141, 3.08%), ACC/GGT (111, 2.43%), AGG/CCT (68, 1.49%), AAAG/CTTT (52, 1.13%), AAAAG/CTTTT (50, 1.09%), and AGC/CGT (47, 1.03%), respectively (Figure 1). The other 306 motifs (1444, 31.55%) not specifically mentioned were also presented to be entered into the database in addition to the above repeats motifs (Figure 1).
Figure 1. Frequency distribution of cannabis EST-SSR based on motif numbers.
One hundred and seventeen primer pairs were randomly selected to evaluate the quality of the SSR markers across 24 cannabis varieties (Table S1). Amplified DNA fragments were observed with 108 markers, while 9 markers failed to generate amplicons. A total of 119 loci were successfully amplified by the 108 SSR markers. Among the 24 cannabis varieties tested, 21 loci showed no polymorphism. Among the polymorphic loci, 62, 24, 8, 1, 2, and 1 each had 2, 3, 4, 5, 6, and 7 alleles, respectively.
Evaluation of genetic relationships among cannabis varieties
Based on these results, 45 high-quality primer pairs (56 loci) were selected to evaluate the genetic diversity and relatedness of the 115 cannabis varieties listed in Table S1. The number of alleles per locus ranged from 2 to 7 among the 115 varieties, with an average of 2.87. Considering 2 varieties as a variety pair, 6555 variety pairs were found among the 115 varieties. Among 6555 variety pairs, the largest polymorphic ratio (99.38%) was observed between 53 and 54 and the two varieties were all from Shanxi1 province. The smallest polymorphic ratio (31.01%) was observed between 107 and Y5, 107 was from Europe and Y5 was from Yunnan province (Table S4).
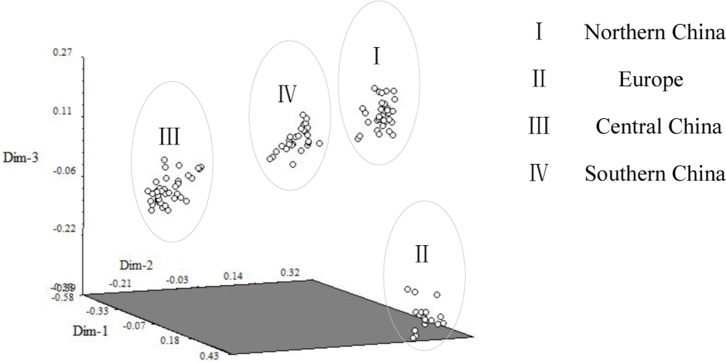
The genetic similarity (GS) coefficients between the cannabis varieties were calculated in the SIMQUAL module of NTSYS-pc [28]. Using a GS score of 0.74 as the threshold, the 115 cannabis varieties could be classified into 4 clusters (Figure 2). The results showed that 34 Northern China (Heilongjiang, Liaoning, Jilin, Neimenggu, and Xinjiang provinces) varieties, 27 Southern China (Anhui, Chongqing, Jiangsu, Zhejiang, Yunnan and Guangxi provinces) varieties, and 39 Central China (Ningxia, Hebei, Henan, Gansu, Shandong, Shanxi1, and Shanxi2 province) varieties fell into clusters I, IV and III, respectively. Fifteen varieties originated from Europe (Ukraine, Poland, and France), and were assigned as individual cluster II.
Figure 2. Dendrogram derived using UPGMA cluster analysis based on the SSR genetic similarity coefficient for 115 cannabis varieties.

In PCoA, the three main axes explained approximately 62% of the total variation, at 36.4%, 16.8%, and 8.7%, respectively. The 115 cannabis varieties could also be distinctly classified into 4 groups,which was consistent with the results of cluster analysis (Figure 3).
Figure 3. Principles coordinate analysis for SSR markers using the genetic similarity matrix for 115 cannabis varieties.
Based on the above results, 115 cannabis varieties could be divided into 4 groups mainly according to the geographical distribution: Northern China, Europe, Central China and Southern China. Then the effective allele number (Ne), Shannon's informative index (I), expected heterozygosity (HE), and the percentage of polymorphic loci (PP) values were calculated for the 4 groups. The results showed that the highest level of genetic diversity was observed in the Central China group. Data in descending order, the Shannon's informative index was III, IV, I, II; the expected heterozygosity was III, IV, I, II; the percentage of polymorphic loci was III, I, IV, II (Table 3).
Table 3. Variability parameters in 4 groups of cannabis.
| Cluster | Origin | Number ofvarieties | Ne | I | HE | PP (%) |
| I | Northern China | 34 | 1.7747 | 0.5518 | 0.3548 | 76.36 |
| II | Europe | 15 | 1.3423 | 0.2776 | 0.1820 | 41.82 |
| III | Central China | 39 | 1.8008 | 0.5800 | 0.3698 | 85.45 |
| IV | Southern China | 27 | 1.7743 | 0.5602 | 0.3565 | 74.55 |
Discussion
Development and characterization of EST-SSR markers of cannabis
Simple sequence repeats (SSRs) are important molecular markers for genetic mapping and variety identification in cannabis and other plants. However, lack of sufficient SSR markers has limited the development of cannabis molecular genetics. In this study, 3,442 EST-SSR markers were identified for the first time. The frequency of EST-contained SSRs was 11.21%. This frequency was higher than the reported fiber crops flax (3.5%) and ramie (3.83%) [29], [30], but lower than other reported plants such as coffee (18.5%) and oilseed rape (15.58%) [31], [32]. The difference was also reflected in the average distance between SSRs, which was 8.49 kb in cannabis, but 16.5 kb and 19.3 kb in flax and ramie [29], [30]. In addition, two of the most abundant motifs were trinucleotide and hexanucleotide repeats in cannabis, while dinucleotide and trinucleotide repeats were the most abundant motifs in flax and ramie. Interestingly, the EST-SSR distribution in cannabis was similar to that in melons [33]. A previous study had revealed that AG/CT was the most abundant class of the isolated SSRs, representing 50% overall in cannabis [2]. However, our data demonstrated that AAG/CTT was the most abundant motif (17.96%). The discrepancy may result from the different techniques utilized in developing the SSR markers. Probe technology was used to develop markers in the previous study, while this study had a transcriptome database (via AutoSSR software) at its disposal. The frequency of the motif here was similar to other dicots, including citrus and melon [23], [33]. Gao hypothesized that AAG/CTT was an advantageous repeat motif in dicots, and that its higher frequency may be related to the increased use of the corresponding proteins [34]. As there is no doubt that cannabis is a dicot, these data suggest that the results presented here may be closer to the actual frequency of the motif.
In order to evaluate the quality of the primers designed, 117 SSRs were randomly selected to amplify their target sequences among 24 cannabis varieties. Of these, 108 (92.31%) SSRs successfully yielded amplicons. Given that the SSRs were randomly selected, the amplification efficiency may be considered to be the actual availability of all 3,442 identified SSRs. Nine SSRs failed to generate amplicons, most likely due to the location of the primer across splice sites, the presence of large introns, and association with poor-quality sequences [20], [29]. In addition, 5 amplicons deviated from their expected sizes. This phenomenon was also noted in other species, and may be due to introns or large insertions, among other reasons [35], [36]. All 115 varietals selected for diversity testing came from the hemp species, while the ESTs came from Cannabis sativa Purple Kush, a marijuana strain that is widely used for its medicinal effects [17]. Species differences between marijuana and hemp may therefore have affected the amplification efficiency. Nevertheless, the observed 92.31% amplification efficiency indicated that the developed EST-SSRs were of high quality, and may serve as a good foundation for future research of cannabis.
Genetic diversity and climate influences of cannabis
Selection and use of genetically diverse genotypes are key factors in any crop breeding program in order to develop cultivars with a broad genetic base. Hemp is a dioecious annual that commences its reproductive cycle when photoperiods are shorter than a critical length [37], such that day length and temperature may determine the floral transition and flowing times [38], [39]. One of the most important traits for hemp is the timing of the transition from a vegetative to flowering state, which can control the growth period, as well as affecting the fiber quality and yield. For example, Northern European hemp grown in Southern Europe performs poorly due to premature flowering, resulting in shorter vegetative periods that limit stem growth, whereas the opposite occurs when Southern European varieties are grown in Northern Europe [38], [40]. Similar phenomena were also found in China. Climate zones are defined as areas with distinct climates, and are classified according to the average and the typical ranges of different variables such as temperature and precipitation. Sunshine intensity and day length in different latitudes are the main factors affecting temperature. Therefore, climate zones are closely related to latitude. Our results showed that 100 hemp varieties from China could be classified into 3 distinct clusters, and that the 3 clusters were consistent with the cool temperate, warm temperate, and subtropical zones in China, respectively. These indicated that the climate, created by latitude, temperature, and day length, is a key factor affecting the germplasm diversity of hemp. Although the three clusters from China were physically closer, hemp in Northern China had a greater similarity coefficient with European hemp than with the other two clusters. This is most likely because these two regions are at similar latitudes, and thus have similar climatic conditions. The varieties from Heilongjiang and Yunnan provinces were individually classified in clusters IA and IVA (Figure 2), perhaps owing to the higher latitude (i.e. the colder environment) and low latitude plateaus, respectively. Our results provide a new insight into the study on germplasm resources and systematic classification in hemp, which may be helpful for the introduction, germplasm development, and utilization in different climates, countries, or continents.
In conclusion, this is the first report on large-scale development of SSR markers in cannabis and provides guidance for germplasm introduction and utilization. Future studies with these SSR markers could be useful for conservation programs, identification activities, and breeding procedures.
Supporting Information
Description of 115 cannabis varieties used in this study.
(DOC)
One hundred and seventeen EST-SSR markers used for PCR amplification.
(XLS)
The information of 3443 EST-SSR markers developed.
(XLS)
The polymorphic ratios among 115 cannabis varieties.
(XLS)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was financially supported by the International Cooperation Project in Science and Technology (2010DFR30620-4), National Modern Agro-industry Technology Research System (nycytx-19-E04), and National Natural Science Foundation of China (no. 31301376). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gilmore S, Peakall R (2003) Isolation of microsatellite markers in Cannabis sativa L. (marijuana). Mol Ecol Notes 3: 105–107. [Google Scholar]
- 2. Alghanim HJ, Almirall JR (2003) Development of microsatellite markers in Cannabis sativa for DNA typing and genetic relatedness analyses. Anal Bioanal Chem 376: 1225–1233. [DOI] [PubMed] [Google Scholar]
- 3. Gillan R, Cole MD, Linacre A, Thorpe JW, Watson ND (1995) Comparison of Cannabis sativa by random amplification of polymorphic DNA (RAPD) and HPLC of cannabinoid: a preliminary study. Sci Justice 35: 169–177. [DOI] [PubMed] [Google Scholar]
- 4. Jagadish V, Robertson J, Gibbs A (1996) RAPD analysis distinguished Cannabis sativa samples from different sources. Foren Sci Int 79: 113–121. [Google Scholar]
- 5. Meijer EP, Bagatta M, Carboni A, Crucitti P, Moliterni VM, et al. (2003) The inheritance of chemical phenotype in Cannabis sativa L. Genetics. 163: 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu ZH, Guo HY, Hu XL, Chen X, Liu XY, et al. (2012) Genetic diversity of Cannabis sativa L. based on AFLP analysis. J Plant Genet Resour 13: 555–561. [Google Scholar]
- 7. Lv J, Zhao L, Zang G, Cheng C, Tang Q, et al. (2010) Screening of the AFLP markers linked to the sex locus of Cannabis sativa . J Hunan Agric Univ 36: 123–127. [Google Scholar]
- 8. Miller CH, Shutler G, Abrams S, Hanniman J, Neylon S, et al. (2003) A simple DNA extraction method for marijuana samples used in amplified fragment length polymorphism (AFLP) analysis. Foren Sci 48: 343–347. [PubMed] [Google Scholar]
- 9. Kojoma M, Iida O, Makino Y, Sekita S, Satake M (2002) DNA fingerprinting of Cannabis sativa using inter-simple sequence repeat (ISSR) amplification. Planta Med 68: 60–63. [DOI] [PubMed] [Google Scholar]
- 10. Rotherham D, Harbison SA (2011) Differentiation of drug and non-drug Cannabis using a single nucleotide polymorphism (SNP) assay. Forensic Sci Int. 207: 193–197. [DOI] [PubMed] [Google Scholar]
- 11. Ramu P, Billot C, Rami JF, Senthilvel S, Upadhyaya HD, et al. (2013) Assessment of genetic diversity in the sorghum reference set using EST-SSR markers. Theor Appl Genet 126: 2051–2064. [DOI] [PubMed] [Google Scholar]
- 12. Kashi Y, King D, Soller M (1997) Simple sequence repeats as a source of quantitative genetic variation. Trends Genet 13: 74–78. [DOI] [PubMed] [Google Scholar]
- 13. Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, et al. (1998) A microsatellite map of wheat. Genetics 149: 2007–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Röder MS, Korzun V, Gill BS, Ganal MW (1998) The physical mapping of microsatellite markers in wheat. Theor Appl Genet 41: 278–283. [Google Scholar]
- 15. Guo W, Wang W, Zhou B, Zhang T (2006) Cross species transferability of G. arboreum- derived EST- SSRs in the diploid species of Gossypium . Theor Appl Genet 112: 1573–1581. [DOI] [PubMed] [Google Scholar]
- 16.Chandra S, Lata H, Techen N, Mehmadic Z, Khan IA, et a1. (2011) Analysis of genetic using SSR markers and cannabinoid contents in different varieties of Cannabis sativa L. Planta Med 77-P_5.
- 17. Bakel HV, Stout JM, Cote AG, Tallon CM, Sharpe AG, et al. (2011) The draft genome and transcriptome of Cannabis sativa . Genome Biology 12: R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei W, Qi X, Wang L, Zhang Y, Hua W, et al. (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12: 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cordeiro GM, Casu R, Melntyre CL, Manners JM, Henry RJ (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160: 1115–1123. [DOI] [PubMed] [Google Scholar]
- 20. Thiel T, Michalek W, Varshney K, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeurm vulgare L.). Theor Appl Genet 106: 411–422. [DOI] [PubMed] [Google Scholar]
- 21. Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM (2000) Analysis of SSRs derived from grapes ESTs. Theor Appl Genet 100: 723–726. [Google Scholar]
- 22. Xin Y, Cui H, Lu M, Yao Y, Jin J, et a1 (2006) Data mining for SSRs in ESTs and EST-SSR marker development in Chinese cabbage. Acta Horti Sinica 33: 549–554. [Google Scholar]
- 23. Jiang D, Zhong G, Hong Q (2006) Analysis of microsatellites in citrus unigenes. Acta Genet Sin 33: 345–353. [DOI] [PubMed] [Google Scholar]
- 24. Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acid Res 8: 4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang C, Guo W, Zhang T, Li Y, Liu H (2009) AutoSSR: an improved automatic software for SSR analysis from large-scale EST sequences. Cotton Sci 2l: 243–247. [Google Scholar]
- 26.Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Totowa: Humana Press, 365–386. [DOI] [PubMed]
- 27.Rohlf FJ (2002) NTSYS-pc. Numerical taxonomy and multivariate analysis system, version 2.10. Exeter Software, New York.
- 28. Yeh FC, Boyle TJ (1997) Population genetic analysis of codominant and dominant markers and quantitative traits. Belg J Bot 129: 157. [Google Scholar]
- 29. Cloutier S, Niu Z, Datla R, Duguid S (2009) Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor Appl Genet 119: 53–63. [DOI] [PubMed] [Google Scholar]
- 30. Liu T, Zhu S, Fu L, Tang Q, Yu Y, et al. (2013) Development and characterization of 1,827 expressed sequence tag-derived simple sequence repeat markers for ramie (Boehmeria nivea L. Gaud). Plos One 8: e60346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, et al. (2007) Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 114: 359–372. [DOI] [PubMed] [Google Scholar]
- 32. Li X, Zhang M, Cui H (2007) Analysis of SSR information in EST resource of oilseed rape. Chin J Oil Crop Sci 29: 20–25. [Google Scholar]
- 33. Hu J, Li J (2009) Information on EST-SSR loci in melon (Cucumis melo L.) and marker exploitation. Acta Horti Sinica 36: 513–520. [Google Scholar]
- 34. Gao L, Tang J, Li H, Jia J (2003) Analysis of microsatellites in major crops assessed by computational and experimental app roaches. Mol Breed 12: 245–261. [Google Scholar]
- 35. Saha M, Mian M, Eujayl I, Zwonitzer J, Wang L, et al. (2004) Tall fescue EST-SSR markers with transferability across several grass species. Theor Appl Genet 109: 783–791. [DOI] [PubMed] [Google Scholar]
- 36. Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23: 48–55. [DOI] [PubMed] [Google Scholar]
- 37. Jack H, Surya PB, David JM (2012) Review of flowering control in industrial hemp. J Nat Fibers 9: 23–36. [Google Scholar]
- 38. Amaducci S, Colauzzi M, Bellocchi G, Venturi G (2008) Modelling postemergent hemp phenology (Cannabis sativa L.): Theory and evaluation. Eur J Agron 28: 90–102. [Google Scholar]
- 39. Lisson SN, Mendham NJ, Carberry PS (2000) Development of a hemp (Cannabis sativa L.) simulation model 2. The flowering response of two hemp cultivars to photoperiod. Aust J Exp Agr 4: 413–417. [Google Scholar]
- 40.Bocsa I, Karus M (1998) The Cultivation of hemp: botany, varieties, cultivation and harvesting. Sebastopol: hemptech. 184 p.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of 115 cannabis varieties used in this study.
(DOC)
One hundred and seventeen EST-SSR markers used for PCR amplification.
(XLS)
The information of 3443 EST-SSR markers developed.
(XLS)
The polymorphic ratios among 115 cannabis varieties.
(XLS)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.