

Abstract
The polymerase (gp43) processivity during T4 replisome mediated DNA replication has been investigated. The size of the Okazaki fragments remains constant over a wide range of polymerase concentrations. A dissociation rate constant of ≈0.0013 sec-1 was measured for the polymerases from both strands, consistent with highly processive replication on both the leading and lagging strands. This processive replication, however, can be disrupted by a catalytically inactive mutant D408N gp43 that retains normal affinity for DNA and the clamp. The inhibition kinetics fit well to an active exchange model in which the mutant polymerase (the polymerase trap) displaces the replicating polymerase. This kinetic model was further strengthened by the observation that the sizes of both the Okazaki fragments and the extension products on a primed M13mp18 template were reduced in the presence of the mutant polymerase. The effects of the trap polymerase therefore suggest a dynamic processivity of the polymerase during replication, namely, a solution/replisome polymerase exchange takes place without affecting continued DNA synthesis. This process mimics the polymerase switching recently suggested during the translesion DNA synthesis, implies the multiple functions of the clamp in replication, and may play a potential role in overcoming the replication barriers by the T4 replisome.
Keywords: DNA replication, polymerase processivity, polymerase exchange
Bacteriophage T4 DNA polymerase is responsible for DNA synthesis on both leading and lagging strands. This enzyme is the gene 43 product (gp43), which, along with seven other T4 replication proteins, constitutes the T4 replisome that carries out coordinated DNA synthesis (1). Among these seven proteins, the clamp loader (gp44/62) and the clamp protein (gp45) are polymerase accessory factors that significantly increase the processivity of gp43 during replication by forming the holoenzyme complex (2). The current working model of T4 DNA replication involves two such holoenzyme complexes acting on leading and lagging strands (3). Moving ahead of the holoenzyme complexes is the T4 primosome generated from the helicase (gp41), the primase (gp61), and the helicase accessory protein (gp59). This unit is required to rapidly unwind the double-stranded DNA in front of the moving fork (4, 5) and to synthesize the pentaribonucleotide primers for Okazaki fragment synthesis (6). The last replisome component is the single-stranded DNA (ssDNA) binding protein (gp32) which is involved in the stabilization of the ssDNA loop structure generated during lagging strand synthesis (7) and in the organization of the replisome (8, 9).
The entire 172-kb T4 genome is fully duplicated within ≈15 min (10). As a result, the high processivity of the polymerase is crucial for efficient DNA replication. The holoenzyme stability has been measured on a short, defined DNA fork to give a half-life of ≈6 min (11). This value is within the same range as the 11-min half-life for the T4 helicase on a moving fork determined by Alberts and coworkers (12). Dissociation halflives of this magnitude suggest that both the helicase and the polymerase within the replisome could potentially finish replicating the entire genome before dissociation. However, several points are worthy of emphasis. First, the replisome cannot be regarded as a rigid body during replication, a process during which every component stays in position. The T4 replisome is subject to continuous remodeling during lagging strand synthesis; namely, the clamp, the clamp loader, and the primase repetitively dissociate from the replisome to be recruited from solution during each round of Okazaki fragment synthesis (13). Second, during in vivo replication, the replisome may potentially interact with protein components from other pathways, such as those involved in transcription (14) and lesion bypass processes (15). In all these cases, changes in the replisome may occur to modulate those interactions. An intriguing question is how the replisome achieves the remodeling flexibility required under these circumstances while maintaining high processivity during normal replication.
In this paper, we report on the processivity of the gp43 polymerase within the replisome during active replication. Our results confirm that gp43 possesses high processivity on both strands during replication with a dissociation half-life of ≈9 min, consistent with the previously reported data (11). Consequently with considerable interest is our discovery that the replicating polymerase can be replaced rapidly (<1 min) by an inactive mutant gp43 (D408N), implying that an active polymerase exchange process takes place during normal replication. Taken together, these results define a “dynamic processivity” of the polymerase. The significance of this finding and its implications for processes such as the polymerase switching during translesion synthesis is also discussed.
Materials and Methods
[α-32P]dGTP, [α-32P]dCTP, and [γ-32P]ATP were purchased from New England Nuclear. [8-3H]dGTP was purchased from ICN. Unlabeled deoxynucleotides and ribonucleotides were purchased from Roche Molecular Biochemicals. Bacteriophage T4 proteins were purified by using established protocols (9). All other chemicals were of analytical grade or better.
The DNA Substrate. The minicircle substrate was designed and synthesized as described in ref. 9. During replication, dGTP is only incorporated into the leading strand, and dCTP into the lagging strand, on this substrate. The tailed replicative form II (TRFII) DNA substrate was also prepared as described (13) with a few modifications. The Stoffel fragment (25 units) of Ampli-Taq DNA polymerase (Applied Biosystems) was used in the roll-around reaction. The reaction was allowed to proceed at 72°C for 40 min, and the product was purified with QIAquick PCR purification kit with one extra washing step with a 17% guanidine hydrochloride aqueous solution. The TRFII DNA obtained was analyzed by gel electrophoresis to be a single band of 7.2 kb and was essentially free of M13 ssDNA (data not shown).
Gp43 Mutagenesis and Characterization. The cloning and characterization of D408N gp43 and the C-terminal deletion (gp43Δ10) mutants are discussed in the supporting information, which is published on the PNAS web site.
The Standard Replication Reactions. Replication reactions were carried out in a complex buffer containing 25 mM Tris-acetate (pH 7.5), 125 mM KOAc, and 10 mM Mg(OAc)2. The standard replication conditions in all minicircle reactions and the assay methods were essentially as described in ref. 9. Reactions carried out on the TRFII substrate consisted of 3 nM TRFII; 16 nM gp43; 100 nM gp45 (as trimer); 18 nM gp44/62, 200 nM each monomers gp41, gp61, and gp59; 4 μM gp32; 50 μM each CTP, GTP, and UTP; 1 mM ATP; 50 μM each dATP, dGTP, dCTP, and dTTP; and [α-32P]dGTP [3,000 Ci/mmol (1 Ci = 37 GBq)], in a typical reaction volume of 50 μl. The reaction procedures and the order of addition were the same as described in ref. 13. In reactions involving the trap polymerase, replication was initiated in the presence of the DNA template (minicircle or TRFII) and eight T4 proteins and allowed to proceed for 1 min (minicircle) or 7 min (TRFII) before the addition of various amounts of the trap polymerase along with radioactive dNTP. Reaction aliquots were withdrawn at various time points and analyzed by the filter binding assay or alkaline agarose gel electrophoresis (9, 13).
The Polymerase Dilution Experiment. Standard replication reactions were allowed to proceed for 1 min (minicircle) or 2.5 min (TRFII) before being diluted by 10- or 20-fold (minicircle) or 5-fold (TRFII) into a dilution mixture in which the radioactive dNTP was included and either DNA or DNA/gp43 were omitted. Aliquots of the reaction mixture were sampled at various time intervals and quenched with an equal volume of 0.5 M EDTA, pH 8.0. The DNA products were analyzed either through 0.8% alkaline agarose gel electrophoresis or through a filter binding assay as described (9, 13).
Primer Extension on the Primed M13 ssDNA. A 46-mer DNA primer (5′-TCT GAC CTG AAA GCG TAA GAA TAC GTG GCA CAG ACA ATA TTT TTG A-3′) was 5′ end-labeled with [γ-32P]ATP by the standard T4 polynucleotide kinase reaction and annealed to M13mp18 ssDNA (position 5,017 to position 5,062). The concentrations listed below represent final reaction concentrations. All reactions were carried out at room temperature by mixing 1.6 nM primed M13 ssDNA, 4.5 μM gp32, 0.5 mM ATP, 25 μM dATP, 40 nM gp43, 250 nM gp45 (as trimer), and 250 nM gp44/62. After 20 sec to allow the complete assembly of the holoenzyme, 0, 160, or 320 nM trap polymerases (or WT gp43 in the control reaction) were added along with 50 μM dNTPs to initiate the reaction. The replication reaction was allowed to proceed for various time lengths (3, 7, 10, and 20 sec), after which it was stopped by rapid addition of quench solution (500 mM EDTA, pH 8.0). Extension products were then analyzed by alkaline agarose gel electrophoresis.
Results
Effect of Trap gp43 on the Rates of Leading and Lagging Strand Synthesis. D408N gp43 was used as a protein trap to test for its inhibition of both leading and lagging strand syntheses. The conserved Asp-408 within the palm region is crucial for dNTP incorporation. Its mutation results in the loss of the nucleotidyl transfer activity. The mutant protein retains the normal binding capabilities of gp43exo- (D219A gp43, henceforth referred to as WT gp43). We observed similar binding affinities of WT and mutant gp43 toward the clamp protein through an intermolecular fluorescence resonance energy transfer experiment (Kd values of 145 ± 15 nM and 148 ± 11 nM for WT gp43 and gp43 trap, respectively; see supporting information). The gp44/62 ATPase assay (16) further demonstrated the ability of this mutant protein to form the holoenzyme with the clamp and the clamp loader on a fork DNA substrate (see supporting information).
If the polymerase dissociates from the replisome during replication, the trap polymerase then could compete with the active enzyme for the re-binding of the replisome and thus inhibit the strand synthesis. In the case where the polymerase is highly processive, such inhibition by trap gp43 should not be observed. The results of adding various levels of trap gp43 to an active replisome carrying out DNA synthesis on the minicircle substrate are shown in Fig. 1A. Synthesis of both strands was inhibited by addition of the trap protein. Several other observations accompany these results. Firstly, the inhibition by the trap polymerase is rapid, generally within 1 min after the addition of the trap and much faster than the holoenzyme off rate (koff = 0.002 sec-1) measured previously on a small fork substrate (11). Secondly, the inhibitory effect was trapconcentration dependent. Thirdly, the leading and lagging strands were inhibited to the same degree at all trap gp43 concentration levels, suggesting that the trap polymerase acts on both strands by the same mechanism. Finally, the trap also inhibited leading strand synthesis even in the absence of lagging strand synthesis by omitting rNTPs and the primase (data not shown), indicating that the trap acts on both strands independently. As will be shown below, the trap also inhibits leading and lagging strand syntheses on the larger TRFII substrate. Therefore the results with the minicircle substrate are not due to defects in replisome structure because of the latter's smaller size.
Fig. 1.

(A) Effect of the trap polymerase on leading and lagging strand synthesis on the minicircle substrate. Standard replication reactions were carried out with [8-3H]dGTP and [α-32P]dCTP to monitor the rate of leading and lagging strand synthesis, respectively. At 0, 180, 360, and 1,080 nM D408N trap gp43 concentrations, the rates of leading strand synthesis (○) were 32.5, 24.2, 14.5, and 0.7 nM/sec, and the rates of lagging strand synthesis (□) were 30.4, 20.4, 12.5, and 4.9 nM/sec, respectively. (B) Measurement of the dissociation rate constants of the leading and lagging strand polymerases during replication on the minicircle substrate. ○, the control reaction in which gp43 was present in the dilution mix; □, leading strand synthesis after 10-fold dilution of gp43 as monitored by the [α-32P]dGTP incorporation; ▵, lagging strand synthesis after 20-fold dilution of gp43 as monitored by the [α-32P]dCTP incorporation. The amount of product formation of each reaction was normalized by the reaction volume and the fold of dilution performed. The data points were fitted into Eq. 1, and koff values of 0.0013 ± 0.00005 and 0.0012 ± 0.00009 sec-1 were generated for the leading and lagging strand polymerases, respectively.
Measurement of Polymerase Dissociation Rate by Dilution. To provide further insights into the polymerase processivity, we sought to experimentally measure the polymerase dissociation rate during active replication. On both the minicircle and TRFII substrates, a dilution experiment was designed to measure the processivity of the replisome polymerases, similar to one devised for evaluating the processivity of the helicase during replication (12). The replisomes were assembled, and replication was initiated. Reactions were then diluted after 1 min (minicircle) or 2.5 min (TRFII) into a dilution mix from which either the DNA substrate alone or both the DNA and gp43 were omitted. The amount of nucleotide incorporation measured after dilution equals the product of the numbers of active replication forks and the rate of fork movement. We found that the fork rate remained the same at different levels of gp43 (data not shown). Therefore, the amount of nucleotide incorporation directly reflects the number of the active forks at any given time after dilution. Control experiments demonstrated that there was no more active complex formation after dilution. We observed the same polymerase dissociation rate constants on the leading and lagging strand after either a 10- or 20-fold dilution. Therefore, the decrease of the number of active replication forks leads to a decrease in the amount of the dNMP incorporation over time and allows a direct measurement of polymerase dissociation. As shown in Fig. 1B, when gp43 was present in the dilution mix, the rate of dNMP incorporation was constant, indicating a replicating polymerase that remains processive up to 16 min after dilution. However, when gp43 was omitted from the dilution mix, a progressive decrease in the rate of dNMP incorporation over time was detected. The data were fitted to the following single exponential equation:
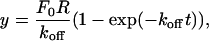 |
[1] |
where y is the amount of dNMP incorporation, F0 is the initial fork number, R is the fork rate (150 nucleotides per sec), and t is the time after dilution (12). The koff measured for both strands on either the minicircle (≈0.0013 sec-1) or the TRFII substrate (≈0.0017 sec-1, data not shown) are in good agreement and are also consistent with the holoenzyme off rate measured earlier on a small fork substrate (11), suggesting that both holoenzymes already possess an intrinsic processivity that is not further enhanced by the presence of primosome proteins. Together, these results indicate that the component polymerases on both strands are highly processive and dissociate slowly with similar rate constants. Further evidence for the high processivity of the polymerases comes from the observation that the Okazaki fragment size remained fairly constant within a wide range of gp43 concentrations (15–240 nM, see supporting information).
These results, however, are in seeming contradiction to those of the trapping experiments described above. This discrepancy prompted us to investigate further the action of the trap polymerase during replication. Several control experiments were first conducted to eliminate certain alternatives by which the trap might act (see supporting information). The results of these experiments indicated that trap gp43 (i) does not act through sequestering the clamp, the clamp loader, or the helicase accessory protein from being recruited by the replisome; (ii) does not inhibit the helicase unwinding activity; and (iii) does not act by interfering with the replisome assembly process under our experimental conditions. We consequently hypothesized an active exchange mechanism (Scheme 1: E, replisome; D, DNA; A, WT polymerase; B, trap polymerase) for the trap polymerase based on the collective results in which the trap displaces the replicating polymerase from the replisome, hence stopping the holoenzyme on its track. We subsequently performed a series of experiments to gain more support for this model in contrast to one of passive exchange in which the trap polymerase only binds the replisome after the WT polymerase dissociates (Scheme 1, k2 = 0).
Scheme 1.

Effect of the Trap Polymerase Concentration on the Okazaki Fragment Size. If an active exchange mechanism is functioning, an early stalling of the holoenzyme will be forced that will lead to the general shortening of the product size in the presence of the trap. As a result, Okazaki fragment size would decrease on the lagging strand upon the addition of the trap polymerase. As shown in Fig. 2, a decrease of the Okazaki size was observed at increasing trap concentrations. On the minicircle substrate, the average Okazaki fragment size dropped from ≈1 kb in the absence of the trap to 0.5 kb when a 6-fold excess of the trap (relative to WT gp43) was present. A decrease from 2.1 kb to 1.1 kb was observed on the TRFII substrate as the trap concentration increased from 0- to 8-fold excess.
Fig. 2.

Effects of D408N trap gp43 on the size of the Okazaki fragments. Lagging strand synthesis was monitored at increasing trap gp43 concentrations (0 and 1.5 μM on the minicircle and 0, 64, and 128 nM on the TRFII). The Okazaki fragments synthesized on the minicircle and the TRFII substrate are shown in A and B, respectively. In B, the DNA products at 20, 40, 60, 90, 120, and 150 sec are shown in the presence of increasing trap gp43 concentrations. The arrows in B indicate the average Okazaki fragment size at each trap concentration. The size of the Okazaki fragments are shown for minicircle (C) at 0 (solid trace) and 1.5 μM (dashed trace) of trap gp43, and for TRFII (D)at 0 (solid trace), 64 (dashed trace), and 128 nM (dotted trace) trap gp43, respectively. The peaks in D with relative Rf (Rf) values of 0.03 and 0.26 are due to leading strand synthesis and strand displacement synthesis, respectively. Relative Rf values are defined as the ratio between the migrated distances and the length of the gel.
Effect of the Trap Polymerase Concentration on the Primer Extension on a Primed M13mp18 ssDNA Substrate. We further tested the effect of the trap on the size of the replication products in a simplified system containing the holoenzyme, gp32, and a primed M13 substrate. A control experiment indicated that the holoenzyme assembly on this substrate was complete within 5 sec and that there was no additional holoenzyme formation between 5 and 30 sec. Reactions were carried out as described under Materials and Methods. Gp32 was included to eliminate the hairpin structures that could stall the holoenzyme. Inclusion of dATP in the premixing allowed the assembled holoenzyme to incorporate the first nucleotide that facilitates the correct assembly of the holoenzyme (11). The results once more demonstrated the ability of the trap to displace the replicating polymerase from the p/t junction (Fig. 3A, lanes 1–9). In the absence of the trap, the holoenzyme progresses steadily with a rate of ≈150 nucleotides per sec (lanes 1–3). The product bands are sharp with minimal smear, indicating a synchronized and processive holoenzyme progression. When the trap was added, the size of the extension product diminished as a result of the prematurely stalled complexes, and the product bands were smeared on the gel, consistent with an increased dissociation rate of the polymerase in the presence of the trap (lanes 4–9). As a control, the extension reactions were conducted under otherwise identical conditions, except that the WT polymerase was added instead of the trap polymerase. No inhibition of the holoenzyme movement was observed (Fig. 3A, lanes 10–15). The results indicate that the inhibition by the trap protein was not due to any nonspecific binding arising from high trap protein concentrations.
Fig. 3.

Effects of the trap gp43 on the primer extension by the holoenzyme complex on a primed M13mp18 ssDNA substrate. Reactions were carried out as described under Materials and Methods.(A) Extension products (at 3, 7, and 10 sec) are shown in the presence of 0 (lanes 1–3), 160 (lanes 4–6), and 320 nM (lanes 7–9) trap gp43 (D408N). Control experiments are shown in lanes 10–12 (160 nM WT gp43) and lanes 13–15 (320 nM WT gp43). (B) Extension products (at 3, 7, 10 and 20 sec) are shown in the presence of 0 (lanes 1–4), 160 (lanes 5–8), and 320 nM (lanes 9–12) trap gp43Δ10.
We then used another mutant of gp43, the C-terminal deletion gp43Δ10, to further test the active exchange model. The C-terminal tail of gp43 has been shown to be crucial for the interaction between the polymerase and the clamp but is dispensable for the nucleotidyl transfer activity and for DNA binding (17). Control experiments showed that gp43Δ10 lost its capability to interact with the clamp. An extremely high off rate limited the primer extension by this mutant under the experimental conditions to 6–7 nucleotides per sec (see supporting information), as compared to the 150 nucleotides per sec extension rate of the WT polymerase. Therefore, gp43Δ10 can be considered as an inactive polymerase trap in this experiment. When this mutant was used in the above primer extension assay, no trapping effect was observed (Fig. 3B). To eliminate the possibility that exchange did occur but that the WT polymerase quickly replaced gp43Δ10 and resumed replication because of the inability of gp43Δ10 to form a stable complex with the clamp, we conducted the same experiments with a 10-fold dilution of the WT gp43 after assembly. Gp43 at such a low concentration cannot form the holoenzyme. Again no trapping was observed (data not shown). These results thus provide strong evidence for the active exchange mechanism involving specific protein–protein interactions between the polymerase C terminus and the clamp.
The Measurement of the Trapping Rate Constant. To measure the active exchange rate, we studied directly the trapping effects on dNMP incorporation at short times for both the minicircle and the TRFII substrates at various trap concentrations. The experimental data generated for both substrates were then fitted according to Scheme 1. The polymerase dissociation rate constant k-1 is set at 0.002 sec-1, a value generated from current and previous studies (11). Because the fitting begins with an equilibrated system and because k-1 is very low, the dissociation of the polymerase from the replisome complex was insignificant. Therefore, the experimental data do not constrain the value of k1. We set k1 at 1 μM-1·sec-1, a common second-order rate constant for macromolecular interactions. The rate of fork movement (kcat) has been shown to average at 150 sec-1 on both substrates (9, 13). The fitting generated a second-order active-exchange rate constant of 0.17 and 0.29 μM-1·sec-1 (k2) on the minicircle and the TRFII, respectively (Fig. 4). These values correspond to an ≈0.02 sec-1 exchange rate at 100 nM trap gp43, a rate 10-fold higher than the polymerase off rate. The high exchange rate obtained here confirms an active polymerase exchange process and eliminates the possibility of the passive exchange mechanism.
Fig. 4.

The trapping rates on the leading strand synthesis on both the minicircle (A) and the TRFII (B) substrate at various trap gp43 concentrations. Standard replication reactions were carried out at 0 (○), 200 (▪), and 500 nM (▴) trap gp43 on the minicircle and 0 (○), 64 (▪), and 128 nM (▴) trap gp43 on the TRFII. Data were fitted into Scheme 1 by the dynafit program (Biokin, Pullman, WA). k1, k-1, and kcat were set at 1 μM-1·sec-1, 0.002 sec-1, and 150 sec-1, respectively. The exchange rate constants (k2) of 0.17 ± 0.07 μM-1·sec-1 and 0.29 ± 0.01 μM-1·sec-1 were generated on the minicircle and the TRFII, respectively. k3 was fitted to be 0.03 ± 0.01 sec-1, which represented an inactivation step on the minicircle substrate.
Whereas a good fit to the data could be generated on the TRFII substrate without involving the k3 step, the fitting of the minicircle data required a real k3 value (0.03 sec-1) that dictated the rate of an irreversible step of fork inactivation. After the exchange takes place, the trap polymerase stalls the holoenzyme while the helicase keeps unwinding, resulting in the collapse of the small 70-bp minicircle substrate. The temporary separation of the holoenzyme and the primosome, however, will not cause a complete unwinding of the TRFII within our experimental time frame, because of its large size.
The Effect of Trap Concentration at Constant WT/Trap Polymerase Ratio. A distinct difference between the active and passive exchange models is that the inhibitory effect remains the same for the passive exchange model as long as the WT/trap ratio is kept constant. However, for the active exchange model, higher concentrations of the trap always induce a greater inhibition even at the same WT/trap ratio.
Two parallel experiments were performed to test this model property. In the first, reactions were carried out with 100 nM WT gp43 in the presence or the absence of 100 nM trap gp43. A 1.7-fold decrease in replication rate was observed in the presence of the trap. In the second, two additional reactions were carried out at the same WT/trap ratio (1:1) except that the protein concentrations of both the trap and the WT polymerase were increased to 500 nM. In this experiment, an enhanced 3.3-fold decrease in rate was observed when the trap was present. The net drop of 1.9-fold (3.3/1.7) in rate is very close to the 1.8-fold decrease predicted by computer simulation of the active exchange model with the kinetic parameters generated in the previous section.
Discussion
T4 gp43 polymerase per se is a distributive enzyme with a dissociation rate constant of 6–8 sec-1 (18). Its processivity is greatly enhanced by the accessory proteins, gp45 and gp44/62 (2, 11). To examine the processivity of the polymerase during active replisomal replication, we measured the dissociation rate constants of the leading and lagging polymerases. A value of ≈0.0013 sec-1 was obtained for both polymerases on the minicircle and the TRFII substrate. This rate constant agrees with the one (0.002 sec-1) measured on a static fork substrate (11) and is also close to the rate (0.001 sec-1) measured for the helicase during replication (12). This high processivity is further substantiated by the observation that the size of the Okazaki fragments does not increase with decreasing polymerase concentrations because diluting a dissociative replisome component would result in the formation of longer Okazaki fragments (13). Thus, the high processivity of the holoenzymes and the helicase constitutes the basis for processive replication by the T4 replisome.
To further study the polymerase processivity during replication, we used an inactive mutant gp43 (D408N) as a protein trap and measured its effects on replication. Such an approach has been used in the investigation of the dissociative properties of the clamp, the clamp loader, and the primase during lagging strand synthesis and has yielded results consistent with those of the concentration titration experiments (13). In this paper, we show that the mutant polymerase, although completely inactive in nucleotidyl transfer activity, retains normal binding properties of the WT polymerase and hence can compete with the WT polymerase for forming the replisome. We studied the effect of this trap protein on leading and lagging strand synthesis under conditions that enabled us to measure the trapping effects on the replication but not on the replisome assembly. We surprisingly observed a rapid inhibition of replication (t1/2 < 1 min) by trap gp43 on both leading and lagging strand in a trap concentration-dependent manner. These results would, at first glimpse, suggest that both replisome polymerases frequently dissociate from the replisome during replication. However, this interpretation would appear to be at odds with the high gp43 processivity during replication demonstrated by the dilution experiments.
After eliminating several possible causes for the trapping effects, such as the depletion of the replisome proteins, the inhibition of the helicase activity, or the inhibition of the replisome assembly, we imagined an active exchange mechanism to explain the trapping effect. A series of experiments provide compelling evidence for this mechanism. Firstly, the lagging strand size decreases in the presence of increasing amount of the trap gp43, a result expected if the trap displaces the lagging strand polymerase during replication. Secondly, the trap leads to a decreased size and a broadened distribution of the primer extension product by the holoenzyme on a primed M13mp18 substrate. No change in either product size or distribution was observed when the same amount of the WT polymerase was added instead of the trap polymerase, excluding trapping caused by nonspecific protein–protein or protein–DNA interactions. Furthermore, no trapping was observed by the gp43Δ10 mutant, revealing the crucial role of the gp43 C terminus–clamp interaction during the polymerase exchange. Thirdly, the kinetics of inhibition at varying trap concentrations on both the minicircle and the TRFII substrates fit well into the active exchange mechanism but poorly into the passive exchange model. An exchange rate of ≈0.2 μM-1·sec-1 was obtained on both minicircle and TRFII substrates. Finally, a stronger inhibition was observed at higher concentrations of both polymerases (trap and WT) while their ratio was kept constant, a result predicted by the active exchange model but not the passive exchange model. Taken together, these results strongly suggest that the trap can displace an active leading or lagging strand polymerase from the replisome. Because the trap retains the binding properties of the WT protein, we infer that such exchange also exists between WT polymerases. Given a ≈600 nM in vivo gp43 concentration (9), we calculate that on any given replisome, the polymerase exchange occurs at an average of approximately once per 10 sec, or ≈90 events per replication fork during the 15-min time-span for the complete replication of the T4 genome.
How does the polymerase exchange take place? Our results (Fig. 3B) indicate that an interaction between the incoming polymerase and the clamp has to be established in order for the former to displace the replicating polymerase. A model of gp45 clamp/RB69 polymerase (a close relative of T4 gp43) interaction has been built based on crystal structure information (19). The polymerase C-terminal region was shown to interact with the interdomain loop of one of the clamp subunits. However, solution studies revealed a clamp structure with one open and two closed interfaces (20) and strongly argued for an interaction between the polymerase C terminus and the open clamp subunit interface (21, 22). The x-ray data and its associated holoenzyme model which used the weaker interdomain binding site of gp45 was predicted, however, to play some role in holoenzyme assembly, DNA replication, translesion bypass, or other replication associated processes (19, 21).
Recent time-resolved fluorescence resonance energy transfer studies indicate that the clamp with one open and two closed subunit interfaces is the only state in solution (D. Millar, M.A.T., and S.J.B., unpublished data). Based on the stable solution structure of the clamp, we considered it less likely for the incoming polymerase to insert its C-tail into one of the two closed clamp subunit interfaces unless its opening is facilitated by the clamp loader. However, our preliminary results suggest that neither ATP hydrolysis nor the clamp loader is required during polymerase exchange. We therefore propose that the incoming polymerase may either transiently attach to the interdomain region of the clamp before the exchange takes place (the interdomain model, Fig. 5A) or directly displace the replicating polymerase by inserting its C-tail into the open clamp interface (the direct displacement model, Fig. 5B). Sterically, the direct displacement model is less favorable than the interdomain model. In the latter, a transient intermediate with two polymerases attaching to the same clamp can form with no major steric clashes given a certain degree of flexibility in the gp43 C-terminal region. Such flexibility has been suggested for the C terminus of the Escherichia coli Pol IV through the crystallographic study of its interaction with the β-clamp (23). Therefore, although the interdomain loop identified in the crystal structure is not the bona fide binding site for the T4 replicating polymerase, it may serve a role in polymerase exchange by providing the initial contact point for the incoming polymerase.
Fig. 5.

Solution structure models of polymerase exchange. Gp45 is colored in orange, the initial gp43 is colored in blue, and the incoming polymerase is colored in green. The initial holoenzyme state was derived from the solution structure holoenzyme model (21). (A) Interdomain binding intermediate model is based on binding of the incoming gp43 at the interdomain binding site (pink) on gp45 as identified by Shamoo and Steitz (19). This semistable intermediate would then require a conformational change to displace the bound polymerase and insert the C-terminal tail of gp43 into the subunit interface of gp45, restoring the holoenzyme solution complex. (B) Direct displacement model disrupts the holoenzyme complex through a short-lived protein transition-state complex that includes two gp43 molecules and one gp45. The incoming C-terminal tail of gp43 displaces the bound gp43 at the open subunit interface of gp45, resulting in a restored holoenzyme solution complex. This model is rotated 120° around the x axis from that in A.
What are the possible roles and implications of the polymerase exchange? Perhaps, bacteriophage T4 has evolved a redundant mechanism of DNA replication in which multiple polymerases from a pool are used to synthesize DNA. Given that the clamp structure and the clamp–polymerase interaction appear to be universally conserved, this idea may be applicable to other replication systems. Furthermore, the replisome frequently encounters structural barriers during replication (24). For example, certain DNA lesions stall the movement of the replisome (15), and the replisome is paused upon encountering the transcription complex (14). It is conceivable that other DNA barriers may exist in vivo, such as those caused by DNA binding proteins or DNA structural distortions (24). These barriers are likely to cause changes within the replisome. Polymerase exchange can perhaps provide a mechanism for resetting the replisome after the structural perturbation to overcome these barriers.
Polymerase exchange may also reflect the clamp function during replication. It is known that both the β-clamp and proliferating cell nuclear antigen make contact with a variety of proteins involved in replication, transcription, DNA repair, and cell cycle control (25). Given the universally conserved clamp architecture, T4 gp45 likely possesses similar binding properties. It appears that the clamp may serve as a platform for multiple protein bindings and thus becomes the major bridging point between the replisome and other proteins or complexes. The clamp may orchestrate multiprotein reactions through the sequential binding of proteins to the clamp in a competitive and temporally regulated manner, as suggested by the interactions of several E. coli polymerases with the β-clamp (26). On the other hand, concurrent binding of several proteins to the clamp could happen if allowed sterically, as in a recently proposed “tool-belt” model for the pivotal role of the clamp during translesion synthesis (27). In this model, several translesion polymerases simultaneously attach to the sliding clamp. Depending on the nature of the lesion and the DNA distortion induced by it, one of them is directed to displace the replicating polymerase to accomplish the polymerase switching process. This concurrent binding model is also supported by the recent finding of a heterotrimeric clamp in archaeon Sulfolobus solfataricus (28), in which the three clamp subunits preferably bind the polymerase, the flap endonuclease 1, and the ligase individually. Such an arrangement allows tight coupling between Okazaki fragment synthesis and processing. Given all these observations, it is not surprising that the polymerase exchange takes place during normal replication. This observation provides further support for the notion of a platform function of the clamp for multiple protein binding, a role crucial for the organization of a variety of pathways with versatile biological functions.
The polymerase exchange during replication enables us to view the polymerase processivity from a new perspective, namely, the polymerase possesses a “dynamic” rather than a “static” processivity during replication. This dynamic processivity reflects the fluidity of the T4 replisome that confers both high processivity during normal DNA synthesis and structural flexibility needed for the interaction between the replisome and other cellular components.
Acknowledgments
This work was supported by National Institutes of Health Grant GM13306.
Abbreviations: TRFII, tailed replicative form II; ssDNA, single-stranded DNA.
See Commentary on page 8255.
References
- 1.Benkovic, S. J., Valentine, A. M. & Salinas, F. (2001) Annu. Rev. Biochem. 70, 181-208. [DOI] [PubMed] [Google Scholar]
- 2.Huang, C. C., Hearst, J. E. & Alberts, B. M. (1981) J. Biol. Chem. 256, 4087-4094. [PubMed] [Google Scholar]
- 3.Morris, C. F., Sinha, N. K. & Alberts, B. M. (1975) Proc. Natl. Acad. Sci. USA 72, 4800-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu, C. C. & Alberts, B. M. (1981) J. Biol. Chem. 256, 2813-2820. [PubMed] [Google Scholar]
- 5.Raney, K. D., Carver, T. E. & Benkovic, S. J. (1996) J. Biol. Chem. 271, 14074-14801. [DOI] [PubMed] [Google Scholar]
- 6.Hinton, D. M. & Nossal, N. G. (1987) J. Biol. Chem. 262, 10873-10878. [PubMed] [Google Scholar]
- 7.Chastain, P. D., II, Makhov, A. M., Nossal, N. G. & Griffith, J. (2003) J. Biol. Chem. 278, 21276-21285. [DOI] [PubMed] [Google Scholar]
- 8.Burke, R. L., Alberts, B. M. & Hosoda, J. (1980) J. Biol. Chem. 255, 11484-11493. [PubMed] [Google Scholar]
- 9.Yang, J., Trakselis, M. A., Roccasecca, R. M., Valentine, A. M. & Benkovic, S. J. (2003) J. Biol. Chem. 278, 49828-49838. [DOI] [PubMed] [Google Scholar]
- 10.Mathews, C. K. (1994) in Molecular Biology of Bacteriophage T4, ed. Karam, J. D. (Am. Soc. Microbiol., Washington, DC), pp. 1-8.
- 11.Kaboord, B. F. & Benkovic, S. J. (1995) Curr. Biol. 5, 149-157. [DOI] [PubMed] [Google Scholar]
- 12.Schrock, R. D. & Alberts, B. (1996) J. Biol. Chem. 271, 16678-16682. [DOI] [PubMed] [Google Scholar]
- 13.Trakselis, M. A., Roccasecca, R. M., Yang, J. & Benkovic, S. J. (2003) J. Biol. Chem. 278, 49839-49849. [DOI] [PubMed] [Google Scholar]
- 14.Liu, B. & Alberts, B. M. (1995) Science 267, 1131-1137. [DOI] [PubMed] [Google Scholar]
- 15.Goodman, M. F. (2000) Trends Biochem. Sci. 25, 189-195. [DOI] [PubMed] [Google Scholar]
- 16.Berdis, A. J. & Benkovic, S. J. (1996) Biochemistry 35, 9253-9265. [DOI] [PubMed] [Google Scholar]
- 17.Berdis, A. J., Soumillion, P. & Benkovic, S. J. (1996) Proc. Natl. Acad. Sci. USA 93, 12822-12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capson, T. L., Peliska, J. A., Kaboord, B. F., Frey, M. W., Lively, C., Dahlberg, M. & Benkovic, S. J. (1992) Biochemistry 31, 10984-10994. [DOI] [PubMed] [Google Scholar]
- 19.Shamoo, Y. & Steitz, T. A. (1999) Cell 99, 155-166. [DOI] [PubMed] [Google Scholar]
- 20.Alley, S. C., Shier, V. K., Abel-Santos, E., Sexton, D. J., Soumillion, P. & Benkovic, S. J. (1999) Biochemistry 38, 7696-7709. [DOI] [PubMed] [Google Scholar]
- 21.Alley, S. C., Trakselis, M. A., Mayer, M. U., Ishmael, F. T., Jones, A. D. & Benkovic, S. J. (2001) J. Biol. Chem. 276, 39340-39349. [DOI] [PubMed] [Google Scholar]
- 22.Alley, S. C., Jones, A. D., Soumillion, P. & Benkovic, S. J. (1999) J. Biol. Chem. 274, 24485-24489. [DOI] [PubMed] [Google Scholar]
- 23.Bunting, K. A., Roe, S. M. & Pearl, L. H. (2003) EMBO J. 22, 5883-5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyrien, O. (2000) Biochimie 82, 5-17. [DOI] [PubMed] [Google Scholar]
- 25.Warbrick, E. (2000) BioEssays 22, 997-1006. [DOI] [PubMed] [Google Scholar]
- 26.Lopez de Saro, F. J., Georgescu, R. E., Goodman, M. F. & O'Donnell, M. (2003) EMBO J. 22, 6408-6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becherel, O. J., Fuchs, R. P. & Wagner, J. (2002) DNA Repair 1, 703-708. [DOI] [PubMed] [Google Scholar]
- 28.Dionne, I., Nookala, R. K., Jackson, S. P., Doherty, A. J. & Bell, S. D. (2003) Mol. Cell. 11, 275-282.p [DOI] [PubMed] [Google Scholar]