

Abstract
Background
Studies have shown a relationship between air pollution and increased risk of cardiovascular morbidity and mortality. Due to the complexity of ambient air pollution composition, recent studies have examined the effects of co-exposure, particularly particulate matter (PM) and gas, to determine whether pollutant interactions alter (e.g. synergistically, antagonistically) the health response. This study examines the independent effects of fine (FCAPs) and ultrafine (UFCAPs) concentrated ambient particles on cardiac function, and determine the impact of ozone (O3) co-exposure on the response. We hypothesized that UFCAPs would cause greater decrement in mechanical function and electrical dysfunction than FCAPs, and that O3 co-exposure would enhance the effects of both particle-types.
Methods
Conscious/unrestrained radiotelemetered mice were exposed once whole-body to either 190 μg/m3 FCAPs or 140 μg/m3 UFCAPs with/without 0.3 ppm O3; separate groups were exposed to either filtered air (FA) or O3 alone. Heart rate (HR) and electrocardiogram (ECG) were recorded continuously before, during and after exposure, and cardiac mechanical function was assessed using a Langendorff perfusion preparation 24 hrs post-exposure.
Results
FCAPs alone caused a significant decrease in baseline left ventricular developed pressure (LVDP) and contractility, whereas UFCAPs did not; neither FCAPs nor UFCAPs alone caused any ECG changes. O3 co-exposure with FCAPs caused a significant decrease in heart rate variability when compared to FA but also blocked the decrement in cardiac function. On the other hand, O3 co-exposure with UFCAPs significantly increased QRS-interval, QTc and non-conducted P-wave arrhythmias, and decreased LVDP, rate of contractility and relaxation when compared to controls.
Conclusions
These data suggest that particle size and gaseous interactions may play a role in cardiac function decrements one day after exposure. Although FCAPs + O3 only altered autonomic balance, UFCAPs + O3 appeared to be more serious by increasing cardiac arrhythmias and causing mechanical decrements. As such, O3 appears to interact differently with FCAPs and UFCAPs, resulting in varied cardiac changes, which suggests that the cardiovascular effects of particle-gas co-exposures are not simply additive or even generalizable. Additionally, the mode of toxicity underlying this effect may be subtle given none of the exposures described here impaired post-ischemia recovery.
Electronic supplementary material
The online version of this article (doi:10.1186/s12989-014-0054-4) contains supplementary material, which is available to authorized users.
Keywords: Air pollution, Cardiac function, Ventricular contraction, Electrocardiogram, Arrhythmia, Particulate matter
Background
Risk assessments of air pollution health effects have become increasingly challenging given the complexity of present-day air pollution mixtures. Epidemiological studies indicate that fine (PM2.5) and ultrafine (PM0.1) particulate matter (PM) are the principal instigators of adverse clinical events, particularly those involving the cardiovascular system [1]. However, air pollution is a mixture of not only PM, but also gaseous irritants, vapors, and biological substances; thus when examining the effects of any given pollutant, the influence of other components must be considered. As such, studies need to determine whether the resultant physiological and biochemical effects of multipollutant exposures represent the simple additive effects of the pollutants, their synergism or antagonism. One particularly relevant interaction is that of PM and the ubiquitous gaseous co-pollutant O3.
Although studies have examined the effects of sequential exposures, for example, ozone (O3) and then PM2.5 causes decreased HRV, systolic blood pressure and heart rate (HR) in rats [2], only a few studies have addressed the health effects of simultaneous exposures with distinct pollutants and the effects are still not fully clear. For instance, Brook et al. demonstrated acute arterial vasoconstriction in healthy subjects co-exposed to PM2.5 and O3 [3], whereas Urch et al. [4] found no significant changes in mean arterial pressure, systolic blood pressure or HR in a similar study population; although constriction was observed with PM2.5 alone. Animal studies also indicate that the effect of combining pollutants does not necessarily yield the expected synergistic response, especially in the case of susceptible models. Wagner et al. recently showed that depression of heart rate and blood pressure during PM2.5 and O3 co-exposure was not as great as either pollutant alone in rats fed a high-fructose diet [5]. The respiratory effects of O3 and PM co-exposure are equally conflicting. For example, rats instilled with ozonized DEP had increased inflammatory cells and protein in the lungs [6], whereas mice co-exposed to O3 and DEP did not have increased cytotoxicity or inflammation [7]. Instead, in this latter study, co-exposed mice had increased bronchoconstriction, which is a measure of lung function. Similar investigations into the effects of simultaneous exposure on cardiac function have not been widely conducted.
Rodent electrocardiograms (ECG) can provide valuable insight into cardiovascular function in air pollution studies, particularly when pollutant concentrations are low and overt inflammation or toxicity are not observed. ECG is now routinely used in rodents for the detection of disturbances in myocardial impulse formation and conduction, as well as abnormal cardiac rhythm and altered autonomic regulation of the heart. As such, a wide range of cardiac responses demonstrated by controlled human and animal PM exposure studies have provided biological plausibility to the health effects of air pollution [3,8-10]. Some of these are responses observed using ECG and have been shown to be similar in humans and animals [11,12]. For instance, some human subjects exposed to PM have decreased heart rate variability (HRV), which is a predictor of increased risk [13-16], and enhanced arrhythmogenesis [17]. Experiments in animals not only show a similar PM-induced decrease in HRV and increased incidence of arrhythmia [1], but also functional decrements in the heart such as diminished left ventricular developed pressure (LVDP) and decreased contractility [18-20]. On the other hand, few studies, if any, have examined the effects of simultaneous PM and O3 exposure on both ECG and mechanical function (e.g. contractility) of the heart.
Thus, the purpose of this study was to determine the effects of concentrated ambient particles (CAPs), with and without O3 co-exposure, on cardiac electrical and mechanical function in mice. Previous data suggests that PM size determines the physiological impact with fine PM causing primarily pulmonary effects and ultrafine PM altering cardiac function [21,22]. We hypothesized (1) that inhalation of either fine (FCAPs) or ultrafine CAPs (UFCAPs) would cause cardiac electrical dysfunction, mechanical decrements and arrhythmogenesis in mice; but (2) that UFCAPs, due to its size, would have a greater effect on the heart than FCAPs; and (3) that O3 co-exposure would potentiate the response elicited by both particle sizes, respectively.
Results
Chamber and exposure characteristics
Table 1 shows the concentration and particle size of CAPs and O3, and chamber characteristics for each exposure group. Table 2 indicates the elemental composition of the particulate matter from each of the exposure groups. Other than iron (Fe), FCAPs and UFCAPs particulate matter were of similar composition with the majority of the elemental fraction composed of SO4.
Table 1.
Chamber and exposure characteristics
| Groups | ||||||
|---|---|---|---|---|---|---|
| FA | UFCAPs | UFCAPs + O 3 | FCAPs | FCAPs + O 3 | O 3 | |
| Temperature (°C) | 22.3 ± 0.1 | 23.0 ± 0.1 | 22.6 ± 0.1 | 22.0 ± 0.1 | 22.2 ± 0.1 | 22.5 ± 0.2 |
| Rel. humidity (%) | 50.2 ± 0.7 | 70.5 ± 4.6 | 56.0 ± 3.6 | 59.8 ± 3.4 | 59.0 ± 5.8 | 52.4 ± 2.2 |
| O3 (ppb) | 4.0 ± 0.0 | 25.7 ± 4.7 | 298.3 ± 0.7 | 33.1 ± 2.0 | 300.0 ± 0.4 | 299.0 ± 1.1 |
| PM Mass (ug/m3) | 4.9 ± 2.2 | 138.8 ± 33.1 | 85.7 ± 6.5 | 190.9 ± 32.8 | 211.5 ± 37.3 | 3.4 ± 1.3 |
| PM Total # (particles/cc) | 24.2 ± 1.4 | 2.1E5 ± 5.6E3 | 1.6E5 ± 2.0E3 | 1.0E4 ± 5.2E1 | 1.1E4 ± 1.6E2 | 20.2 ± 4.2 |
| Particle size (um) | - | 0.076 | 0.072 | 0.246 | 0.235 | - |
| Geo. Std. Dev. | - | 1.67 | 1.66 | 1.96 | 1.67 | - |
| PM CARBON | ||||||
| TC (μg/m3) | 3.4 ± 0.2 | 67.6 ± 6.5(48.7%) | 46.4 ± 3.4(54.1%) | 53.8 ± 4.4 (28.2%) | 47.8 ± 7.3 (22.6%) | 4.3 ± 0.4 |
| OC (μg/m3) | 3.7 ± 0.2 | 64.5 ± 5.9 (46.5%) | 44.6 ± 3.1 (52.0%) | 50.3 ± 4.0 (26.3%) | 45.7 ± 6.6 (21.6%) | 4.6 ± 0.4 |
| EC (μg/m3) | ** | 3.1 ± 0.6(2.2%) | 1.8 ± 0.3(2.1%) | 3.4 ± 0.5(1.8%) | 2.2 ± 0.7(1.0%) | ** |
Reported values are mean ± SEM for each group over all exposure days.
**below detection limit.
Carbon percentages are by mass.
Table 2.
Elemental composition of particulate matter in exposure groups
| Element(μg/m 3 ) | Groups | |||||
|---|---|---|---|---|---|---|
| FA | UFCAPs | UFCAPs + O 3 | FCAPs | FCAPs + O 3 | O 3 | |
| Al | --- | bdl | bdl | 1.8364 | 0.8428⋄ | --- |
| As | --- | 0.0020 | 0.0040 | 0.0044 | 0.0077 | --- |
| Ba | --- | 0.0070 | 0.0047 | 0.0865 | 0.0480⋄ | --- |
| Ca | --- | bdl | bdl | 1.1741 | 0.6475 | --- |
| Cd | --- | 0.0005 | 0.0005 | 0.0015 | 0.0010 | --- |
| Co | --- | 0.0002 | 0.0002 | 0.0008 | 0.0006 | --- |
| Cr | --- | 0.0088 | 0.0198 | 0.0129 | 0.0134 | --- |
| Cu | --- | 0.0119 | 0.0727Δ | 0.1550 | 0.0537⋄ | --- |
| Fe | --- | 0.0723 | 0.0848 | 2.1031 | 1.0555⋄ | --- |
| K | --- | 0.7448 | 0.4325 | 2.6025 | 1.3867 | --- |
| Li | --- | bdl | bdl | 0.0017 | 0.0013 | --- |
| Mg | --- | 0.0282 | 0.0254 | 0.5585 | 0.6145 | --- |
| Mn | --- | 0.0048 | 0.0058 | 0.0604 | 0.0348 | --- |
| Mo | --- | 0.0014 | 0.0032 | 0.0033 | 0.0036 | --- |
| Na | --- | 0.1491 | 0.2550 | 0.8990 | 3.7144 | --- |
| Ni | --- | 0.0053 | 0.0348Δ | 0.0079 | 0.0084 | --- |
| P | --- | bdl | bdl | 0.1278 | 0.1565 | --- |
| Pb | --- | 0.0128 | 0.0124 | 0.0373 | 0.0271 | --- |
| SO4 | --- | 56.4662 | 39.8730 | 39.4038 | 49.3026 | --- |
| Sb | --- | 0.0035 | 0.0040 | 0.0146 | 0.0108 | --- |
| Se | --- | bdl | bdl | 0.0246 | 0.0248 | --- |
| SiO2 | --- | Bdl | bdl | 4.9640 | 2.8436⋄ | --- |
| Sn | --- | 0.0182 | 0.0239 | 0.0151 | 0.0121 | --- |
| Sr | --- | 0.0016 | 0.0006 | 0.0179 | 0.0101 | --- |
| Ti | --- | bdl | bdl | 0.0691 | 0.0429 | --- |
| V | --- | 0.0023 | 0.0052 | 0.0092 | 0.0122 | --- |
| Zn | --- | 0.1527 | 0.0989 | 0.2171 | 0.2057 | --- |
bdl - Below Detection Level.
--- Very low PM concentrations, insufficient sample mass for elemental analysis.
Δ Significantly different from UFCAPs.
⋄Significantly different from FCAPs.
Estimated particle doses
The following particle doses were calculated for the mice in each of the PM-exposed group: (1) UFCAPs - 0.418 μg (2) FCAPs - 0.426 μg (3) UFCAPs + O3 - 0.264 μg and (4) FCAPs + O3 - 0.446 μg. Using the same model and exposure characteristics the estimated human doses were determined to be: (1) UFCAPs - 103.4 μg (2) FCAPs - 81.3 μg (3) UFCAPs + O3 - 65.8 μg and (4) FCAPs + O3 - 85.0 μg.
Heart rate
Although all animals experienced an increase in HR while in the exposure chamber before the start of the exposure (Baseline) and a progressive decrease during the 4-hour exposure (Exp1, Exp2, Exp3 and Exp4), there were no significant differences in HR among any of the exposure groups during any time period (Figure 1).
Figure 1.
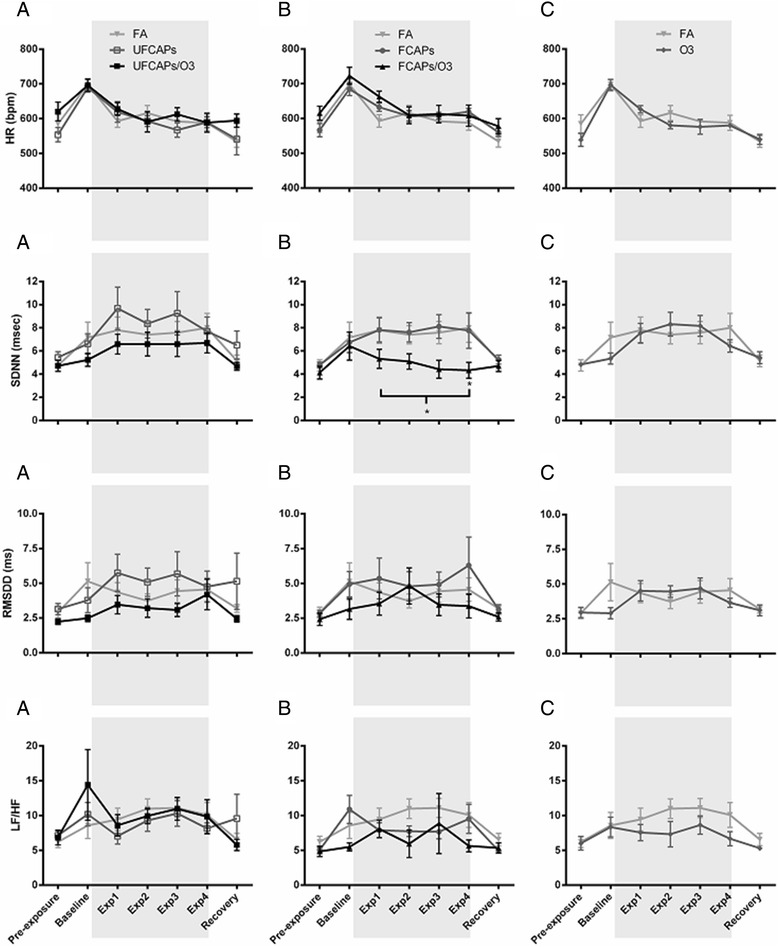
The effect of CAPs with and without O 3 on heart rate and heart rate variability. All animals were placed in the chambers and allowed to acclimate for 1 hr before the exposure began (Baseline) and then exposed for 4 hrs (shaded area). When compared to pre-exposure, all animals experienced an increase in HR during baseline, and then a progressive decrease from baseline during hour-1, 2, 3 and 4 of exposure (Exp1, Exp2, Exp3, and Exp4). Exposure to UFCAPs with (UF/O3) or without (UF) ozone did not cause any significant changes in HR or HRV at any time point (Column A.). Similarly, there was no effect of FCAPs alone (F) on any parameter; only exposure to FCAPs + O3 (F/O3) significantly decreased SDNN when compared to FA (Column B.). Exposure to ozone alone did not cause any significant effects (Column C.). Bracket indicates that each hour of the exposure period is significantly different. Values are mean ± SEM; *p < 0.05, significantly different from FA (n = 6).
Heart rate variability (HRV)
Exposure to FCAPs + O3 caused a significant decrease in the SDNN (4.8 ± 0.4 ms) compared with FA controls (7.7 ± 0.5 ms) (Figure 1). No other significant differences in time-domain HRV measurements were found among any of the exposure groups pre-, during or post-exposure. There were also no significant differences in the LF/HF between any exposure groups.
Electrocardiogram
Figure 2 shows the electrocardiogram data before, during and after exposure. There were no significant differences in ECG between any of the groups during pre-exposure or recovery. All animals experienced a decrease in PR interval, QRS, ST interval, and QTc during the baseline, which was likely related to the increase in HR. Thereafter, PR interval and ST interval increased in all animals during the exposure; though there were no significant differences. In contrast, QRS and QTc were significantly increased in mice exposed to UFCAPs + O3 when compared to FA. Exposure to O3 alone demonstrated a trend towards decreased QTc when compared with FA.
Figure 2.
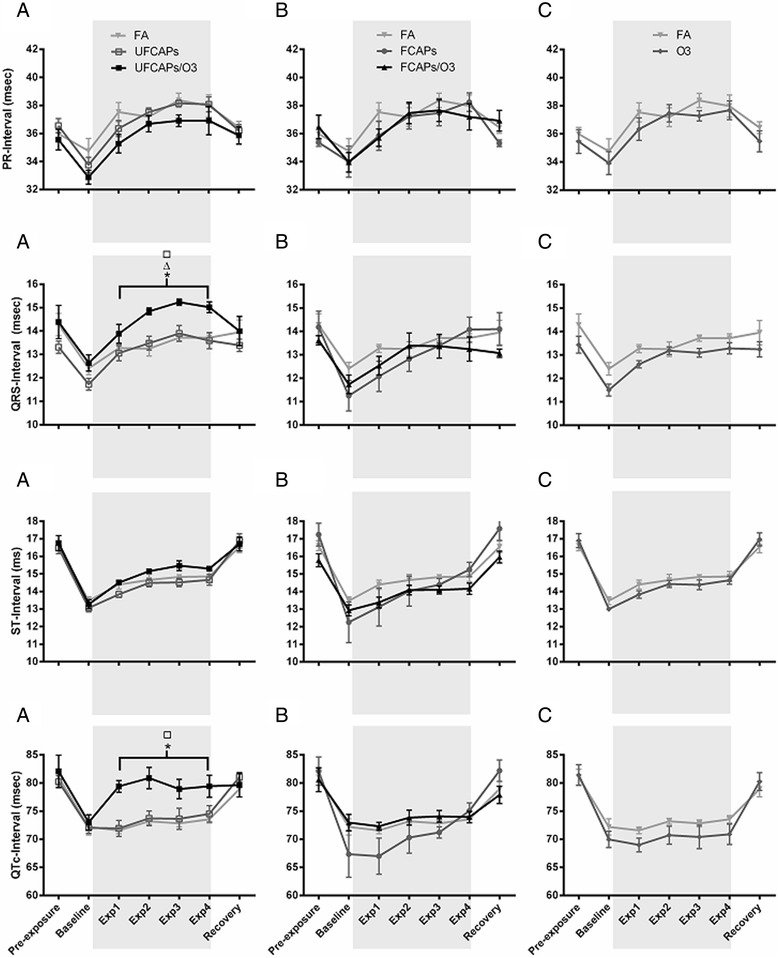
Electrocardiogram effects before, during and after exposure to CAPs alone or with O 3 . All animals were placed in the chambers and allowed to acclimate for 1 hr before the exposure began (Baseline) and then exposed for 4 hrs (shaded area). All animals experienced a decrease in PR interval, QRS complex duration, ST interval and QTc from pre-exposure to baseline; these changes likely corresponded to the change in HR. Although both PR and ST intervals increased in all groups during hour-1, 2, 3, and 4 of exposure (Exp1, Exp2, Exp3 and Exp4), there were no differences in either of these parameters among any of the groups. QRS also showed an increasing trend during exposure in all groups; however only mice exposed to UFCAPs + O3 had a significant increase in QRS and QTc when compared to FA (Column A.). There were no significant effects of FCAPs, with or without O3, or O3 alone at any time points (Column B. and C., respectively). Bracket indicates that each hour of the exposure period is significantly different. Values are mean ± SEM. p < 0.05; *significantly different from FA, ∆ significantly different from UFCAPs alone, □ significantly different from O3 alone (n = 6).
Cardiac arrhythmia
There was a significant increase in the number of non-conducted P-wave arrhythmias during the 4-hour exposure period to UFCAPs + O3 when compared with FA (Figure 3C). No other significant differences in arrhythmias were observed among any of the exposure groups. Although other types of arrhythmias were noted, they were few in number and not statistically different between any of the groups.
Figure 3.
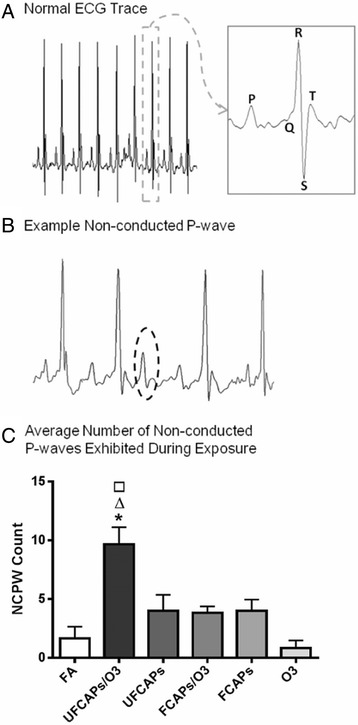
Typical mouse electrocardiogram and arrhythmia count during exposure. A. Typical mouse ECG during normal sinus rhythm and B. a non-conducted p-wave (NCPW) - represents a sudden loss of conduction from the atria to the ventricles. C. Non-conducted p-waves were significantly increased only in mice exposed to UFCAPs + O3. Values are mean ± SEM. p < 0.05; *significantly different from FA, ∆ significantly different from UFCAPs alone, □ significantly different from O3 alone (n = 6).
Cardiac effects before ischemia
Post-exposure (baseline) hemodynamics and the onset time to ischemic contracture for each of the exposure groups are listed in Table 3. As shown in Figure 4, there was a significant decrease in LVDP in the FCAPs (31.9 ± 6.7 cmH2O), O3 (54.7 ± 12.6 cmH2O) and UFCAPs + O3 (45.0 ± 9.2 cmH2O) groups compared to FA (96.7 ± 9.6 cmH2O) 24 hours after exposure and before ischemia. Left ventricular contractility was also significantly depressed in the UFCAPs, FCAPs, O3 and UFCAPs + O3 groups compared to the FA control group. The maximum dP/dt was significantly lower in FCAPs (1397 ± 296 cmH2O/sec), O3 (2483 ± 480 cmH2O/sec) and UFCAPs + O3 (1975 ± 306 cmH2O/sec) when compared to FA (3880 ± 208 cmH2O/sec) and the minimum dP/dt before ischemia was also significantly lower in the UFCAPs (-1452 ± 395 cmH2O/sec), FCAPs (-982 ± 259 cmH2O/sec), O3 (-1520 ± 318 cmH2O/sec) and UFCAPs + O3 (-1323 ± 286 cmH2O/sec) groups when compared to FA (-2744 ± 317 cmH2O/sec) (Figure 5; Table 3). There was no difference in HR, coronary flow rate or ischemic contracture between any exposure groups before ischemia (Table 3).
Table 3.
Baseline hemodynamic properties and the onset time to ischemic contracture
| Group (n = 6-8) | LVDP (cmH 2 O) | HR (bpm) | Flow rate (mL/min) | d P /d t max (cmH 2 O/sec) | d P /d t min (cmH 2 O/sec) | Time to contracture (min) |
|---|---|---|---|---|---|---|
| FA | 96.7 ± 9.6 | 306.6 ± 17.5 | 1.7 ± 0.3 | 3880 ± 208 | -2744 ± 317 | 14.3 ± 1.9 |
| UFCAPs | 59.7 ± 15.4 | 270.3 ± 19.6 | 1.7 ± 0.5 | 2564 ± 825 | -1452 ± 395 | 14.6 ± 1.8 |
| UFCAPs + O3 | 45.0 ± 9.3* | 295.1 ± 18.5 | 2.0 ± 0.5 | 1975 ± 306* | -1323 ± 286* | 15.1 ± 1.6 |
| FCAPs | 31.9 ± 6.7* | 301.3 ± 27.6 | 5.7 ± 2.9 | 1397 ± 296* | -981 ± 259* | 11.1 ± 0.5 |
| FCAPs + O3 | 88.0 ± 18.4 | 277.6 ± 15.7 | 1.7 ± 0.2 | 3034 ± 528 | -2219 ± 394 | 12.9 ± 1.3 |
| O3 | 54.7 ± 12.6 | 246.3 ± 36.4 | 2.5 ± 0.6 | 2483 ± 480 | -1520 ± 318 | 15.3 ± 1.6 |
Values are means ± SEM. Flow rate = coronary flow rate; dP/dt max = maximum 1st derivative of the change in left ventricular pressure/time; dP/dt min = minimum 1st derivative of the change in left ventricular pressure/time; time to contracture = onset time to ischemic contracture. *Significantly different from FA; p < 0.05; n = 5-8.
Figure 4.
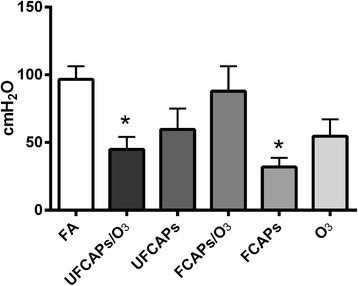
Effect of CAPs exposure on left ventricular developed pressure (LVDP). Exposure to FCAPs alone (F) significantly decreased LVDP at baseline (24 hrs after exposure - prior to ischemia) when compared to FA, however there was no effect with O3 co-exposure. In contrast, UFCAPs alone had no effect on LVDP but with O3 co-exposure caused a significant decrease when compared to FA. Values are means ± SEM (n = 5-8/group). *Significantly different from FA; p < 0.05.
Figure 5.
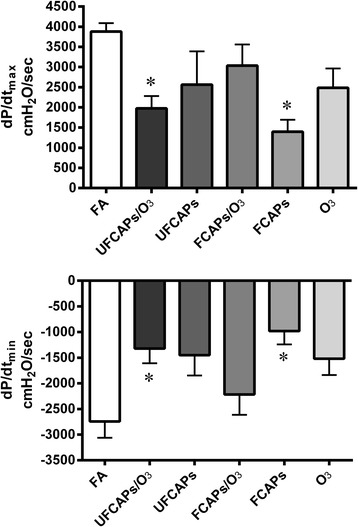
Effect of CAPs exposure with and without O 3 on rate of left ventricle contractility and relaxation. Assessment of contractility (dP/dt max - upper panel) and lusitropy (dP/dt min - lower panel) were carried out at baseline 24 hrs after exposure - prior to ischemia. Exposure to FCAPs alone significantly decreased dP/dt max and dP/dt min at baseline when compared to FA, however there was no effect with O3 co-exposure (FCAPs/O3). In contrast, UFCAPs alone had no effect; but, O3 co-exposure with UFCAPs (UFCAPs/O3) caused both dP/dt max and dP/dt min to significantly decrease when compared to FA. Values are means ± SEM (n = 5-8 in each group). *Significantly different from FA; p < 0.05.
Multivariate analysis of variance demonstrated that differences in LVDP, maximum dP/dt and minimum dP/dt between the FCAPs alone and FCAPs + O3 groups could be accounted for by the decrease in aluminum (Al), barium (Ba), copper (Cu), iron (Fe) or silicon dioxide (SiO2) compositions (Table 2); these elements clustered together however the analysis could not determine which element specifically was responsible. There were no apparent differences in elemental composition between UFCAPs alone and UFCAP + O3, except nickel (Ni), which were linked to any cardiac response changes, nor were there any other significant linkages with any other cardiac endpoints.
Cardiac effects post-ischemia
After ischemia there were minimal differences among the groups. There was a significant decrease in HR 20 min after reperfusion in the O3 group (213.8 ± 14.2 bpm) compared to FA (285.3 ± 17.5 bpm) (Figure 6). There were no differences in the post-ischemia coronary flow rates of any of the groups. Although all groups experienced a significant decrease in LVDP recovery when compared to pre-ischemia, there was no significant difference in post-ischemia recovery of LVDP (Figure 7), dP/dtmax, and dP/dtmin between any exposure groups.
Figure 6.
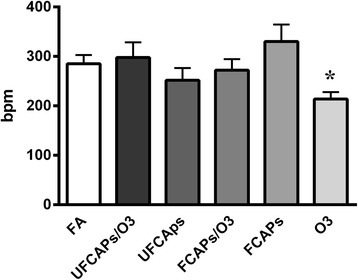
Heart rate twenty minutes after ischemia-reperfusion. After 20 mins of reperfusion, heart rate (HR) was significantly lower in animals exposed to O3 when compared to FA. There were no other significant differences in post-ischemia HR between any groups or at any other time point. Values are means ± SEM (n = 5-8 in each group). *Significantly different from FA; p < 0.05.
Figure 7.
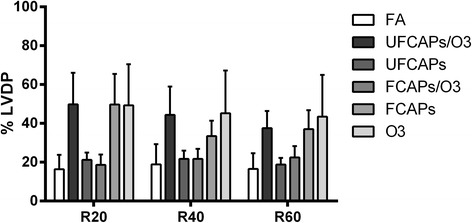
Post-ischemia recovery of LVDP. Following ischemia, all animals experienced a significant decrease in recovery LVDP (expressed as a percentage of pre-ischemia) when compared to pre-ischemia. There were no significant differences among any groups in the recovery LVDP at 20 (R20), 40 (R40) or 60 (R60) minutes post-ischemia; however there was a trend towards increased % LVDP in the FCAPs, O3 and UFCAPs + O3 groups at R20, R40 and R60 when compared to FA; consequently, these groups were the same ones demonstrating pre-ischemia changes. Values are means ± SEM (n = 5-8 in each group). *Significantly different from FA; p < 0.05.
Biochemical markers and inflammatory cells in BAL and blood
Exposure to O3 alone or UFCAPs + O3 caused a significant decrease in glutathione S-transferase (GTR) when compared to controls. There were no other significant differences in any other BAL cells or markers, or any of the serum or plasma markers (Table 4).
Table 4.
Biochemical markers in the bronchoalveolar lavage and serum
| Bronchoalveolar lavage | Blood | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | LDH(U/L) | MIA(μg/ml) | NAG(U/L) | Protein(μg/ml) | ACE(U/L) | CK(U/L) | CRP(μg/dl) | HBDH(U/L) | GTR(IU/ml) | LDH(U/L) | SOD(U/ml) | Protein(g/dl) |
| Air | 67.2 ± 12.4 | 10.3 ± 0.4 | 6.0 ± 0.2 | 131.0 ± 9.5 | 193.9 ± 26.4 | 2.3E3 ± 891.5 | 64.8 ± 10.0 | 216.3 ± 51.8 | 0.042 ± 0.014 | 551.4 ± 153.7 | 1.94 ± 0.2 | 4.4 ± 0.4 |
| UFCAPs | 50.7 ± 3.7 | 9.7 ± 0.2 | 6.7 ± 0.4 | 87.0 ± 11.8 | 250.3 ± 33.5 | 1.0E3 ± 32.9 | 73.2 ± 5.6 | 172.9 ± 6.5 | 0.022 ± 0.002 | 390.6 ± 30.2 | 1.6 ± 0.02 | 4.8 ± 0.2 |
| UFCAPs + O3 | 77.6 ± 20.3 | 10.6 ± 0.8 | 5.3 ± 0.5 | 120.2 ± 31.4 | 158.7 ± 25.3 | 1.0E3 ± 178.5 | 65.1 ± 6.6 | 239.5 ± 59.1 | 0.015 ± 0.004* | 441.8 ± 61.4 | 2.0 ± 0.2 | 4.7 ± 0.2 |
| FCAPs | 58.5 ± 13.7 | 9.8 ± 0.5 | 6.0 ± 0.1 | 94.9 ± 14.3 | 191.3 ± 23.3 | 1.2E3 ± 124.2 | 86.9 ± 12.9 | 190.1 ± 32.0 | 0.023 ± 0.005 | 396.9 ± 71.5 | 1.8 ± 0.1 | 5.0 ± 0.2 |
| FCAPs + O3 | 78.2 ± 313.7 | 10.2 ± 0.3 | 6.4 ± 0.3 | 127.4 ± 11.1 | 167.5 ± 22.2 | 1.9E3 ± 499.5 | 55.6 ± 9.6 | 170.1 ± 20.5 | 0.019 ± 0.003 | 382.4 ± 53.6 | 1.9 ± 0.2 | 4.3 ± 0.1 |
| O3 | 53.9 ± 7.5 | 9.5 ± 0.4 | 5.0 ± 0.2 | 83.7 ± 6.3 | 142.4 ± 30.3 | 1.7E3 ± 334.0 | 71.9 ± 18.0 | 203.9 ± 36.4 | 0.016 ± 0.004* | 431.9 ± 86.1 | 2.6 ± 0.3 | 4.5 ± 0.3 |
Values are mean ± SEM. *p < 0.05; significantly different from FA.
LDH – Lactate dehydrogenase.
MIA – Microalbumin.
NAG - N-acetyl-b-d-glucosaminidase.
ACE – angiotensin converting enzyme.
CK – Creatine Kinase.
CRP – C-reactive protein.
HBDH - α-hydroxybutyrate dehydrogenase.
GTR – glutathione-S-transferase.
SOD – Superoxide dismutase.
Discussion
This study demonstrates that a single inhalation exposure to either FCAPs or UFCAPs differentially affects cardiac mechanical and electrical responses in mice, and that the effect of O3 co-exposure on the response varies for each particle size. FCAPs alone caused decreased ventricular contractility but contrary to our original hypothesis UFCAPs alone had no effect. However, introduction of O3 as a co-pollutant with UFCAPs caused a significant decrease in cardiac contractility 24 hours after exposure and blunted the effects of FCAPs. In contrast, although exposure to either FCAPs or UFCAPs alone did not cause any significant electrocardiogram effects, co-exposure to each with O3 caused electrical and HRV changes that might indicate increased cardiac risk. Overall, our results demonstrate that UFCAPs + O3 produces the most significant effects across both mechanical and electrical cardiac function (Table 5). Thus, these data suggest there is a differential effect of particle size, which holds true in the presence or absence of O3, confirming the health effects resulting from a PM-gas co-exposure are not simply the sum of both pollutants. Instead, it appears each interaction (FCAPs + O3 vs. UFCAPs + O3) is complex and needs to be examined separately, particularly when exposure concentrations are low and the responses are subtle.
Table 5.
Summary of effects
| Group | LVDP | dP/dt max | dP/dt min | LVDP recovery (post-ischemia) | Coronary flow rate (post-ischemia) | Heart rate | SDNN | RMSSD | LF/HF | QTc | NCPW |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FA | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- |
| UFCAPs | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE |
| UFCAPs + O3 | ↓ | ↓ | ↓ | NE | NE | NE | NE | NE | NE | ↑ | ↑ |
| FCAPs | ↓ | ↓ | ↓ | NE | NE | NE | NE | NE | NE | NE | NE |
| FCAPs + O3 | NE | NE | NE | NE | NE | NE | ↓ | NE | NE | NE | NE |
| O3 | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE |
NE = No Effect.
↓ = significant decrease.
↑ = significant increase.
NOTE: the above responses are compared to Air.
Our previous findings [21] suggested that UFCAPs would cause greater cardiac effects than FCAPs. Ultrafine black carbon particles have been shown to translocate into the blood circulation and have the potential to cause direct effects on the cardiovascular system [23,24]. UF particles cause heterogeneity of repolarization and decreased HRV in humans [25], whereas mechanical assessments in animals reveal decreased LVDP, contractility and coronary flow [20,21,26,27]. In this study, animals were exposed via whole-body inhalation as opposed to instillation [21,26], direct perfusion [20,27], or nasal inhalation [22], which could have resulted in a comparatively lower effective dose and milder response [28,29]. However, among our animals, calculations of estimated total dose indicated that there was no difference between UFCAPs and FCAPs suggesting that in the absence of O3 more than just particle burden was responsible for the cardiac decrements. Instead, the site of deposition (i.e. pulmonary vs. extra-thoracic) may have played a more important role. FCAPs, which we estimated had a higher extra-thoracic deposition when compared to UFCAPs (0.232 μg vs. 0.142 μg, respectively), may have caused its effects through the activation of upper airway sensory mechanisms. Previous studies have shown that PM2.5 can cause irritation and subsequent activation of autonomic reflex arcs, particularly due to the presence of acidic components; UF particles did not produce the same response [30]. Thus, the higher relative exposure concentrations and differential deposition of FCAPs may have resulted in variable epithelial injury, inflammation, clearance and thus toxicological presentation [31].
On the other hand, it is not entirely surprising that on their own FCAPs and UFCAPs did not cause any significant changes in ECG given our previous negative results with a more toxic pollutant [32]. Similarly, Campen et al. [33] found that Apolipoprotein E (ApoE) -/- mice on a high fat diet, which are assumed to be susceptible to the cardiotoxic effects of inhaled pollutants, did not have any ECG changes when exposed to high concentrations of road dust PM or the vapor phase of gasoline engine exhaust. As far as arrhythmias are concerned, spatial dispersion of cardiac repolarization, which contributes to arrhythmogenesis, is increased in people after co-exposure to CAPs and O3 with each pollutant causing minimal effects on their own [34]. Even in the presence of O3, it is clear from not only our results, but the previously mentioned human data and other humans studies [35], that relatively low CAPs exposures will likely only cause mild electrical and HRV changes in healthy populations. Thus, a significant ECG effect due to acute exposure may not necessarily be direct evidence of serious cardiovascular morbidity or premature mortality; rather, it may reflect a transient instability that can worsen if exposure continues over a longer period.
Co-exposure to UFCAPs and O3 produced electrophysiological changes indicative of increased heterogeneity of repolarization, as well as an increased incidence of non-conducted p-wave arrhythmias, which suggest atrioventricular block. In humans, this form of arrhythmia is usually seen with a wide QRS complex [36], which was also observed in our mice exposed to UFCAPs + O3; this sometimes indicates that conduction is impaired in the ventricles, particularly when observed with a block. These results corroborate findings from human studies of PM exposure [37,38] as well as human studies of PM and O3 co-exposure [34]. Similarly, a long QTc due to prolonged repolarization suggests increased risk of early after-depolarizations, which can trigger arrhythmias and potentially myocardial infarction when propagated. Indeed it is not unusual that electrical and mechanical dysfunction were both observed in mice exposed to UFCAPs + O3 given increased arrhythmogenesis has been shown to be associated with changes in myocardial stretch [39].
Consequently, clarifying the role of each pollutant in the health response is challenging. It has been suggested that PM may be the main driver of the cardiovascular response in some instances with O3 acting as a modifier. Brook et al. previously showed that PM and O3 together cause acute arterial vasoconstriction in healthy humans subjects, but so does PM alone [3,40]. However, we observed depression of mechanical function with O3 alone; although much of the current research is focused on PM as a cardiotoxicant, several studies have also noted the adverse cardiovascular effects of O3 inhalation; which include decreased HR, alteration of cardiac repolarization, and increased inflammation [9,41-43]. As such, the type of cardiac responses following air pollution may be dependent on the type of pollutant, or combination of pollutants, with some degree of overlapping effects. Tankersley et al. [44] showed that both carbon black particles and O3 caused reduced cardiac output in mice but due to two different mechanisms. Thus, we speculate that although both particles and gases produce similar cardiac decrements, the mechanisms mediating the response may not be the same (e.g. translocation vs. airway sensory irritation). Combinations of pollutants only complicates the assessment due the involvement of various separate or overlapping mechanisms. Regardless, the responses appear to be independent of total particle dose or even pulmonary deposition given UFCAPs was estimated to be less than FCAPs (pulmonary dose - 0.104 μg vs. 0.135 μg, respectively).
The role of the autonomic nervous system cannot be entirely discounted either; as demonstrated through HRV, the responses observed here and in other studies with respect to PM exposure appear to be dependent on the size of the particles. The interpretation and importance of HRV in air pollution studies is still not entirely agreed upon, particularly when examining populations with underlying cardiovascular disease. Mills et al. [45] and Peretz et al. [46] did not observe any HRV changes in humans exposed to diesel exhaust, however this lack of effect does not necessarily imply that there are no autonomic changes, instead a trigger (e.g. stress, exercise, etc) may be necessary to reveal any HRV differences. On the other hand, some studies show that particles, especially fine, cause HRV effects in humans. Several studies have demonstrated that exposure to FCAPs causes decreased HRV in young healthy or elderly adults [15,47,48] with O3 co-exposure only potentiating the response [15]. In healthy young adults, there was no dose-response relationship between FCAPs mass and HRV, however when combined with O3, increases in CAPs mass decreased HRV in a dose-dependent manner [35]. On the other hand, UFCAPs either have no effect [49] or increase HRV [17,25] or the results are less conclusive across all studies. Long-term exposure to UFCAPs, or a higher concentration, may have caused a significant change in HRV given these particles have the ability to penetrate deep into the lung, cause inflammation and activate autonomic reflex pathways [50].
Some of these pathways may lead to subsequent ischemic damage, which has been shown to be increased by PM. Cozzi et al. showed that in mice intra-tracheally instilled with ultrafine PM, infarct size and oxidative stress in the myocardium were significantly increased [51]. This corroborates our previous PM instillation studies which also demonstrated an increase in post-ischemia infarct size and decreased recovery of LVDP [21]. It appears that the method of exposure significantly impacts the post-ischemia response because even though exposure to FCAPs or UFCAPs + O3 caused significant pre-ischemia functional decrements, there was no change in coronary flow post-ischemia and there appeared to be an improvement of LVDP recovery (Figure 7). These findings are similar to what we observed with inhalation of multipollutant mixtures [43] and may represent activation of some compensatory mechanism post-exposure that actually protects the heart during ischemic injury. Lastly, although infarct size was not measured in our animals, we theorize that there was probably minimum to no increase particularly given we previously observed a decrease in infarct size with multipollutant mixture inhalation [43]. Thus, acute inhalation of fine or ultrafine PM alone or in combination with O3 may not be potent enough to cause serious ischemia-related damage and that a higher concentration is necessary to overcome this apparent response threshold.
Other than the mode of exposure, the chemical and physical characteristics of the PM might also account for some of the differences in response observed in this study. Indeed it is a limitation that exposure to CAPs alone could not be done on the same days as CAPs + O3; this accounts for the variation in not only particle numbers but composition as well. However, it is our assertion that the responses to these “real-world” particle concentrations are important, especially given the daily fluctuation of particulate air pollution and the ubiquitousness of O3. It is also important to note that although we compare these results to our previous study [21], the composition of the current FCAPs and UFCAPs is different. Our CAPs, particularly the UFCAPs, had a higher organic (OC) and total carbon (TC) content; thus possibly explaining the differences in response.
As such, there was a three-fold decrease in Cu and a two-fold decrease in Fe in the FCAPs + O3 exposure when compared to FCAPs alone which may have contributed to the lack of effect in the former. There was also an increase in Ni and Cu, which have been shown to be two of the most toxic metals found in PM [52], in the UFCAPs + O3 exposure when compared to UFCAPs alone. In contrast, even though it appears that mass was not a factor in the observed decrements because there was less PM in the UFCAPs + O3 exposure than UFCAPs alone and the opposite for the FCAPs and O3 co-exposures, there was a significantly higher sulfate and OC/TC content in the UFCAPs, especially the UFCAPs + O3, when compared to FCAPs, which may explain the larger cardiac effect [53]. On the other hand, responses to FCAPs and UFCAPs combined with O3 might also be partially explained by the chemical changes occurring in PM upon ozonization. Ozone is highly reactive and therefore it has the potential to react with certain components of PM such as the aromatic compounds [6]. It has been documented that ozonization of aromatic substances can result in the formation of carbonyls, carboxylic acids, quinones, and epoxides, which can be more toxic than the parent compound [54,55], but also less potent due to “over-ozonization” [56]. It is yet unclear which mechanism is at play here.
Additionally, O3 may cause epithelial injury and oxidative stress, which facilitate the PM effects [57]. Adamson et al. [58] showed that O3 and urban particulate co-exposure resulted in greater epithelial injury and interstitial inflammation than for either component alone; not to mention UFP did not have a large biological effect without O3. As such, co-exposures may produce differential responses due to toxicological interactions within the host. Thomson et al. [59] showed that on their own, PM and O3 increased expression of the potent vasoconstrictor endothelin-1 (ET-1) in the lungs and its circulating levels in the plasma, however, together they only caused an upregulation (i.e. without plasma “spill-over”). Although there were no significant changes in inflammatory cells or markers in the blood or lavage, we found that O3 alone and UFCAPs + O3, but not FCAPs or UFCAPs alone, caused significantly decreased serum glutathione S-transferase (GTR) levels, which is indicative of increased oxidative stress; direct measurement of oxidative stress in the myocardium may have revealed a greater involvement as was shown by Cozzi et al. [51]. Wang et al. previously showed that PM2.5 and O3 increased several markers of inflammation and oxidative stress in rats however their exposure concentrations were significantly higher than those used here [60]. Regardless, synergistic interactions between inhalable PM and O3 can increase the generation of reactive oxygen species due to the porous surface of particles which provides ample surface area for reactivity, but that the potency still depends on particle concentration, size and other factors [61,62].
Conclusion
The results of this study demonstrate that fine and ultrafine CAPs differentially alter cardiac responses, which include both mechanical and electrical effects. More importantly, these data clearly show that the effects of co-exposure may not be simply additive or synergistic, nor even generalizable. Although only fine CAPs had significant effects on its own, O3 co-exposure with FCAPs caused decreased HRV whereas with UFCAPs caused electrical changes and arrhythmia. Interestingly, O3 co-exposure only caused mechanical decrements with UFCAPs and to our surprise blunted the effects of FCAPs. This indicates that the size, and potentially the chemical composition, of the particle is an important determinant of the type of cardiac response, particularly when gaseous co-pollutants are present. Although the responses were subtle, the important message may be that latent underlying changes are occurring post-exposure and that the deleterious effects of even a single exposure to air pollution needs to be considered. Some of these might not manifest as overt symptoms, however the latent effect might not be any less serious, instead increasing the susceptibility to subsequent triggered adverse responses (i.e. due to loss of compensatory capacity), particularly in people with existing cardiovascular disease.
Materials and methods
Animals
Ten to twelve-week old female C57BL/6 mice (body weight = 21.6 ± 0.1 g) were used in this study (Jackson Laboratory - Bar Harbor, ME). Mice were initially housed five per cage and maintained on a 12-hr light/dark cycle at approximately 22°C and 50% relative humidity in an AAALAC–approved facility. Food (Prolab RMH 3000; PMI Nutrition International, St. Louis, MO) and water were provided ad libitum. Each mouse implanted with a radiotelemeter was singly housed after surgery. All protocols were approved by the Institutional Animal Care and Use Committee of the U.S. Environmental Protection Agency and are in accordance with the National Institutes of Health Guides for the Care and Use of Laboratory Animals. The animals were treated humanely and with regard for alleviation of suffering.
Experimental groups
Mice were randomly assigned to one of six exposure groups: (1) fine concentrated ambient particles (FCAPs); (2) ultrafine CAPs (UFCAPs); (3) ozone (O3); (4) FCAPs and O3 co-exposure (FCAPs + O3); (5) UFCAPs and O3 co-exposure (UFCAPs + O3); and (6) filtered air (FA). Each group had n = 6. Separate groups (same as above) of mice were used for Langendorff cardiac perfusion experiments (n = 5-8).
Surgical implantation of radiotelemeters
Animals were weighed and then anesthetized using inhaled isoflurane (Isothesia, Butler Animal Health Supply, Dublin OH). Anesthesia was induced by spontaneous breathing of 2.5% isoflurane in pure oxygen at a flow rate of 1 L/min and then maintained by 1.5% isoflurane in pure oxygen at a flow rate of 0.5 L/min; all animals received the analgesic Buprenorphrine (0.03 mg/kg, i.p. manufacturer). Briefly, using aseptic technique, each animal was implanted subcutaneously with a radiotelemeter (ETA-F10, Data Sciences International, St Paul, MN); the transmitter was placed under the skin to the right of the midline on the dorsal side. The two electrode leads were then tunneled subcutaneously across the lateral dorsal sides; the distal portions were fixed in positions that approximated those of the lead II of a standard electrocardiogram (ECG). Body heat was maintained both during and immediately after the surgery. Animals were given food and water post-surgery and were housed individually. All animals were allowed 7-10 days to recover from the surgery and reestablish circadian rhythms.
Radiotelemetry data acquisition
Radiotelemetry methodology (Data Sciences International, Inc., St. Paul, MN) was used to track changes in cardiovascular function by monitoring heart rate (HR), and ECG waveforms immediately following telemeter implantation, through exposure until 24 hours post-exposure. This methodology provided continuous monitoring and collection of physiologic data from individual mice to a remote receiver. Sixty-second ECG segments were recorded every 5 minutes during the pre- and post-exposure periods and continuously during exposure (baseline and hours 1-4); HR was automatically obtained from the waveforms (Dataquest ART Software, version 3.01, Data Sciences International, St. Paul, MN, USA).
Electrocardiogram analysis
ECGAuto software (EMKA Technologies USA, Falls Church VA) was used to visualize individual ECG waveforms, analyze and quantify ECG segment durations and areas, as well as identify cardiac arrhythmias as previously described [63]. Briefly, using ECGAuto, P-wave, QRS complex, and T-wave were identified for individual ECG waveforms and compiled into a library. Analysis of all experimental ECG waveforms was then based on established libraries. The following parameters were determined for each ECG waveform: PR interval (Pstart-R), QRS complex duration (Qstart-S), ST segment interval (S-Tend) and QT interval (Qstart-Tend). QT interval was corrected for HR using the correction formula for mice QTc = QT/(RR/100)1/2 [64]. Figure 3A and B show a typical ECG trace as well as a typical non-conducted p-wave (NCPW) arrhythmia, which indicates an intermittent atrioventricular block, as observed in mice, respectively.
HRV analysis
Heart rate variability (HRV) was calculated as the mean of the differences between sequential RRs for the complete set of ECG waveforms using ECGAuto. For each 1-min stream of ECG waveforms, mean time between successive QRS complex peaks (RR interval), mean HR, and mean HRV-analysis–generated time-domain measures were acquired. The time-domain measures included standard deviation of the time between normal-to-normal beats (SDNN), and root mean squared of successive differences (RMSSD). HRV analysis was also conducted in the frequency domain using a fast-Fourier transform. The spectral power obtained from this transformation represents the total harmonic variability for the frequency range being analyzed. In this study, the spectrum was divided into low-frequency (LF) and high-frequency (HF) regions. The ratio of these two frequency domains (LF/HF) provides an estimate of the relative balance between sympathetic (LF) and vagal (HF) activity.
Concentrated ambient particle and ozone exposure
See Additional file 1 for full exposure details. Briefly, concentrated ambient particles (CAPs) and ozone (O3) were generated in the U.S. EPA’s Concentrated Air Particles Laboratory, Research Triangle Park, NC. All exposures were carried out in the summer months of June and July and under sunny and warm climate conditions. Ambient air containing PM from outside the facility entered the systems and passed through a size selective inlet removing PM > 2.5 μm so that remaining particles were in the size fractions of interest. The largest source of PM was from mobile sources (≈20%), wood combustion (≈21%), road dust (≈4%) and other minor sources such as brake wear and marine salt; the remaining PM was from secondary sulfates (≈50-55%).
Incoming air was then split into two streams and particles were selectively concentrated into either the fine (0.1 to 2.5 μm) or ultrafine mode (<300 nm) and then delivered into two separate chambers. Real time measurements of number concentration and particle size distribution were performed using a scanning mobility particle sizer (SMPS) and an Aerodynamic Particle Sizer (APS). A generator was used to produce O3 (0.3 ppm), which was then delivered to a third chamber. Chamber plumbing was altered to allow different configurations of concentrated PM and/or O3 including: FCAPs alone, UFCAPs alone, FCAPs + O3, UFCAPs + O3, O3 alone, or filtered air (FA). Exposure to FCAPs/UFCAPs alone had to be done on separate days from FCAPs/UFCAPs co-exposures with O3 due to limitations in the exposure system (i.e. exposure to CAPs alone and CAPs + O3 could not be done on the same day); day-to-day variations in particle concentrations and composition were expected due to this.
The study protocol included two days of animal-to-chamber acclimatization prior to exposure. A normal four-hour exposure (Exp1 (exposure hour 1), Exp2, Exp3, and Exp4) started with one hour of additional chamber acclimatization (Baseline). All mice were moved back to their home-cages after the exposure (Recovery). The Multiple Pathway Particle Dosimetry (MPPD; Version 3.0) model was used to predict particle doses [65,66] for mice and humans; ventilatory parameters were estimated using typical values [67].
Cardiac perfusion
The procedure for cardiac perfusion has been previously described [21]. Briefly, 24 hours after exposure, mice were anesthetized with sodium pentobarbital (80 mg/kg, i.p.). Heparin (100 units) was injected intravenously before removal of heart. The hearts were rapidly removed and placed in ice-cold Krebs-Henseleit buffer, after which the aortas were cannulated. Retrograde perfusion via the aorta was performed under constant pressure (100 cmH2O) above the heart. The non-recirculating perfusate was a Krebs-Henseleit buffer containing (in mmol/L) 120 NaCl, 5.9 KCl, 1.2 MgSO4, 1.75 CaCl2, 25 NaHCO3, and 11 glucose. The buffer was aerated with 95% O2—5% CO2 and maintained at pH 7.4 and a temperature of 37°C.
For assessment of contractile function, a latex balloon on the tip of a polyethylene catheter was inserted through the left atrium into the left ventricle. The catheter was connected to a pressure transducer (Argon Medical Devices, Athens, TX) at the same height as the heart. The pressure of the left ventricular balloon was inflated to 0-5 cmH2O. A PowerLab system was used to collect and process the heart rate, left ventricular developed pressure (LVDP = LV peak minus end-diastolic pressure (LVEDP)), and contractility (dP/dt) data (AD Instruments, Milford, MA). All hearts were perfused for 25 min; we then initiated 20 min of global no-flow ischemia by stopping the flow of oxygenated perfusion buffer, followed by 1 h of reperfusion. Onset of ischemic contracture was measured as the time from the start of ischemia until initial contracture (at least 5 cmH2O increase in left ventricular pressure). Recovery of LVDP, expressed as a percentage of the initial pre-ischemic LVDP, was measured at 20, 40 and 60 min of reperfusion after 20 min of ischemia.
Tissue collection and analysis
See Additional file 1 for full details, procedures were performed as previously described [32]. Briefly, 24 hrs after exposure, mice were euthanized and blood and lung lavage fluid (BAL) were collected, processed and analyzed. Multiple biochemical markers (e.g. lactate dehydrogenase, protein, etc) were assessed in the BAL, and serum or plasma supernatants were analyzed for creatine kinase, C-reactive protein (CRP), and other markers to assess cardiopulmonary inflammation, injury and oxidative stress.
Statistics
All data are expressed as means ± SEM. Statistical analyses of the data were performed with GraphPad Prism 5 (GraphPad software, San Diego CA). For HR, ECG intervals and HRV, two-way analysis of variance (ANOVA) for repeated-measures and Bonferroni post hoc tests were used to determine statistical differences. A one-way ANOVA was used to analyze arrhythmia counts. For Langendorff cardiac perfusion data, comparisons between groups were performed by one-way ANOVA followed by Bonferroni post hoc test for multiple comparisons. Comparisons were made across all groups taking into account the multiple endpoints, exposure groups and time points as well as any interactions. An oblique principal component cluster analysis and multivariate analysis of variance (MANOVA – GLM procedure and least squares means post hoc test) were performed using SAS version 9.3 software, (SAS Institute Inc, Cary, NC) to determine whether the elements found in the CAPs on their own or in combination with one another had an effect on the cardiac responses. The objective of this approach was to reduce the large number of variables (i.e. elements) to a smaller set that still retain the information in the original data set and then examine for effects. Five clusters were revealed and elements belonging to the same cluster had strong correlations. A p-value < 0.05 was considered statistically significant.
Additional file
Supplementary Material.
Acknowledgments
We would like to thank Dr. Eugene Flournoy-Gibbs, Dr. Ilona Jaspers, Dr. Stephen Gavett, Dr. M. Ian Gilmour, Dr. Daniel L. Costa and Dr. Wayne Cascio for reviewing the manuscript, Dr. James Brown for his assistance with the particle dosing estimates, Mrs. Najwa Haykal-Coates for her assistance with the statistical analysis and Judy Richards and Elizabeth Boykins for the chemical analysis.
Funding
EPA-UNC Toxicology Training Agreement (CR 83515201) for NK and CEMALB cooperative agreement with EPA (CR 83346301 - Health Effects of Environmental Pollutants in Humans) for RMK.
Disclaimer
This paper has been reviewed and approved for release by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency. Approval does not signify that the contents necessarily reflect the views and policies of the U.S. EPA, nor does mention of trade names.
Abbreviations
- ACE
Angiotensin converting enzyme
- BAL
Bronchoalveolar lavage
- CRP
c-reactive protein
- BP
Blood pressure
- BRS
Baroreflex sensitivity
- DEP
Diesel exhaust particles
- dP/dt
1st derivative of the change in left ventricular pressure/time
- ECG
Electrocardiogram
- EC
Elemental carbon
- ET-1
Endothelin-1
- FA
Filtered air
- FCAPs
Fine concentrated ambient particles
- FMD
Flow-mediated dilation
- GTR
Glutathione S-transferase
- HF
High frequency
- HR
Heart rate
- HRV
Heart rate variability
- I/R
Ischemia/reperfusion
- LVDP
Left ventricular developed pressure
- MAP
Mean arterial pressure
- MDA
Malondialdehyde
- NAG
N-acetyl-b-d-glucosaminidase
- NCPW
Non-conducted p-wave
- OC
Organic carbon
- O3
Ozone
- PM
Particulate matter
- RMSSD
Root mean square of successive differences of NNs
- SDNN
Standard deviation of NN intervals
- SOD
Superoxide dismutase
- Tco
Core body temperature
- TC
Total carbon
- TRPA1
Transient receptor potential cation channel, member A1
- UFCAPs
Ultrafine concentrated ambient particles
Footnotes
Nicole Kurhanewicz and Rachel McIntosh-Kastrinsky contributed equally to this work.
Competing interests
The authors declare that they have competing interests.
Authors’ contribution
NK and RMK carried out all the radiotelemetry and Langendorff experimentations and data analysis. HT participated in the Langendorff experiments. LW carried out all the exposures and monitoring. AKF and MSH participated in the study design and statistical analysis. All authors read and approved the final manuscript.
Contributor Information
Nicole Kurhanewicz, nak@email.unc.edu.
Rachel McIntosh-Kastrinsky, ramcinto@email.unc.edu.
Haiyan Tong, Email: tong.haiyan@epa.gov.
Leon Walsh, Email: walsh.leon@epa.gov.
Aimen K Farraj, Email: farraj.aimen@epa.gov.
Mehdi S Hazari, Email: hazari.mehdi@epa.gov.
References
- 1.Brook RD, Rajagopalan S, Pope CA, 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC, Jr, Whitsel L, Kaufman JD, American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 2.Wang G, Jiang R, Zhao Z, Song W. Effects of ozone and fine particulate matter (PM(2.5)) on rat system inflammation and cardiac function. Toxicol Lett. 2013;217:23–33. doi: 10.1016/j.toxlet.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F. Inhalation of fine particulate air pollution and ozone causes acute arterial vasoconstriction in healthy adults. Circulation. 2002;105:1534–1536. doi: 10.1161/01.CIR.0000013838.94747.64. [DOI] [PubMed] [Google Scholar]
- 4.Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, Brook RD. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113:1052–1055. doi: 10.1289/ehp.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner JG, Allen K, Yang HY, Nan B, Morishita M, Mukherjee B, Dvonch JT, Spino C, Fink GD, Rajagopalan S, Sun Q, Brook RD, Harkema JR. Cardiovascular depression in rats exposed to inhaled particulate matter and ozone: effects of diet-induced metabolic syndrome. Environ Health Perspect. 2014;122:27–33. doi: 10.1289/ehp.122-A27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madden MC, Richards JH, Dailey LA, Hatch GE, Ghio AJ. Effect of ozone on diesel exhaust particle toxicity in rat lung. Toxicol Appl Pharmacol. 2000;168:140–148. doi: 10.1006/taap.2000.9024. [DOI] [PubMed] [Google Scholar]
- 7.Farraj AK, Boykin E, Ledbetter A, Andrews D, Gavett SH. Increased lung resistance after diesel particulate and ozone co-exposure not associated with enhanced lung inflammation in allergic mice. Inhal Toxicol. 2010;22:33–41. doi: 10.3109/08958370902862434. [DOI] [PubMed] [Google Scholar]
- 8.Dockery DW, Pope CA, 3rd, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Jr, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 9.Kodavanti UP, Thomas R, Ledbetter AD, Schladweiler MC, Shannahan JH, Wallenborn JG, Lund AK, Campen MJ, Butler EO, Gottipolu RR, Nyska A, Richards JE, Andrews D, Jaskot RH, McKee J, Kotha SR, Patel RB, Parinandi NL. Vascular and cardiac impairments in rats inhaling ozone and diesel exhaust particles. Environ Health Perspect. 2011;119:312–318. doi: 10.1289/ehp.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters A. Particulate matter and heart disease: evidence from epidemiological studies. Toxicol Appl Pharmacol. 2005;207:477–482. doi: 10.1016/j.taap.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 11.Hazari MS, Callaway J, Winsett DW, Lamb C, Haykal-Coates N, Krantz QT, King C, Costa DL, Farraj AK. Dobutamine "stress" test and latent cardiac susceptibility to inhaled diesel exhaust in normal and hypertensive rats. Environ Health Perspect. 2012;120:1088–1093. doi: 10.1289/ehp.1104684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills NL, Tornqvist H, Gonzalez MC, Vink E, Robinson SD, Soderberg S, Boon NA, Donaldson K, Sandstrom T, Blomberg A, Newby DE. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med. 2007;357:1075–1082. doi: 10.1056/NEJMoa066314. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji H, Venditti FJ, Jr, Manders ES, Evans JC, Larson MG, Feldman CL, Levy D. Reduced heart rate variability and mortality risk in an elderly cohort. The Framingham Heart Study. Circulation. 1994;90:878–883. doi: 10.1161/01.CIR.90.2.878. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji H, Larson MG, Venditti FJ, Jr, Manders ES, Evans JC, Feldman CL, Levy D. Impact of reduced heart rate variability on risk for cardiac events. The Framingham Heart Study. Circulation. 1996;94:2850–2855. doi: 10.1161/01.CIR.94.11.2850. [DOI] [PubMed] [Google Scholar]
- 15.Gold DR, Litonjua A, Schwartz J, Lovett E, Larson A, Nearing B, Allen G, Verrier M, Cherry R, Verrier R. Ambient pollution and heart rate variability. Circulation. 2000;101:1267–1273. doi: 10.1161/01.CIR.101.11.1267. [DOI] [PubMed] [Google Scholar]
- 16.Gong H, Jr, Linn WS, Terrell SL, Clark KW, Geller MD, Anderson KR, Cascio WE, Sioutas C. Altered heart-rate variability in asthmatic and healthy volunteers exposed to concentrated ambient coarse particles. Inhal Toxicol. 2004;16:335–343. doi: 10.1080/08958370490439470. [DOI] [PubMed] [Google Scholar]
- 17.Zareba W, Couderc JP, Oberdorster G, Chalupa D, Cox C, Huang LS, Peters A, Utell MJ, Frampton MW. ECG parameters and exposure to carbon ultrafine particles in young healthy subjects. Inhal Toxicol. 2009;21:223–233. doi: 10.1080/08958370802492407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen LC, Hwang JS. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. IV. Characterization of acute and chronic effects of ambient air fine particulate matter exposures on heart-rate variability. Inhal Toxicol. 2005;17:209–216. doi: 10.1080/08958370590912789. [DOI] [PubMed] [Google Scholar]
- 19.Gurgueira SA, Lawrence J, Coull B, Murthy GG, Gonzalez-Flecha B. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect. 2002;110:749–755. doi: 10.1289/ehp.02110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang H, Kloner RA, Kleinman MT, Simkhovich BZ. Direct and acute cardiotoxic effects of ultrafine air pollutants in spontaneously hypertensive rats and Wistar–Kyoto rats. J Cardiovasc Pharmacol Ther. 2008;13:189–198. doi: 10.1177/1074248408321569. [DOI] [PubMed] [Google Scholar]
- 21.Tong H, Cheng WY, Samet JM, Gilmour MI, Devlin RB. Differential cardiopulmonary effects of size-fractionated ambient particulate matter in mice. Cardiovasc Toxicol. 2010;10:259–267. doi: 10.1007/s12012-010-9082-y. [DOI] [PubMed] [Google Scholar]
- 22.Amatullah H, North ML, Akhtar US, Rastogi N, Urch B, Silverman FS, Chow CW, Evans GJ, Scott JA. Comparative cardiopulmonary effects of size-fractionated airborne particulate matter. Inhal Toxicol. 2012;24:161–171. doi: 10.3109/08958378.2011.650235. [DOI] [PubMed] [Google Scholar]
- 23.Takenaka S, Karg E, Roth C, Schulz H, Ziesenis A, Heinzmann U, Schramel P, Heyder J. Pulmonary and systemic distribution of inhaled ultrafine silver particles in rats. Environ Health Perspect. 2001;109(Suppl 4):547–551. doi: 10.1289/ehp.01109s4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furuyama A, Kanno S, Kobayashi T, Hirano S. Extrapulmonary translocation of intratracheally instilled fine and ultrafine particles via direct and alveolar macrophage-associated routes. Arch Toxicol. 2009;83:429–437. doi: 10.1007/s00204-008-0371-1. [DOI] [PubMed] [Google Scholar]
- 25.Samet JM, Rappold A, Graff D, Cascio WE, Berntsen JH, Huang YC, Herbst M, Bassett M, Montilla T, Hazucha MJ, Bromberg PA, Devlin RB. Concentrated ambient ultrafine particle exposure induces cardiac changes in young healthy volunteers. Am J Respir Crit Care Med. 2009;179:1034–1042. doi: 10.1164/rccm.200807-1043OC. [DOI] [PubMed] [Google Scholar]
- 26.Bagate K, Meiring JJ, Gerlofs-Nijland ME, Cassee FR, Wiegand H, Osornio-Vargas A, Borm PJ. Ambient particulate matter affects cardiac recovery in a Langendorff ischemia model. Inhal Toxicol. 2006;18:633–643. doi: 10.1080/08958370600742706. [DOI] [PubMed] [Google Scholar]
- 27.Simkhovich BZ, Marjoram P, Kleinman MT, Kloner RA. Direct and acute cardiotoxicity of ultrafine particles in young adult and old rat hearts. Basic Res Cardiol. 2007;102:467–475. doi: 10.1007/s00395-007-0681-0. [DOI] [PubMed] [Google Scholar]
- 28.Costa DL, Lehmann JR, Winsett D, Richards J, Ledbetter AD, Dreher KL. Comparative pulmonary toxicological assessment of oil combustion particles following inhalation or instillation exposure. Toxicol Sci: Off J Soc Toxicol. 2006;91:237–246. doi: 10.1093/toxsci/kfj123. [DOI] [PubMed] [Google Scholar]
- 29.Osier M, Oberdorster G. Intratracheal inhalation vs intratracheal instillation: differences in particle effects. Fund Appl Toxicol: Off J Soc Toxicol. 1997;40:220–227. doi: 10.1006/faat.1997.2390. [DOI] [PubMed] [Google Scholar]
- 30.Nadziejko C, Fang K, Nadziejko E, Narciso SP, Zhong M, Chen LC. Immediate effects of particulate air pollutants on heart rate and respiratory rate in hypertensive rats. Cardiovasc Toxicol. 2002;2:245–252. doi: 10.1385/CT:2:4:245. [DOI] [PubMed] [Google Scholar]
- 31.Johnson RL., Jr Relative effects of air pollution on lungs and heart. Circulation. 2004;109:5–7. doi: 10.1161/01.CIR.0000110643.19575.79. [DOI] [PubMed] [Google Scholar]
- 32.Farraj AK, Haykal-Coates N, Winsett DW, Hazari MS, Carll AP, Rowan WH, Ledbetter AD, Cascio WE, Costa DL. Increased non-conducted P-wave arrhythmias after a single oil fly ash inhalation exposure in hypertensive rats. Environ Health Perspect. 2009;117:709–715. doi: 10.1289/ehp.0800129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campen MJ, McDonald JD, Reed MD, Seagrave J. Fresh gasoline emissions, not paved road dust, alter cardiac repolarization in ApoE-/- mice. Cardiovasc Toxicol. 2006;6:199–210. doi: 10.1385/CT:6:3:199. [DOI] [PubMed] [Google Scholar]
- 34.Sivagangabalan G, Spears D, Masse S, Urch B, Brook RD, Silverman F, Gold DR, Lukic KZ, Speck M, Kusha M, Farid T, Poku K, Shi E, Floras J, Nanthakumar K. The effect of air pollution on spatial dispersion of myocardial repolarization in healthy human volunteers. J Am Coll Cardiol. 2011;57:198–206. doi: 10.1016/j.jacc.2010.08.625. [DOI] [PubMed] [Google Scholar]
- 35.Fakhri AA, Ilic LM, Wellenius GA, Urch B, Silverman F, Gold DR, Mittleman MA. Autonomic effects of controlled fine particulate exposure in young healthy adults: effect modification by ozone. Environ Health Perspect. 2009;117:1287–1292. doi: 10.1289/ehp.0900541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelter MM, Adams MG. Non-conducted P waves. Am J Crit Care: Off Pub Am Assoc Crit-Care Nurses. 2003;12:567–568. [PubMed] [Google Scholar]
- 37.Baja ES, Schwartz JD, Wellenius GA, Coull BA, Zanobetti A, Vokonas PS, Suh HH. Traffic-related air pollution and QT interval: modification by diabetes, obesity, and oxidative stress gene polymorphisms in the normative aging study. Environ Health Perspect. 2010;118:840–846. doi: 10.1289/ehp.0901396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong H, Rappold AG, Diaz-Sanchez D, Steck SE, Berntsen J, Cascio WE, Devlin RB, Samet JM. Omega-3 fatty acid supplementation appears to attenuate particulate air pollution-induced cardiac effects and lipid changes in healthy middle-aged adults. Environ Health Perspect. 2012;120:952–957. doi: 10.1289/ehp.1104472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milan DJ, MacRae CA. Animal models for arrhythmias. Cardiovasc Res. 2005;67:426–437. doi: 10.1016/j.cardiores.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Brook RD, Urch B, Dvonch JT, Bard RL, Speck M, Keeler G, Morishita M, Marsik FJ, Kamal AS, Kaciroti N, Harkema J, Corey P, Silverman F, Gold DR, Wellenius G, Mittleman MA, Rajagopalan S, Brook JR. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54:659–667. doi: 10.1161/HYPERTENSIONAHA.109.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitsel EA, Quibrera PM, Christ SL, Liao D, Prineas RJ, Anderson GL, Heiss G. Heart rate variability, ambient particulate matter air pollution, and glucose homeostasis: the environmental epidemiology of arrhythmogenesis in the women's health initiative. Am J Epidemiol. 2009;169:693–703. doi: 10.1093/aje/kwn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farraj AK, Hazari MS, Winsett DW, Kulukulualani A, Carll AP, Haykal-Coates N, Lamb CM, Lappi E, Terrell D, Cascio WE, Costa DL. Overt and latent cardiac effects of ozone inhalation in rats: evidence for autonomic modulation and increased myocardial vulnerability. Environ Health Perspect. 2012;120:348–354. doi: 10.1289/ehp.1104244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McIntosh-Kastrinsky R, Diaz-Sanchez D, Sexton KG, Jania CM, Zavala J, Tilley SL, Jaspers I, Gilmour MI, Devlin RB, Cascio WE, Tong H. Photochemically altered air pollution mixtures and contractile parameters in isolated murine hearts before and after ischemia. Environ Health Perspect. 2013;121:1344–1348. doi: 10.1289/ehp.1306609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tankersley CG, Peng RD, Bedga D, Gabrielson K, Champion HC. Variation in echocardiographic and cardiac hemodynamic effects of PM and ozone inhalation exposure in strains related to Nppa and Npr1 gene knock-out mice. Inhal Toxicol. 2010;22:695–707. doi: 10.3109/08958378.2010.487549. [DOI] [PubMed] [Google Scholar]
- 45.Mills NL, Finlayson AE, Gonzalez MC, Tornqvist H, Barath S, Vink E, Goudie C, Langrish JP, Soderberg S, Boon NA, Fox KA, Donaldson K, Sandström T, Blomberg A, Newby DE. Diesel exhaust inhalation does not affect heart rhythm or heart rate variability. Heart. 2011;97:544–550. doi: 10.1136/hrt.2010.199042. [DOI] [PubMed] [Google Scholar]
- 46.Peretz A, Kaufman JD, Trenga CA, Allen J, Carlsten C, Aulet MR, Adar SD, Sullivan JH. Effects of diesel exhaust inhalation on heart rate variability in human volunteers. Environ Res. 2008;107:178–184. doi: 10.1016/j.envres.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devlin RB, Ghio AJ, Kehrl H, Sanders G, Cascio W. Elderly humans exposed to concentrated air pollution particles have decreased heart rate variability. Eur Respir J Suppl. 2003;40:76s–80s. doi: 10.1183/09031936.03.00402403. [DOI] [PubMed] [Google Scholar]
- 48.Pope CA, 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- 49.Schneider A, Hampel R, Ibald-Mulli A, Zareba W, Schmidt G, Schneider R, Ruckerl R, Couderc JP, Mykins B, Oberdorster G, Wölke G, Pitz M, Wichmann HE, Peters A. Changes in deceleration capacity of heart rate and heart rate variability induced by ambient air pollution in individuals with coronary artery disease. Part Fibre Toxicol. 2010;7:29. doi: 10.1186/1743-8977-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costa SK, Kumagai Y, Brain SD, Teixeira SA, Varriano AA, Barreto MA, de Lima WT, Antunes E, Muscara MN, Costa SK. Involvement of sensory nerves and TRPV1 receptors in the rat airway inflammatory response to two environment pollutants: diesel exhaust particles (DEP) and 1,2-naphthoquinone (1,2-NQ) Arch Toxicol. 2010;84:109–117. doi: 10.1007/s00204-009-0427-x. [DOI] [PubMed] [Google Scholar]
- 51.Cozzi E, Hazarika S, Stallings HW, 3rd, Cascio WE, Devlin RB, Lust RM, Wingard CJ, Van Scott MR. Ultrafine particulate matter exposure augments ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol. 2006;291:H894–903. doi: 10.1152/ajpheart.01362.2005. [DOI] [PubMed] [Google Scholar]
- 52.Riley MR, Boesewetter DE, Kim AM, Sirvent FP. Effects of metals Cu, Fe, Ni, V, and Zn on rat lung epithelial cells. Toxicology. 2003;190:171–184. doi: 10.1016/S0300-483X(03)00162-8. [DOI] [PubMed] [Google Scholar]
- 53.Chuang KJ, Chan CC, Su TC, Lin LY, Lee CT. Associations between particulate sulfate and organic carbon exposures and heart rate variability in patients with or at risk for cardiovascular diseases. J Occup Environ Med / Am College of Occup Environ Med. 2007;49:610–617. doi: 10.1097/JOM.0b013e318058205b. [DOI] [PubMed] [Google Scholar]
- 54.Kozumbo WJ, Agarwal S. Induction of DNA damage in cultured human lung cells by tobacco smoke arylamines exposed to ambient levels of ozone. Am J Respir Cell Mol Biol. 1990;3:611–618. doi: 10.1165/ajrcmb/3.6.611. [DOI] [PubMed] [Google Scholar]
- 55.Pitts JN, Jr, Lokensgard DM, Ripley PS, KA VANC LVANV, Shaffer SD, Thill AJ, Belser WL., Jr "Atmospheric" Epoxidation of Benzo[a]pyrene by Ozone: formation of the Metabolite Benzo[a]pyrene-4,5-Oxide. Sci (New York, NY) 1980;210:1347–1349. doi: 10.1126/science.210.4476.1347. [DOI] [PubMed] [Google Scholar]
- 56.Madden MC, Friedman M, Hanley N, Siegler E, Quay J, Becker S, Devlin R, Koren HS. Chemical nature and immunotoxicological properties of arachidonic acid degradation products formed by exposure to ozone. Environ Health Perspect. 1993;101:154–164. doi: 10.1289/ehp.93101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elder AC, Gelein R, Finkelstein JN, Cox C, Oberdorster G. Pulmonary inflammatory response to inhaled ultrafine particles is modified by age, ozone exposure, and bacterial toxin. Inhal Toxicol. 2000;12(Suppl 4):227–246. doi: 10.1080/089583700750019585. [DOI] [PubMed] [Google Scholar]
- 58.Adamson IY, Prieditis H, Vincent R. Pulmonary toxicity of an atmospheric particulate sample is due to the soluble fraction. Toxicol Appl Pharmacol. 1999;157:43–50. doi: 10.1006/taap.1999.8658. [DOI] [PubMed] [Google Scholar]
- 59.Thomson E, Kumarathasan P, Goegan P, Aubin RA, Vincent R. Differential regulation of the lung endothelin system by urban particulate matter and ozone. Toxicol Sci: Off J Soc Toxicol. 2005;88:103–113. doi: 10.1093/toxsci/kfi272. [DOI] [PubMed] [Google Scholar]
- 60.Watkinson WP, Campen MJ, Nolan JP, Costa DL. Cardiovascular and systemic responses to inhaled pollutants in rodents: effects of ozone and particulate matter. Environ Health Perspect. 2001;109(Suppl 4):539–546. doi: 10.1289/ehp.01109s4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valavanidis A, Loridas S, Vlahogianni T, Fiotakis K. Influence of ozone on traffic-related particulate matter on the generation of hydroxyl radicals through a heterogeneous synergistic effect. J Hazard Mater. 2009;162:886–892. doi: 10.1016/j.jhazmat.2008.05.124. [DOI] [PubMed] [Google Scholar]
- 62.Park JH, Troxel AB, Harvey RG, Penning TM. Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem Res Toxicol. 2006;19:719–728. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hazari MS, Haykal-Coates N, Winsett DW, Costa DL, Farraj AK. A single exposure to particulate or gaseous air pollution increases the risk of aconitine-induced cardiac arrhythmia in hypertensive rats. Toxicol Sci: Off J Soc Toxicol. 2009;112:532–542. doi: 10.1093/toxsci/kfp214. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- 65.Anjilvel S, Asgharian B. A multiple-path model of particle deposition in the rat lung. Fund Appl Nematol: Off J Soc Toxicol. 1995;28:41–50. doi: 10.1006/faat.1995.1144. [DOI] [PubMed] [Google Scholar]
- 66.Cassee FR, Muijser H, Duistermaat E, Freijer JJ, Geerse KB, Marijnissen JC, Arts JH. Particle size-dependent total mass deposition in lungs determines inhalation toxicity of cadmium chloride aerosols in rats. Application of a multiple path dosimetry model. Arch Toxicol. 2002;76:277–286. doi: 10.1007/s00204-002-0344-8. [DOI] [PubMed] [Google Scholar]
- 67.Mendez LB, Gookin G, Phalen RF. Inhaled aerosol particle dosimetry in mice: a review. Inhal Toxicol. 2010;22(Suppl 2):15–20. doi: 10.3109/08958378.2010.541337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material.