

Abstract
Background
Age changes in expression of inflammatory, synaptic, and neurotrophic genes are not well characterized during human brain development and senescence. Knowing these changes may elucidate structural, metabolic, and functional brain processes over the lifespan, as well vulnerability to neurodevelopmental or neurodegenerative diseases.
Hypothesis
Expression levels of inflammatory, synaptic, and neurotrophic genes in the human brain are coordinated over the lifespan and underlie changes in phenotypic networks or cascades.
Methods
We used a large-scale microarray dataset from human prefrontal cortex, BrainCloud, to quantify age changes over the lifespan, divided into Development (0 to 21 years, 87 brains) and Aging (22 to 78 years, 144 brains) intervals, in transcription levels of 39 genes.
Results
Gene expression levels followed different trajectories over the lifespan. Many changes were intercorrelated within three similar groups or clusters of genes during both Development and Aging, despite different roles of the gene products in the two intervals. During Development, changes were related to reported neuronal loss, dendritic growth and pruning, and microglial events; TLR4, IL1R1, NFKB1, MOBP, PLA2G4A, and PTGS2 expression increased in the first years of life, while expression of synaptic genes GAP43 and DBN1 decreased, before reaching plateaus. During Aging, expression was upregulated for potentially pro-inflammatory genes such as NFKB1, TRAF6, TLR4, IL1R1, TSPO, and GFAP, but downregulated for neurotrophic and synaptic integrity genes such as BDNF, NGF, PDGFA, SYN, and DBN1.
Conclusions
Coordinated changes in gene transcription cascades underlie changes in synaptic, neurotrophic, and inflammatory phenotypic networks during brain Development and Aging. Early postnatal expression changes relate to neuronal, glial, and myelin growth and synaptic pruning events, while late Aging is associated with pro-inflammatory and synaptic loss changes. Thus, comparable transcriptional regulatory networks that operate throughout the lifespan underlie different phenotypic processes during Aging compared to Development.
Introduction
The human brain changes structurally, functionally, and metabolically throughout the lifespan [1], [2]. Programmed dendritic growth followed by pruning, neuronal loss, shifts in energy metabolism from ketone body to glucose consumption, and rapid myelination occur during development [3], [4], [5], [6]. Many of these changes are completed within the first two decades of life, although myelination can continue into the fourth decade [5], [7], [8]. In middle age, the brain reaches a level of homeostatic stability, but neuronal and synaptic loss associated with cognitive changes can appear later on [2], [9], [10], [11], [12]. Aging also is a risk factor for progressive brain disorders in which neuroinflammation plays a prominent role [13], [14], [15], [16], [17].
Neuroinflammation involves activation of resident brain microglia and astrocytes, and can be produced by different internal or external stresses [14], [18], [19], [20]. Microglial activation via toll-like receptors (TLRs) or cluster of differentiation (CD)14 receptors releases cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor-α (TNFα) and interferon gamma (IFNγ), chemokines such as fractalkine (CX3CL1), and nitric oxide (NO), following activation of inducible nitric oxide synthase (iNOS), thereby creating response cascades that can negatively impact brain structure and function [21]. Downstream activation of IL-1 receptors (IL-1R) and TNFα receptors on astrocytes and other cell types alters levels of transcription factors such as nuclear factor-kappa B (NF-κB) and activator protein (AP)-2, which can increase expression of a number of inflammatory genes [21], [22], [23], [24], [25].
The term “inflamm-aging” has been proposed to describe the progressive increase in proinflammatory status in the brain with senescence [26]. Inflamm-aging may prime brain microglia and astrocytes to respond excessively to different stressors, including neurodegenerative, traumatic or infectious insults [27], [28], [29], [30], [31]. Increased inflammatory response markers with late-state brain aging have been documented in rodents [27], [32], nonhuman primates [33], and humans [34]. Increases have been noted in proinflammatory cytokines IL-1α, IL-1β, IL-18 and IFNγ, major histocompatibility complex class II (MHC II), CD11b, scavenger receptors CD68, CD86 and CD40, and TLRs 1, 2, 4, 5, 7, and 9. In the human brain, increased TNFα and interferon gamma-inducible protein 16 (IFI-16) were reported, as were increased mRNA and protein levels of CD11b, glial fibrillary associated protein (GFAP), IL-1β, iNOS, NF-κB p50, cytosolic phospholipase A2 (cPLA2) Type IVA and cyclooxygenase (COX)-2, while levels of brain derived neurotropic factor protein (BDNF) and synaptophysin (SYP) were reduced [34], [35].
Cytokines, chemokines, growth and other microglial and astrocytic factors that change with age in the adult brain also have important regulatory actions during neurodevelopment [36], [37]. For example, microglia participate postnatally in synaptic pruning and apoptosis, and produce nerve growth factor (NGF), BDNF, neurotrophin (NT)-3 and cytokines that influence neuronal path finding, synaptogenesis and experience dependent plasticity [38], [39], [40], [41].
Multiple metabolic and protein networks have been described that underlie brain structure and function, and brain vulnerability to disease [42], [43], [44], [45], [46]. The extent to which these phenotypic networks or “cascades” are regulated at the transcriptional level, particularly during brain development, maturity, and aging are not well understood. To address this limitation, in the present study we analyzed age changes over the lifespan in brain mRNA levels of 39 genes whose protein products have been reported to be involved in neuroinflammation, synaptic integrity, neurotrophic effects, and related processes. As in our prior report on age expression of brain lipid metabolic markers [47], we employed the large-scale microarray dataset called BrainCloud, which contains genome-wide expression levels in frontal cortex from non-pathological individuals, in the fetal period to postnatal 78 years of age [48]. Similarly, and consistent with the literature, we considered gene expression in two distinct postnatal age intervals, Development (0 to 21 years) and Aging (22 to 78 years) [35], [47], [49], [50], henceforth identified by capitalizations.
Based on prior studies, we hypothesized that expression of genes linked to neuroinflammation would increase with age in the Aging interval [29], [34], [51], [52], while expression of genes coding for synaptic integrity and plasticity would decrease [9]. Furthermore, expression of these same genes would change during Development to reflect reported roles of their protein products in synaptic and neuronal growth, pruning, myelination, and other events in this period [1].
We also hypothesized that expression of genes coding for products that belong to common growth and neuroinflammatory cascades would be coordinately regulated during the lifespan, in relation to the specific phenotypic networks in which their proteins interacted. Coordinated or synchronized gene transcription underlying changes in phenotype networks has been demonstrated in cell culture, rodent brain, and artificial systems [53], [54], [55], and in the human brain in relation to age [47], [48], [56], [57], [58].
Methods
BrainCloud
BrainCloud (http://braincloud.jhmi.edu/) is a publically available software program that contains gene expression and methylation microarray datasets of more than 30,000 probes [48]. We analyzed data from 231 dorsolateral prefrontal cortex samples (Brodmann Areas 46/49) from subjects ranging in age from birth to 78 years. Results were transferred to an accessible and easily operated interface. Subjects had no history of significant psychiatric disorder, as determined by telephone screening and medical examiner reports [48]. Cause of death was listed as accident, homicide, or natural cause. Toxicology measurements were taken post-mortem and history of drug abuse and neuropathology tests were assessed [48]. Samples were genotyped with Illumina Infinium HD Gemini 1M Duo BeadChips or with Illumina Infinium II 650 K. mRNA was quantified with the Illumina Human 49K Oligo array (HEEBO-7 set) [48].
Based on the literature and to remain consistent with our prior publication [35], [47], [49], [50], we divided our sample into a Development interval (0 to 21 years, n = 87, mean age: 10.99) and an Aging interval (22 to 78 years, n = 144, mean age: 45.38), capitalized to distinguish these defined intervals from the developmental and aging processes themselves. The brains were from 73 females and 158 males.
When multiple probes in BrainCloud represented a gene, we selected the probe sequence that covered all exon-coding regions of the gene's identified transcripts. The highest intensity probe (average log2 intensity of fluorescent signal for this probe across all subjects) was taken when two probes covered all exon-coding regions of the transcripts. Most probes that met the first criterion also were the highest intensity probes of that gene. Genes were selected based on their availability in the BrainCloud dataset. Gene expression levels remained consistent with BrainCloud's calculated expression as log2 (Sample/Reference), reference being the pooled RNA from all subjects [48].
Statistical Analyses
Pearson's r correlations were used to correlate expression levels of pairs of genes. Linear regression was used to relate expression to age within the separate Development and Aging intervals. Nonlinear best-fit comparisons with each gene were performed using ‘The extra sum-of-squares F test’ to compare one-phase decay to a first-order linear model. A four-way ANOVA that included relevant factors provided by BrainCloud software, such as gender, race and batch number, was performed to calculate fold-differences and p-values comparing the Aging with Development group. The p-values from the ANOVA results include correction of significance for multiple comparisons using the Benjamini-Hochberg [59] control of False Discovery Rate (FDR), also known as the “step-up” FDR procedure. Batch effect removal on factors (array batch, race, and sex) was performed using the sva package (version 3.8.0) in Bioconductor, based on the method of Johnson et al. [60]. No other normalization was performed on the raw data. Differential gene expression would be a result of age and not race, sex, or array batch. Pearson's r correlations and multiple ANOVAs were performed in Partek Genomics Suite (Version 6.6, Partek, St. Louis, MO, USA). Linear regression analyses and scatter plots were performed using GraphPad Prism version 6.0 for Mac OS X (GraphPad Software, La Jolla, CA, USA, www.graphpad.com).
Heat Map
Heat maps, which are graphical representations of data where individual values are represented in a matrix on a color scale, were created using Partek Genomics Suite 6.6 to visualize correlated expression levels across all genes in the separate Development and Aging periods.
Results
Differences in Gene Expression during Development and Aging
Correlations with age
Table S1 presents the 39 genes that were selected in this study, with their corresponding chromosomal location, protein name, and reported general functions [61], [62], [63], [64]. While many of their protein functions may be unrelated, many of their proteins have been reported to participate in phenotypic cascades involving cell growth, inflammation, synaptic maintenance and gene transcription, and other processes. For example, Figure 1, generated from the literature [65], [66], [67], [68], [69] and string interactions in GeneCards [64], illustrates several abbreviated cascades initiated by activation of TLR-2 and TLR-4 commonly found on microglia, and of IL-1R and TNFα receptors commonly found on neurons and astrocytes. These cascades are abnormal in a number of progressive neuroinflammatory diseases [13], [15], [69], [70], [71].
Figure 1. Pathways associated with activation of toll-like receptors TLR-2 and TLR-4, the IL-1 receptor IL-1R1, and the TNFα receptor, TNFRSF1A.

IL-1β binding to the IL-1R1 leads to recruitment of IL-1 receptor accessory protein (IL-1RAP), and can be blocked by the naturally occurring IL-1 receptor antagonist, IL-1RN. The IL-1R1/1β/RAP signaling complex is capable of recruiting interleukin receptor-associated kinase (IRAK), IL-1 receptor accessory protein (IL-1RAP), and myeloid differentiation factor 88 (MYD88). IRAK can be phosphorylated and subsequently dissociate from the receptor complex to interact with tumor necrosis factor receptor-associated factor (TRAF6) and TGF-β activated kinase 1 (TAK1)/MAP3K7 binding protein 2 (TAB2) complex. The TLR2 and TLR4 cascades are simplified in Figure 1 into a single cascade. However, gram-negative bacterial lipopolysaccharide (LPS) can activate TLR4, which associates with lymphocyte antigen-96 (MD-2) while gram-positive bacteria are recognized by TLR2 [115]. Their activation effects converge with those of the IL-1R and TNFα receptor on the TRAF6 complex. IL-1R and all TLRs except TLR3 exhibit the same Toll/interleukin-1 receptor (TIR) region that allows recruitment of MYD88 upon activation [116]. Nuclear I kappa kinase (NIK) and various mitogen-activated protein kinases (MAPKs) can promote activation of cytoplasmic nuclear factor-kappa B (NF-κB). Activated NF-κB then can enter the nucleus of the cell to regulate transcription of various genes by binding to their promoter regions. RELA (p65) and p50, proteins in NF-κB family; AP-2: transcription factor AP-2 alpha (activating enhancer binding protein 2 alpha); A20: tumor necrosis factor, alpha-induced protein 3; NFKBIA: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; COX-2: cyclooxygenase-2; cPLA2: phospholipase A2 (cytosolic, calcium-dependent); sPLA2: phospholipase A2 (secretory).
Of the genes listed in Table S1, Figure 2 summarizes those whose expression levels correlated significantly with age in the Development and/or Aging interval, and indicates by quadrant the direction of the correlation. Age-related expression of MOBP (myelin-associated oligodendrocyte basic protein) and of NFKB1 increased during both Development and Aging (right upper quadrant), while expression of NGF and CX3CL1 (Chemokine (C-X3-C Motif) Ligand 1) decreased during both Development and Aging (left lower quadrant). Expression of SNCA, coding for α-synuclein, increased during Development but decreased during Aging (right lower quadrant). Additionally, expression of TRAF6 (TNF receptor associated factor 6), PLA2G4A, PTGS1, TLR4, APP, MAP2, and IL1R1 increased while expression of MYD88, DBN1, RELA, PDGFA, and IFI16 decreased during Development but not Aging. Conversely, expression of GFAP, TSPO, and IL1RN increased and of PTGS2, BDNF, IL1RAP, SYP, CX3CR1, and NOS2 decreased during Aging, without changing significantly with age during the Development interval.
Figure 2. Statistically significant correlations with age in Development and/or Aging intervals.

Graphical identification of genes with statistically significant (p<0.05) Pearson r correlations in expression level during Development (x axis) and Aging (y axis) intervals. Gene expressions negatively correlated with age during Development are to the left, while genes positively correlated are to the right of the vertical line. Genes that were negatively correlated with age in the Aging group are below the horizontal line, while genes positively correlated are above line. Development: n = 87; Aging: n = 144.
Figure 3 gives examples of genes whose expression levels correlated significantly with age during both the Development and Aging periods. Expression of SNCA, increased significantly with age during Development while declining during Aging. Expression of MOBP and NFKB1 increased significantly with age during both intervals, while expression CX3CL1 and NGF decreased. Expression of CX3CR1, and PTGS2 (COX-2), declined with age whereas GFAP and TSPO increased during both intervals.
Figure 3. Significant linear regressions of gene expression during both Development and Aging intervals (top), and Aging interval alone (bottom).
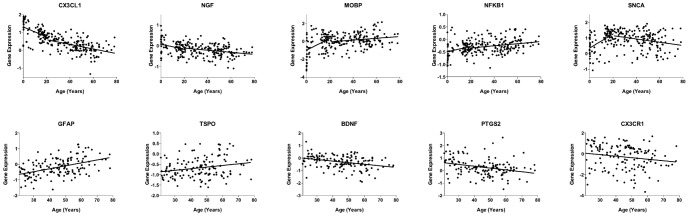
Scatterplots illustrating log2 gene expression over age in years. An increase or decrease of 1 on the log2 scale (y-axis) represents a two-fold change in gene expression in the positive or negative direction, respectively. Each data point represents observation from one brain (Development: n = 87; Aging: n = 144). Gene name (p-value during Development, p-value during Aging) - CX3CR1 (p<0.0001, p<0.0001), NGF (p = 0.006, p = 0.002), MOBP (p<0.0001, p = 0.02), NFκB1 (p = 0.01, p = 0.01), SNCA (p<0.0001, p = 0.002). Genes significant in only Aging interval – GFAP (p<0.0001), TSPO (p = 0.006), BDNF (p<0.0001), PTGS2 (p = 0.0003), CX3CR1 (p = 0.03).
Visual observation of several gene expression levels during Development suggested non-linearity, with initial levels in the first year of life being higher or lower than later plateaus. To test this, we compared goodness of fit with a non-linear equation, Y = (Y0−Plateau)*exp(−K*A)+Plateau (where Y = expression level at age A, and Y0 = expression level at A = 0 years) to that of a linear regression during Development for each gene studied. As illustrated in Figure 4, expression of TLR4, IL1R1, NFKB1, MOBP, PLA2G4A, and PTGS2 increased in the first years of life and reached a plateau, while expression of synaptic genes GAP43 and DBN1 decreased before reaching a plateau.
Figure 4. Nonlinear fits for expression levels with age of eight genes during Development.

Fitted line added to expression data following equation for 0 to 21 years, Y = (Y0−Plateau)*exp(−K*A)+Plateau, where Y = expression level at age A, and Y0 expression level at A = 0 years). An increase or decrease of 1 on the log2 scale (y-axis) represents a two-fold change in expression in the positive or negative direction, respectively.
Mean expression level differences between Aging and Developmental Periods
As summarized in Table 1, mean expression levels were significantly (adjusted p<0.05) lower during Aging than Development for CX3CR1, CX3CL1, PTGS2, BDNF, CASP1, CD68, AIF1, MYD88, NGF, PDGFA, DBN1, IL1B, and SYP, and significantly higher for GFAP, TSPO, MOBP, TRAF6, and NFKB1 (highlights show significance after correction for multiple comparisons).
Table 1. Statistically significant mean gene expression differences between Aging and Development Periods (Multiple ANOVA results).
| Reduced with Aging | Increased with Aging | ||||
| Gene | Adjusted p-value | Fold-Difference | Gene | Adjusted p-value | Fold-Difference |
| CX3CL1 | 5.07E-21 | −1.75 | GFAP | 4.33E-05 | 1.37 |
| CX3CR1 | 8.00E-06 | −2.00 | TSPO | 1.87E-03 | 1.23 |
| BDNF | 1.53E-05 | −1.39 | MOBP | 2.77E-03 | 1.43 |
| CD68 | 1.56E-04 | −1.26 | TRAF6 | 3.41E-03 | 1.17 |
| CASP1 | 1.63E-04 | −1.35 | NFKB1 | 7.76E-03 | 1.11 |
| AIF1 (IBA1) | 1.63E-04 | −1.31 | TLR4 | 6.16E-02 | 1.12 |
| PTGS2 (COX-2) | 1.75E-04 | −1.44 | IL1R1 | 9.70E-02 | 1.21 |
| PDGFA | 1.34E-03 | −1.14 | |||
| DBN1 | 1.34E-03 | −1.13 | |||
| MYD88 | 1.34E-03 | −1.24 | |||
| NGF | 1.34E-03 | −1.14 | |||
| SYP | 2.11E-03 | −1.18 | |||
| IL1B | 7.33E-03 | −1.22 | |||
| BACE1 | 5.33E-02 | −1.11 | |||
Multiple ANOVA results, giving fold changes, showing that mean expression was significantly lower (left) or higher (right) in Aging compared with Development. A negative fold-change represents decreased expression in the Aging compared to Development group, and vice versa. Italicized p-values are nonsignificant after correction for multiple comparisons. In p-values, term E-number = ×10−number. (Development: n = 87; Aging: n = 144).
Correlated group expression changes during Development and Aging
Pearson's correlation matrices relating all combinations of the genes were visualized using unsupervised hierarchical clustering and heat maps within the Development (Figure 5a) and Aging (Figure 5b) intervals. Gene order based on hierarchical clustering are not conserved between Development and Aging heat maps, as they represent the highest probability of correctly clustering genes based on Pearson's r correlation in the individual intervals. In Figures 5a and 5b, genes that are highly positively correlated within a cluster are highlighted in green; those that are negatively correlated in red.
Figure 5. Similarity matrices (hierarchically clustered heat maps) of Pearson's r correlations of gene expression levels with age in Development (A) and Aging (B) groups.

Red indicates negatively correlated associations; green are positively correlated associations, while black represents non-significant associations between gene pairs. Genes are clustered hierarchically along the left y-axis, which is mirrored above in each heat map.
Three different clusters of genes whose expression levels were highly intercorrelated were identified in both the Development and Aging periods. As illustrated in Figure 5a, Development clusters were: Cluster 1: GAP43, SYP, BDNF, IL1RAP, NOS2, MAPK14, MAP2, APP, SNCA; Cluster 2: BACE1, TRAF6, PTGS1, CX3CR1; Cluster 3: TSPO, TNFRSF1A, MYD88, IL1R1, NFKB1, AIF1, CD68, CASP1, TLR2. As illustrated in Figure 5b, Aging clusters were: Cluster 1: NOS2, CX3CL1, SYP, TRAP1, GAP43, BDNF, SNCA; Cluster 2: MAP2, APP, IL1RAP, PTGS1, CX3CR1, PLA2G4A; Cluster 3: GFAP, TSPO, TNFRSF1A, IL1R1, MYD88, CD68, AIF1, CASP1, TLR2.
Figures S1a and S1b show pairwise correlation values between gene expression levels in the Development and Aging periods, respectively. Green coloring highlights statistical significance at p≤0.0001. The global clusters identified in Figures 5a and 5b, and the highly significant pairwise correlations illustrated in Figures S1a and S1b, show plausible synchronization of gene transcription within clusters or networks throughout the lifespan. The most significant pairwise correlations taken from Figures S1a and S1b, at p<10−10 and r≥|0.6|, are given in Table 2.
Table 2. Highly significant (r≥|0.6|) pair-wise correlations in age-related gene expression during Development and Aging intervals.
| Development | Aging | ||||||
| Gene1 | Gene2 | Correlation | p-value | Gene1 | Gene2 | Correlation | p-value |
| MAP2 | APP | 0.77 | 1.53E-18 | MAP2 | APP | 0.80 | 5.61E-34 |
| AIF1 | CD68 | 0.75 | 3.86E-17 | IL1R1 | TNFRSF1A | 0.76 | 3.38E-29 |
| TLR2 | CASP1 | 0.67 | 8.13E-13 | AIF1 | CD68 | 0.75 | 1.49E-27 |
| APP | SNCA | 0.67 | 9.95E-13 | AIF1 | CASP1 | 0.71 | 4.82E-24 |
| MOBP | DBN1 | -0.63 | 3.20E-11 | TLR2 | CASP1 | 0.69 | 1.84E-22 |
| MAP2 | SNCA | 0.63 | 3.37E-11 | CD68 | CASP1 | 0.68 | 1.54E-21 |
| TSPO | MYD88 | 0.63 | 5.75E-11 | CX3CR1 | PTGS1 | 0.68 | 1.68E-21 |
| IL1R1 | NFKB1 | 0.61 | 2.88E-10 | TLR2 | IL1R1 | 0.68 | 3.63E-21 |
| CX3CR1 | PTGS1 | 0.60 | 4.02E-10 | IL1RAP | APP | 0.67 | 1.25E-20 |
| MYD88 | CASP1 | 0.60 | 6.18E-10 | IL1RAP | MAP2 | 0.67 | 1.87E-20 |
| IL1R1 | TNFRSF1A | 0.60 | 7.41E-10 | TSPO | TNFRSF1A | 0.66 | 6.52E-20 |
| TSPO | MYD88 | 0.66 | 6.98E-20 | ||||
| TLR2 | CD68 | 0.66 | 1.16E-19 | ||||
| AIF1 | CX3CR1 | 0.66 | 2.20E-19 | ||||
| TNFRSF1A | MYD88 | 0.65 | 5.94E-19 | ||||
| AIF1 | TLR2 | 0.65 | 1.12E-18 | ||||
| TSPO | IL1R1 | 0.64 | 2.63E-18 | ||||
| TSPO | GFAP | 0.63 | 1.26E-17 | ||||
| MAP2 | SNCA | 0.63 | 1.64E-17 | ||||
| TLR2 | MYD88 | 0.62 | 3.05E-17 | ||||
| TSPO | MAP2 | −0.60 | 7.67E-16 | ||||
| MYD88 | CASP1 | 0.60 | 1.10E-15 | ||||
| IL1R1 | MYD88 | 0.60 | 1.16E-15 | ||||
| TSPO | SNCA | −0.60 | 1.41E-15 | ||||
In p-values, term E-number = ×10−number.
Gene Expression and Chromosome Proximity
Four pairs of genes have the same band number on a chromosome: PLA2G4A and PTGS2 (1q25); IL1β and IL1RN (2q14); CX3CR1 and MOBP (3p21.3); and AIF1 and MAPK14 (6p21.3) (cf. Table S1). At p<0.001, expression levels of PLA2G4A and PTGS2 were correlated positively during both Development and Aging, while expression levels of IL1β and IL1RN were correlated positively during Development only. Expression levels of the other two gene pairs were not correlated significantly in either period.
Discussion
We used the BrainCloud database for human prefrontal cortex [48] to examine age variations in mRNA levels of 39 genes reported to be involved in pathways of neuroinflammation, cytokine signaling, arachidonic acid metabolism, neuronal and myelin integrity, synaptic function, neurotrophic action, and related processes. We divided the postnatal lifespan into Development (0–21 years) and Aging (22 to 78 years) intervals, on the basis of reported distinct functional and structural brain changes in these periods [4], [5], [47], [49], [72].
Confirming this division, expression patterns and age correlations of many of the chosen genes frequently differed significantly between the two intervals. Genes with higher expression during Aging include TSPO, associated with microglial activation and cholesterol transport [73], [74]; GFAP, associated with glial activation [71]; TRAF6, associated with TNFα signaling; MOBP, associated with myelin integrity; and NFKB1, coding for NF-κB, a major transcription factor of inflammatory genes involved in innate immunity [25]. More genes were expressed at lower levels during Aging than Development, reflecting a reduced intensity of early developmental events during Aging. These genes are related to synaptic integrity, neuronal growth, neurotrophic, glial changes and other development modifications (CX3CR1, CX3CL1, PTGS2, BDNF, CASP1, CD68, AIF1, MYD88, NGF, PDGFA, DBN1, IL1B, SYP, and BACE1).
Neurodevelopment is influenced largely by programmed transcriptional changes involving neuronal, glial and synaptic integrity, and myelination, whereas after 21 years of age, gene expression reaches a homeostatic state that depends more on factors such as health status, environment, and nutrition [48], [57], [75], [76]. Late senescence becomes a risk factor for neurodegenerative diseases such as Alzheimer's and Parkinson's disease, when the brain shows increased inflammatory, apoptotic, and arachidonic cascade markers, but reduced neurotrophic and synaptic markers [9], [13], [15], [77], [78], [79]. Our data in the Aging interval indicate many significant positive age correlations in expression of genes in the former category (GFAP, TSPO, TRAF6, NFKB1, TLR4, IL1R1) but decreased correlations for genes in the latter category (BDNF, SYP, SNCA, NGF) (Figure 2). These findings support the proposition that aging increases vulnerability to neurodegenerative disease and is a priming factor for it [9], [13], [15], [26], [27], [28], [29], [30], [31], [77], [78], [79].
The significant age changes in mRNA levels during the Aging interval do not always correspond to reported protein changes. TNFα and IFI-16 protein levels were reported to increase between 26 to 106 years in postmortem human brain [35], whereas we did not find age increases in TNFRSF1A or IFI16 expression in the Aging interval (Figure 2), or on average between Aging and Development (Table 1). On the other hand, and consistent with our expression changes, positive age correlations between 42 and 70 years were reported in brain mRNA and protein levels of GFAP, IL-1β, iNOS, NF-κB p50, cPLA2 IVA and COX-2, while levels of BDNF and SYP declined in this period [34]. Some of the changes correlated with promoter hypermethylation of BDNF and cyclic AMP responsive element binding protein (CREB), and hypomethylation of Bcl-2 associated X protein (BAX), suggesting epigenetic influence [80]. Another study also reported a decrease in BDNF protein during the Aging interval [81], while another reported decreased protein levels of DBN1, GAP-43, and SYN [9]. In general agreement, we found significant age increases in expression of GFAP, IL1R1, and NFKB1, and reductions in expression of BDNF and SYP (Table 1).
Age correlations often differed between the Development and Aging intervals, indicating different roles for the gene products over the lifespan (Figures 2 and 3, Table 1). Expression of SNCA increased during Development but decreased during Aging; expression of PLA2G4A, PTGS1, TRAF6, TLR4, APP, and IL1R1 increased and expression of DBN1, MYD88, RELA, PDGF, and IFI16 decreased during Development alone. Expression of MOBP increased during both intervals, consistent with continued myelination into the fourth decade in frontal cortex [5], as did expression of NFKB1, while expression of NGF decreased during both periods, suggesting reduced neuroplasticity [82]. The NFκB1 system can be stimulated by a number of cell surface receptors (Figure 1), as well as by oxidative stress, hypoxia, and genotoxic stress [25]. In non-stimulated cells, NF-κB complexes are bound in cytoplasm to inhibitory I-kappa-B (IκB) proteins. Stimulation phosphorylates IκB proteins, which are ubiquitinated and broken down, allowing the NF-κB complex to enter the nucleus and activate transcription of multiple genes, particularly related to inflammatory cascades [25].
In the first six months of life, a number of genes showed non-linear expression changes that later reach a plateau (Figure 4). In this same period, neuronal density in layers 2–3 of human frontal cortex falls by 80% [3]. However, dendritic spine densities at different levels of prefrontal cortical pyramidal neurons rise from birth to about 5–10 years of age, and then decline [56], [83]. Metabolic changes also occur, as the brain shifts from using ketone bodies to glucose in the first months of life [6]. Expression of GAP43 and DBN1, coding for presynaptic GAP43 and postsynaptic dendritic spine drebrin, decreased nonlinearly. As dendritic spine density increases in the first 5–10 years (see above), DBN1 and GAP43 likely change in this period. Expression increased for PLA2G4A and PTGS2 coding for postsynaptic functionally-coupled cPLA2 Type IVA and COX-2 [84], [85], [86] suggesting a growing role for arachidonic acid signaling in neurotransmission at dendritic spines [87], [88].
Increases in TLR4 and IL1R1 in the first years of life may reflect increased receptivity of microglia, on which these receptors are located [89], as microglia participate postnatally in synaptic pruning and apoptosis [38], [39], [40], [41]. In this regard, higher (2-fold) levels of expression of CX3CR1 (chemokine (C-X3-C motif) receptor 1) and of its ligand fractalkine (CX3CL1) during Development than Aging highlight the importance their protein products in early neuronal-glial interactions. CX3CR1 is expressed exclusively by microglia in brain [90], whereas CX3CL1 is highly expressed in neurons. Knocking out CX3CR1 reduced neuron loss [91] and amyloid-beta deposition [92] in Alzheimer's disease mouse models, and interfered with formation of thalamocortical synapses during development, when fractalkine is overexpressed.
Gene expression clusters in the heat map matrices of Figures 5a and 5b identify genes having high intercorrelated expression patterns as the brain ages. The clusters were similar in the Development and Aging intervals. Thus, comparable transcriptional regulatory networks operate throughout the life span, but underlie different phenotypic processes during Aging compared to Development. Gene products within Cluster 1 of both Development and Aging (GAP43, SYN, BDNF, NOS2, SNCA) are involved in synaptic signaling and integrity, cellular stress, and neurogenesis. Gene products in Cluster 2 of both periods (BACE1, CX3CR1, PTGS1, and PLA2G4A) are involved in the arachidonic acid cascade, protease activity, APP processing, and inflammatory processing. Gene products in Cluster 3 of both groups (TSPO, TNFRSF1A, IL1R1, MYD88, CD68, AIF1, CASP1, and TLR2) are involved in microglial, inflammasome, NF-κB signaling, and various neuroinflammatory responses.
Pairwise correlations, whether positive or negative, are more frequently significant during the Aging than Development interval (Figures S1a and S1b, Table 2). This may reflect our selective choice of genes, but if confirmed would suggest a more stable state of synchronized gene expression in the Aging than Development interval. This is likely since genome-wide promoter DNA methylation of CpG dinucleotides in human prefrontal cortex changes less in adulthood than early childhood [57]. Mean cortical global methylation is increased in the Aging interval, which may reflect some gene silencing [57], but global methylation is higher in late senescence [34].
Since the subjects were considered to be healthy, some presumably deleterious expression changes in the Aging interval had not progressed sufficiently to produce noticeable functional deficits, although they may have increased vulnerability to stress and other disease factors. Upregulated translator protein (TSPO) has been imaged using positron emission tomography in patients with mild cognitive impairment (MCI) and Alzheimer's disease [73], [93], [94]. Thus, the higher TSPO expression in Aging than Development is consistent with late-stage “inflamm-aging” in presumably healthy subjects (Table 1) [26]. In support, expression levels of TRAF6, NFKB1, TLR4, and IL1R1, important in the initial inflammatory response involving NF-κB (Figure 1), and of GFAP, were upregulated in the Aging interval. Nevertheless, not all gene markers of inflammation or microglial/astrocytic activation were upregulated. Expression levels of inflammation-related genes CASP1, PTGS2, MYD88, and IL1B, and of microglia genes CD68 and AIF1 (also known as IBA1), were reduced in the Aging group.
Our age correlations identified unexpected relations between different genes. Highly significant pairwise correlations were found between expression levels of APP and MAP2, of AIF1 (IBA1) and CD68, and of IL1R1 and TNFRSF1A. APP, a component in Alzheimer's disease pathogenesis, normally helps to maintain functional synapses [95]. Microtubule-associated protein 2 (MAP2) is found in post-synaptic dendrites and is functionally similar to tau protein, whose abnormal phosphorylation is another key component in Alzheimer's disease. AIF1 (IBA1), expressed in macrophages and microglia, and CD68, expressed in macrophages and monocytes, contribute to the inflammatory response in brain. Also, IL1R1 and the TNFα receptor (TNFRSF1A) are important in initial signaling in inflammation (Figure 1). Among the other highly correlated genes, CASP1 is involved in inflammasome formation [96], and TSPO is upregulated during neuroinflammation. Disturbed α-synuclein (SNCA) and APP occurs in Parkinson's disease and Alzheimer's disease respectively, and expression of SNCA and APP was very highly correlated in Development (r = 0.67) and in Aging (r = 0.56) (Figure S1). The Aging interval had a greater number of significant correlations between genes, and many significant correlations occurred consistently in Development and Aging. The correlated expression of genes in the canonical pathway of NF-κB (NFKB1, MYD88, TLR4, IL1R1, TRAF6) also showed a highly integrated network of genes with similar expression patterns with age [25].
Significant correlations in gene expression that corresponded to chromosome proximity (Table S1) for PLA2G4A (cPLA2 IVA) and PTGS2 (COX-2) at locus 1q25, and for IL1R1, IL1B, and IL1RN at locus 2q14 indicate robust co-regulatory elements and possible coevolution [47], [97], [98], [99]. The IL-1 receptor type 1 (IL1R1), a key cytokine receptor in innate immunity, interacts with IL1B and the IL-1 receptor antagonist (IL1RN) in a complex cascade (Figure 1) [36]. During Development, expression of IL1B and IL1RN exhibited a strong positive correlation (p<0.001), but their expression was not correlated significantly during Aging. Increased IL-1R signaling has been implicated in bipolar disorder, Alzheimer's disease, and HIV-1 encephalopathy [100], [101], [102]. Our changes during Development and Aging are consistent with neuroregulatory as well as neuroinflammatory functions involving the NF-κB transcription system.
Some of the expression changes with senescence in this study correspond to changes reported in postmortem frontal cortex from Alzheimer's disease, bipolar disorder, and schizophrenia patients, compared to age-matched non-pathological cortex. Alzheimer's disease cortex showed significantly increased mRNA and protein levels of IL-1β, TNFα, GFAP, CD11b, cPLA2 IVA sPLA2 IIA COX-1 and COX-2 [13], [103], [104], but decreased levels of pre-synaptic synaptophysin (SYP) and post-synaptic drebrin (DBN1) [9], [13]. In bipolar disorder, protein and mRNA levels of neuroinflammatory markers (IL-1β, IL-1R, MYD88, NF-kB1) and of activated microglia and astroglial markers (GFAP, NOS2, c-Fos, and CD11b) also were significantly higher than in control cortex [69], [105]. These changes were accompanied by reduced expression of anti-apoptotic factors B cell lymphoma (Bcl)-2, BDNF, SYN, and DBN1, but increased expression of pro-apoptotic Bax, BAD, and active caspase (CASP)-3 and CASP-9 [15]. Ca2+-dependent cPLA2 IVA, secretory sPLA2 IIA and COX-2 also were overexpressed [70]. Similar changes were noted in schizophrenic frontal cortex [71].
This study has several limitations. Selection criteria for the microarray gene probes were a way to biologically standardize all probes by their protein-coding regions. There are other criteria of selection; however, we found that taking mean expression data of all probes for a given gene was not a good representation of gene's expression (data not shown), as expression levels differed between probes of the same gene.
BrainCloud contains expression data selective to the prefrontal cortex [48]. The prefrontal cortex shows prolonged development and preliminary degradation associated with aging earlier than other brain regions [5], [106], [107]. Other studies have found differential gene expressed based on cell type, such as the Allen Brain Atlases and the Loerch study; however, BrainCloud did not differentiate between cell types [108], [109], [110]. Favorably, BrainCloud has a large number of samples (n = 269) compared with other aging databases with time points through a lifespan. For further discussion on BrainCloud and its application, see publications [47], [48].
In the future, it would be of interest to consider mechanisms underlying the age-related expression changes. These may involve histone acetylation and methylation, transcription factors, miRNAs, DNA sequences of cis-elements (transcription factor binding sites), all of which can influence mRNA expression [57], [111], [112], [113]. In this regard, many genes whose expression decreases with age appear to have higher promoter GC content than other genes [49], suggesting differences in methylation state, and human brain aging is associated with changes in global methylation [34], [57]. As we considered only two transcription factors in this study, TFAP2A (AP-2) and NFKB1 (NF-κB), future aging studies may consider more.
Supporting Information
Matrices showing Pearson correlation coefficients between expression levels of individual gene pairs during Development (A) and Aging (B). Green highlights coefficients that are significant at p<0.0001. Hierarchy in gene order corresponds to hierarchy in Figures 5A and 5B.
(XLSX)
Selected genes, chromosomal locations, protein description, and major reported functions. Based on the literature, genes whose protein products participate in major functions may be categorized as follows: (1) Synaptic function, SYP, DBN1, SNCA; (2) Growth and maintenance, BDNF, NGF, GAP43, PDGFA; (3) Myelin integrity, MOBP; (4) Neuroinflammation: (a) Microglial activation, CD68, TSPO, TLR4, TLR2, NOS2, AIF1; (b) Cytokine and chemokine processes, CX3CR1, CX3CL1, IFI16, IL1B, IL1R1, IL1RN, IL1RAP, IL6, TNFRSF1A, TRAF6, TRAP1, MAPK14, MYD88; (c) Glial activation, GFAP; (d) Apoptosis, CASP1; (5) Arachidonic acid cascade, PLA2G4A, PLA2G10, PTGS1, PTGS2; (5) Amyloid membrane processing, APP, BACE; (6) Microtubules, MAP2; (7) Transcription factors, NFKB1, TFAP2A [61], [62], [64], [114].
(DOCX)
Acknowledgments
We thank Drs. Barbara K. Lipska and Chuck T. Chen for their suggestions and comments.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All BrainCloud files are available from the GEO Accession database (accession number: GSE30272).
Funding Statement
The Intramural Research Program of the National Institute on Aging at the National Institutes of Health supported this research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. de Graaf-Peters VB, Hadders-Algra M (2006) Ontogeny of the human central nervous system: what is happening when? Early human development 82: 257–266. [DOI] [PubMed] [Google Scholar]
- 2. Hedden T, Gabrieli JDE (2004) Insights into the ageing mind: a view from cognitive neuroscience. Nature reviews Neuroscience 5: 87–96. [DOI] [PubMed] [Google Scholar]
- 3. Huttenlocher PR (1984) Synapse elimination and plasticity in developing human cerebral cortex. American journal of mental deficiency 88: 488–496. [PubMed] [Google Scholar]
- 4. Huttenlocher PR (1990) Morphometric study of human cerebral cortex development. Neuropsychologia 28: 517–527. [DOI] [PubMed] [Google Scholar]
- 5.Yakovlev PI, Lecours AR (1967) The myelogenetic cycles of regional maturation of the brain. In: Minkowski A, editor. Regional Development of the Brain in Early Life. Philadelphia: F. A. Davis. pp. 3–70. [Google Scholar]
- 6. Chugani HT, Hovda DA, Villablanca JR, Phelps ME, Xu WF (1991) Metabolic maturation of the brain: a study of local cerebral glucose utilization in the developing cat. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 11: 35–47. [DOI] [PubMed] [Google Scholar]
- 7. Sowell ER, Thompson PM, Toga AW (2004) Mapping changes in the human cortex throughout the span of life. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry 10: 372–392. [DOI] [PubMed] [Google Scholar]
- 8. Rapoport SI (1990) Integrated phylogeny of the primate brain, with special reference to humans and their diseases. Brain research Brain research reviews 15: 267–294. [DOI] [PubMed] [Google Scholar]
- 9. Hatanpää K, Hatanpää K, Isaacs KR, Isaacs KR, Shirao T, et al. (1999) Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. Journal of neuropathology and experimental neurology 58: 637–643. [DOI] [PubMed] [Google Scholar]
- 10. Azari NP, Rapoport SI, Salerno JA, Grady CL, Gonzalez-Aviles A, et al. (1992) Interregional correlations of resting cerebral glucose metabolism in old and young women. Brain research 589: 279–290. [DOI] [PubMed] [Google Scholar]
- 11. Creasey H, Rapoport SI (1985) The aging human brain. Annals of neurology 17: 2–10. [DOI] [PubMed] [Google Scholar]
- 12. Koss E, Haxby JV, DeCarli CS, Schapiro MB, Friedland RP, et al. (1991) Patterns of performance preservation and loss in healthy aging. Dev Neuropsychol 7: 99–113. [Google Scholar]
- 13. Rao JS, Rapoport SI, Kim H-W (2011) Altered neuroinflammatory, arachidonic acid cascade and synaptic markers in postmortem Alzheimer's disease brain. Translational psychiatry 1: e31. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Rao JS, Kellom M, Kim H-W, Rapoport SI, Reese EA (2012) Neuroinflammation and synaptic loss. Neurochemical research 37: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim H-W, Rapoport SI, Rao JS (2010) Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiology of disease 37: 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wingo AP, Harvey PD, Baldessarini RJ (2009) Neurocognitive impairment in bipolar disorder patients: functional implications. Bipolar disorders 11: 113–125. [DOI] [PubMed] [Google Scholar]
- 17. McGeer PL, McGeer EG (2004) Inflammation and the Degenerative Diseases of Aging. Annals of the New York Academy of Sciences 1035: 104–116. [DOI] [PubMed] [Google Scholar]
- 18. Perry VH, Bolton SJ, Anthony DC, Betmouni S (1998) The contribution of inflammation to acute and chronic neurodegeneration. Research in immunology 149: 721–725. [DOI] [PubMed] [Google Scholar]
- 19. Kraft AD, Kraft AD, Harry GJ, Harry GJ (2011) Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International journal of environmental research and public health 8: 2980–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain, behavior, and immunity 26: 1191–1201. [DOI] [PubMed] [Google Scholar]
- 21. Choi SH, Aid S, Bosetti F (2009) The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends in pharmacological sciences 30: 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoeck WG, Hoeck WG, Ramesha CS, Ramesha CS, Chang DJ, et al. (1993) Cytoplasmic phospholipase A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proceedings of the National Academy of Sciences of the United States of America 90: 4475–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spriggs DR, Sherman ML, Imamura K, Mohri M, Rodriguez C, et al. (1990) Phospholipase A2 activation and autoinduction of tumor necrosis factor gene expression by tumor necrosis factor. Cancer research 50: 7101–7107. [PubMed] [Google Scholar]
- 24. Bauer MK, Bauer MK, Lieb K, Lieb K, Schulze-Osthoff K, et al. (1997) Expression and regulation of cyclooxygenase-2 in rat microglia. European journal of biochemistry/FEBS 243: 726–731. [DOI] [PubMed] [Google Scholar]
- 25. Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, et al. (2008) Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing research reviews 7: 83–105. [DOI] [PubMed] [Google Scholar]
- 26. Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, et al. (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mechanisms of ageing and development 128: 92–105. [DOI] [PubMed] [Google Scholar]
- 27. VanGuilder HD, Bixler GV, Brucklacher RM (2011) Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J … [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dilger RN, Johnson RW (2008) Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. Journal of leukocyte biology 84: 932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Letiembre M, Hao W, Liu Y, Walter S, Mihaljevic I (2007) Innate immune receptor expression in normal brain aging. Neuroscience [DOI] [PubMed] [Google Scholar]
- 30. Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, et al. (2006) The age-related attenuation in long-term potentiation is associated with microglial activation. Journal of neurochemistry 99: 1263–1272. [DOI] [PubMed] [Google Scholar]
- 31. Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS (2009) Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. The Journal of clinical psychiatry 70: 1078–1090. [DOI] [PubMed] [Google Scholar]
- 32. Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW (2006) mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiology of … [DOI] [PubMed] [Google Scholar]
- 33. Sheffield LG, Berman N (1998) Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiology of Aging [DOI] [PubMed] [Google Scholar]
- 34. Keleshian VL, Modi HR, Rapoport SI, Rao JS (2013) Aging is associated with altered inflammatory, arachidonic acid cascade, and synaptic markers, influenced by epigenetic modifications, in the human frontal cortex. Journal of Neurochemistry 125: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Lu T, Pan Y, Kao S-Y, Li C, Kohane I, et al. (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429: 883–891. [DOI] [PubMed] [Google Scholar]
- 36. Dinarello CA (2002) The IL-1 family and inflammatory diseases. Clin Exp Rheumatol 20: S1–13. [PubMed] [Google Scholar]
- 37. Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, et al. (2001) Maternal cytokine levels during pregnancy and adult psychosis. Brain, behavior, and immunity 15: 411–420. [DOI] [PubMed] [Google Scholar]
- 38. Garden GA, Moller T (2006) Microglia biology in health and disease. Journal of neuroimmune pharmacology: the official journal of the Society on NeuroImmune Pharmacology 1: 127–137. [DOI] [PubMed] [Google Scholar]
- 39. Kim SU, de Vellis J (2005) Microglia in health and disease. Journal of neuroscience research 81: 302–313. [DOI] [PubMed] [Google Scholar]
- 40. Deverman BE, Patterson PH (2009) Cytokines and CNS development. Neuron 64: 61–78. [DOI] [PubMed] [Google Scholar]
- 41. Kaneko M, Stellwagen D, Malenka RC, Stryker MP (2008) Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58: 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnstone DM, Graham RM, Trinder D, Riveros C, Olynyk JK, et al. (2012) Changes in brain transcripts related to Alzheimer's disease in a model of HFE hemochromatosis are not consistent with increased Alzheimer's disease risk. Journal of Alzheimer's disease: JAD 30: 791–803. [DOI] [PubMed] [Google Scholar]
- 43. Guet CC, Elowitz MB, Hsing W, Leibler S (2002) Combinatorial synthesis of genetic networks. Science 296: 1466–1470. [DOI] [PubMed] [Google Scholar]
- 44. Crespo I, Roomp K, Jurkowski W, Kitano H, del Sol A (2012) Gene regulatory network analysis supports inflammation as a key neurodegeneration process in prion disease. BMC systems biology 6: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dieci G, Fermi B, Bosio MC (2014) Investigating transcription reinitiation through in vitro approaches. Transcription 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hashimshony T, Yanai I (2010) Revealing developmental networks by comparative transcriptomics. Transcription 1: 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ryan VH, Primiani CT, Rao JS, Ahn K, Rapoport SI, et al. (2014) Coordination of Gene Expression of Arachidonic and Docosahexaenoic Acid Cascade Enzymes during Human Brain Development and Aging. PLoS ONE 9 6: e100858 doi:10.1371/journal.pone.0100858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, et al. (2011) Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478: 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Somel M, Guo S, Fu N, Yan Z, Hu HY, et al. (2010) MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome research 20: 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodwell GEJ, Sonu R, Zahn JM, Lund J, Wilhelmy J, et al. (2004) A transcriptional profile of aging in the human kidney. PLoS biology 2: e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Norden DM, Godbout JP (2013) Review: Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathology and Applied Neurobiology 39: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nichols NR, Day JR, Laping NJ, Johnson SA, Finch CE (1993) GFAP mRNA increases with age in rat and human brain. Neurobiology of Aging 14: 421–429. [DOI] [PubMed] [Google Scholar]
- 53. Iwama H (2013) Coordinated networks of microRNAs and transcription factors with evolutionary perspectives. Advances in experimental medicine and biology 774: 169–187. [DOI] [PubMed] [Google Scholar]
- 54. Jo YH, Chua S Jr (2009) Transcription factors in the development of medial hypothalamic structures. American journal of physiology Endocrinology and metabolism 297: E563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uffenbeck SR, Krebs JE (2006) The role of chromatin structure in regulating stress-induced transcription in Saccharomyces cerevisiae. Biochemistry and cell biology = Biochimie et biologie cellulaire 84: 477–489. [DOI] [PubMed] [Google Scholar]
- 56. Goyal MS, Raichle ME (2013) Gene expression-based modeling of human cortical synaptic density. Proceedings of the National Academy of Sciences of the United States of America 110: 6571–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, et al. (2012) DNA methylation signatures in development and aging of the human prefrontal cortex. American journal of human genetics 90: 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, et al. (2011) Spatio-temporal transcriptome of the human brain. Nature 478: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benjamini Y, Y H (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Statistical Soc Ser B 57: 289–300. [Google Scholar]
- 60. Johnson WE, Li C, Rabinovic A (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (Oxford, England) 8: 118–127. [DOI] [PubMed] [Google Scholar]
- 61. Boeckmann B, Blatter MC, Famiglietti L, Hinz U, Lane L, et al. (2005) Protein variety and functional diversity: Swiss-Prot annotation in its biological context. C R Biol 328: 882–899. [DOI] [PubMed] [Google Scholar]
- 62. Pruitt KD, Tatusova T, Brown GR, Maglott DR (2012) NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pruitt KD, Tatusova T, Klimke W, Maglott DR (2009) NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res 37: D32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D (1997) GeneCards: integrating information about genes, proteins and diseases. Trends in Genetics 13: 163. [DOI] [PubMed] [Google Scholar]
- 65. De Trez C, Ware CF (2008) The TNF receptor and Ig superfamily members form an integrated signaling circuit controlling dendritic cell homeostasis. Cytokine & growth factor reviews 19: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Troutman TD, Bazan JF, Pasare C (2012) Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell cycle 11: 3559–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, et al. (2003) Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A 100: 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Basu A, Krady JK, Levison SW (2004) Interleukin-1: a master regulator of neuroinflammation. Journal of neuroscience research 78: 151–156. [DOI] [PubMed] [Google Scholar]
- 69. Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Molecular psychiatry 15: 384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim HW, Rapoport SI, Rao JS (2011) Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Molecular psychiatry 16: 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rao JS, Kim HW, Harry GJ, Rapoport SI, Reese EA (2013) Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. Schizophrenia research 147: 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, et al. (2007) Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proceedings of the National Academy of Sciences of the United States of America 104: 19649–19654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hommet C, Mondon K, Camus V, Ribeiro MJ, Beaufils E, et al. (2013) Neuroinflammation and β Amyloid Deposition in Alzheimer's Disease: In vivo Quantification with Molecular Imaging. Dementia and geriatric cognitive disorders 37: 1–18. [DOI] [PubMed] [Google Scholar]
- 74. Taylor JM, Allen AM, Graham A (2014) Targeting mitochondrial 18 kDa Translocator protein (TSPO) regulates macrophage cholesterol efflux and lipid phenotype. Clinical science [DOI] [PubMed] [Google Scholar]
- 75. Lipska BK, Weinberger DR (1995) Genetic variation in vulnerability to the behavioral effects of neonatal hippocampal damage in rats. Proc Natl Acad Sci U S A 92: 8906–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shankar SK (2010) Biology of aging brain. Indian journal of pathology & microbiology 53: 595–604. [DOI] [PubMed] [Google Scholar]
- 77. Glass CK, Saijo K, Winner B, Winner B, Marchetto MC, et al. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Allan SM, Rothwell NJ (2003) Inflammation in central nervous system injury. Philosophical transactions of the Royal Society of London Series B, Biological sciences 358: 1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Caldeira GL, Ferreira IL, Rego AC (2013) Impaired transcription in Alzheimer's disease: key role in mitochondrial dysfunction and oxidative stress. Journal of Alzheimer's disease: JAD 34: 115–131. [DOI] [PubMed] [Google Scholar]
- 80. Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, et al. (2013) Global epigenomic reconfiguration during mammalian brain development. Science 341: 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rao J, Chiappelli J, Kochunov P, Rapoport SI, Hong LE (In preparation) Age Changes in Brain Derived Neurotrophic Factor in Postmortem Gray and White Matter from Schizophrenia and Control Subjects. [Google Scholar]
- 82. Terry AV Jr, Kutiyanawalla A, Pillai A (2011) Age-dependent alterations in nerve growth factor (NGF)-related proteins, sortilin, and learning and memory in rats. Physiology & behavior 102: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, et al. (2011) Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America 108: 13281–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P (1996) COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A 93: 2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ong WY, Yeo JF, Ling SF, Farooqui AA (2005) Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J Neurocytol 34: 447–458. [DOI] [PubMed] [Google Scholar]
- 86. Murakami M, Kambe T, Shimbara S, Kudo I (1999) Functional coupling between various phospholipase A2s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J Biol Chem 274: 3103–3105. [DOI] [PubMed] [Google Scholar]
- 87. Rapoport SI (2008) Arachidonic acid and the brain. The Journal of nutrition 138: 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ramadan E, Rosa AO, Chang L, Chen M, Rapoport SI, et al. (2010) Extracellular-derived calcium does not initiate in vivo neurotransmission involving docosahexaenoic acid. J Lipid Res 51: 2334–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Harry GJ (2013) Microglia during development and aging. Pharmacology & therapeutics 139: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, et al. (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nature neuroscience 9: 917–924. [DOI] [PubMed] [Google Scholar]
- 91. Fuhrmann M, Bittner T, Jung CKE, Burgold S, Page RM, et al. (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nature neuroscience 13: 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, et al. (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. The American journal of pathology 177: 2549–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, et al. (2012) Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [(1)(1)C]DAA1106. Psychiatry research 203: 67–74. [DOI] [PubMed] [Google Scholar]
- 94. Kumar A, Muzik O, Shandal V, Chugani D, Chakraborty P, et al. (2012) Evaluation of age-related changes in translocator protein (TSPO) in human brain using (11)C-[R]-PK11195 PET. Journal of neuroinflammation 9: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, et al. (2006) Synapse formation and function is modulated by the amyloid precursor protein. The Journal of neuroscience: the official journal of the Society for Neuroscience 26: 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, et al. (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430: 213–218. [DOI] [PubMed] [Google Scholar]
- 97. Huising MO, Stet RJ, Savelkoul HF, Verburg-van Kemenade BM (2004) The molecular evolution of the interleukin-1 family of cytokines; IL-18 in teleost fish. Developmental and comparative immunology 28: 395–413. [DOI] [PubMed] [Google Scholar]
- 98. Tay A, Simon JS, Squire J, Hamel K, Jacob HJ, et al. (1995) Cytosolic phospholipase A2 gene in human and rat: chromosomal localization and polymorphic markers. Genomics 26: 138–141. [DOI] [PubMed] [Google Scholar]
- 99. Gutierrez EG, Banks WA, Kastin AJ (1994) Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. Journal of neuroimmunology 55: 153–160. [DOI] [PubMed] [Google Scholar]
- 100. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, et al. (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493: 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Youm Y-H, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, et al. (2011) Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 152: 4039–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhao Y, Kerscher N, Eysel U, Funke K (2001) Changes of contrast gain in cat dorsal lateral geniculate nucleus by dopamine receptor agonists. Neuroreport 12: 2939–2945. [DOI] [PubMed] [Google Scholar]
- 103. Stephenson DT, Lemere CA, Selkoe DJ, Clemens JA (1996) Cytosolic Phospholipase A2(cPLA2) Immunoreactivity Is Elevated in Alzheimer's Disease Brain. Neurobiology of disease 3: 51–63. [DOI] [PubMed] [Google Scholar]
- 104. Moses GSD, Jensen MD, Lue L-F, Walker DG, Sun AY, et al. (2006) Secretory PLA2-IIA: a new inflammatory factor for Alzheimer's disease. Journal of neuroinflammation 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rao JS, Harry GJ, Rapoport SI, Kim H-W (2009) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Molecular psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Casey BJ, Giedd JN, Thomas KM (2000) Structural and functional brain development and its relation to cognitive development. Biological psychology 54: 241–257. [DOI] [PubMed] [Google Scholar]
- 107. Morrison JH, Baxter MG (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nature reviews Neuroscience 13: 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176. [DOI] [PubMed] [Google Scholar]
- 109. Loerch PM, Lu T, Dakin KA, Vann JM, Isaacs A, et al. (2008) Evolution of the aging brain transcriptome and synaptic regulation. PloS one 3: e3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, et al. (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Somel M, Liu X, Khaitovich P (2013) Human brain evolution: transcripts, metabolites and their regulators. Nature reviews Neuroscience 14: 112–127. [DOI] [PubMed] [Google Scholar]
- 112. Rhie SK, Coetzee SG, Noushmehr H, Yan C, Kim JM, et al. (2013) Comprehensive functional annotation of seventy-one breast cancer risk Loci. PloS one 8: e63925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Persengiev S, Kondova I, Bontrop R (2013) Insights on the functional interactions between miRNAs and copy number variations in the aging brain. Frontiers in molecular neuroscience 6: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Veenman L, Bode J, Gaitner M, Caballero B, Pe'er Y, et al. (2012) Effects of 18-kDa translocator protein knockdown on gene expression of glutamate receptors, transporters, and metabolism, and on cell viability affected by glutamate. Pharmacogenetics and genomics 22: 606–619. [DOI] [PubMed] [Google Scholar]
- 115. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388: 394–397. [DOI] [PubMed] [Google Scholar]
- 116. Takeda K, Akira S (2005) Toll-like receptors in innate immunity. International immunology 17: 1–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Matrices showing Pearson correlation coefficients between expression levels of individual gene pairs during Development (A) and Aging (B). Green highlights coefficients that are significant at p<0.0001. Hierarchy in gene order corresponds to hierarchy in Figures 5A and 5B.
(XLSX)
Selected genes, chromosomal locations, protein description, and major reported functions. Based on the literature, genes whose protein products participate in major functions may be categorized as follows: (1) Synaptic function, SYP, DBN1, SNCA; (2) Growth and maintenance, BDNF, NGF, GAP43, PDGFA; (3) Myelin integrity, MOBP; (4) Neuroinflammation: (a) Microglial activation, CD68, TSPO, TLR4, TLR2, NOS2, AIF1; (b) Cytokine and chemokine processes, CX3CR1, CX3CL1, IFI16, IL1B, IL1R1, IL1RN, IL1RAP, IL6, TNFRSF1A, TRAF6, TRAP1, MAPK14, MYD88; (c) Glial activation, GFAP; (d) Apoptosis, CASP1; (5) Arachidonic acid cascade, PLA2G4A, PLA2G10, PTGS1, PTGS2; (5) Amyloid membrane processing, APP, BACE; (6) Microtubules, MAP2; (7) Transcription factors, NFKB1, TFAP2A [61], [62], [64], [114].
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All BrainCloud files are available from the GEO Accession database (accession number: GSE30272).