

Abstract
Introduction
Although it has been suggested that rare coding variants could explain the substantial missing heritability, very few sequencing studies have been performed in rheumatoid arthritis (RA). We aimed to identify novel functional variants with rare to low frequency using targeted exon sequencing of RA in Korea.
Methods
We analyzed targeted exon sequencing data of 398 genes selected from a multifaceted approach in Korean RA patients (n = 1,217) and controls (n = 717). We conducted a single-marker association test and a gene-based analysis of rare variants. For meta-analysis or enrichment tests, we also used ethnically matched independent samples of Korean genome-wide association studies (GWAS) (n = 4,799) or immunochip data (n = 4,722).
Results
After stringent quality control, we analyzed 10,588 variants of 398 genes from 1,934 Korean RA case controls. We identified 13 nonsynonymous variants with nominal association in single-variant association tests. In a meta-analysis, we did not find any novel variant with genome-wide significance for RA risk. Using a gene-based approach, we identified 17 genes with nominal burden signals. Among them, VSTM1 showed the greatest association with RA (P = 7.80 × 10−4). In the enrichment test using Korean GWAS, although the significant signal appeared to be driven by total genic variants, we found no evidence for enriched association of coding variants only with RA.
Conclusions
We were unable to identify rare coding variants with large effect to explain the missing heritability for RA in the current targeted resequencing study. Our study raises skepticism about exon sequencing of targeted genes for complex diseases like RA.
Electronic supplementary material
The online version of this article (doi:10.1186/s13075-014-0447-7) contains supplementary material, which is available to authorized users.
Introduction
Rheumatoid arthritis (RA (MIM 180300)) is a complex autoimmune disorder that results from both genetic and environmental risk factors [1,2]. Strong evidence regarding the existence of a genetic predisposition for RA has been supported by several familial studies including twin studies, in which the heritability of RA has been estimated to be approximately 65% [1,2].
Nearly 60 RA risk loci were identified in several large studies including genome-wide association studies (GWAS) [3–6] and immunochip (iCHIP) [7,8] arrays using common single nucleotide variants (SNVs). The largest genetic contribution effect size has been identified for the major histocompatibility complex (MHC) locus with evidence for three independent association signals on HLA-B, HLA-DRB1, and HLA-DPB1 affecting five amino acid positions [9,10]. The total variance of the MHC region explained 13.03% of the RA risk [11]. The other non-MHC genes identified were primarily immune pathway genes, though their effect sizes were quite modest. To date, the known RA risk loci can explain only about 25% of the total genetic heritability [12].
It has been suggested that rare or low-frequency variants could explain the substantial unexplained heritability of many complex diseases, most of which were not fully captured using the previous conventional genotyping technology. Recently, Stahl et al. [11] inferred a highly polygenic model that attempted to explain the missing heritability of RA. In this model, it was suggested that a small number of rare variants with large effect sizes may contribute to heritability in addition to hundreds of common variants.
New genomic technologies, including next-generation sequencing (NGS), can provide a new approach for identification of rare variants. With advances in NGS technology, the role of rare or low-frequency variants in many complex diseases like RA can be investigated to better characterize the genetic architecture of the disease. Recently, several sequencing studies that have investigated common autoimmune diseases have shown that rare variants within genes containing common variants are associated with complex diseases [13–16]. For RA, Diogo et al. [17] performed deep exon sequencing of 25 biological candidate genes discovered by GWASs in 500 RA cases and 650 controls of European ancestry and subsequent dense genotyping in larger samples, in which they found accumulation of a few rare nonsynonymous variants with nominal significance instead of variants with large effect of genome-wide significance.
Here, we aimed to identify novel functional variants with rare to low frequency using targeted exon sequencing in Korean RA, which dealt with hundreds of selected genes that were related to RA in various aspects such as previous identified genes from GWASs and immunochip data, literature reviews, and related pathways.
Materials and methods
Patients and controls
A total of 1,252 RA cases were enrolled from the BAE cohort of Hanyang University Hospital for Rheumatic Diseases and satisfied the American College of Rheumatology 1987 classification criteria [18] for RA. The ethnically matched 745 healthy controls, excluding those with a personal or familial history of any autoimmune disease, were recruited at the same institute. Informed consent was obtained from all individuals via a questionnaire at the time of enrollment, when clinical information was also collected. The study was approved by the institutional review board of Hanyang University (HYG-11-015-1).
Targeted gene selection
We selected candidate genes using a comprehensive approach that included previous genetic and biological research, pathway databases, text-mining analysis, and animal-model databases (Figure S1 in Additional file 1). Of the non-MHC candidate genes, we included (a) 106 known RA risk loci identified via literature review [3,4,6,9,19], (b) 519 genes associated with RA in our Korean iChip dataset [8], (c) 155 genes shared by both RA and systemic lupus erythematosus (SLE) in our previous Korean GWAS datasets [4,20], (d) 18 genes in RA-related pathways, (e) 65 genes identified via text mining using GRAIL from recent GWAS data [3,4], and (f) 8 human homologs of mouse genes that induced an RA-like phenotype from the Mouse Genome Database (MGD) [21]. Altogether, 666 designable target genes were selected for exon sequencing.
Exon sequencing
We enriched the target exons with Agilent’s SureSelect capture kit (target region = 1.36 Mb) and performed high-throughput paired-end sequencing using a HiSeq2000 (Agilent Technologies, Santa Clara, CA, USA). The sequencing reads were mapped to the human reference genome, where the reference sequence was UCSC assembly hg19 (NCBI build 37.1) using Burrows-Wheeler Aligner (BWA) software [22]. We then applied programs packaged in Picard-tools in order to convert the previous SAM file into a format that was sorted by mapping coordinates and to remove PCR duplicates. We created another SAM file that included only reads that uniquely mapped to the reference genome, and transformed this into a BAM file using Samtools [23]. Those variants are annotated by ANNOVAR (Figure S2 in Additional file 1).
We then performed stringent quality control for 666 target gene by which we selected only high coverage genes that were sequenced coding-region based on the public database (db). We obtained an initial dataset with 50,247 variants from 666 targeted genes. We filtered the original genotype matrix by single nucleotide polymorphism (SNP) quality and depth coverage. The filtered genotype data contains genotype calls satisfying with practical guidance in rare variants analysis of complex trait association studies [24]; coding sites sequenced with >20× coverage and quality score >30 in at least 80% of cases and controls in the public database (db) or in the designed targeting genes. As a result, a total of 398 genes (mean 92.0% coverage of the captured exon) in 1,997 individuals were used in the subsequent variant-calling analysis (Table S1 in Additional file 1). When we called the variants if SNVs had minimal depth coverage >20× and a quality score >30 in more than 80% of the subjects sequenced, a total of 12,916 variants within 398 genes were identified. We then eliminated SNVs that had insufficient call rates (<90%) in cases and controls, Hardy-Weinberg disequilibrium with P <0.01 in controls, and also eliminated samples that had insufficient call rates (<90%). We finally analyzed the 10,588 exonic variants of 1,934 samples for further single-variant association test, gene-based test, and pathway-based association enrichment test (Table 1, Table S2 in Additional file 1). There was no evidence of skewed genotyping between cases and controls in the principle component (PC) analysis (Figure S3 in Additional file 1).
Table 1.
Characteristics of RA patients and controls included in targeted exon sequencing
| RA cases (n = 1,217) * | Controls (n = 717) * | |
|---|---|---|
| Age of onset (mean ± SD years) | 41.9 ± 12.8 | 35.1 ± 10.7 |
| Disease duration (mean ± SD years) | 9.9 ± 11.1 | - |
| Female (%) | 87.4 | 85.3 |
| Rheumatoid factor (%) | 97.8 | - |
| Anti-cyclic citrullinated peptide autoantibodies (%) | 98.1 | - |
*Cryptic relatedness with duplicate or first-degree relatives using KING software, outlier (deviating >8 SEM on any of the five principal components), or samples with less than 80% of the data sequenced were removed. A total of 1,934 samples were included for further analysis. SD, standard deviation; SEM, standard error of the mean.
Statistical analysis
To analyze for single-marker association with RA in targeted exon sequencing data, odds ratios (OR) and P values were calculated using PLINK v1.07 software [25] with adjustments for the top 10 PCs in logistic regression. Fisher’s exact tests were also used for association tests of each rare variant.
For a meta-analysis (3,580 RA cases and 7,938 controls) using a Korean RA GWAS [4] and iCHIP [8] data generated from ethnically matched independent sample collections in addition to the current NGS results, we applied several quality-control filters on Korean RA GWAS (n = 4,799) and iCHIP (n = 4,722) data to select high-quality SNVs (minor allele frequency (MAF) ≥1%, P value of Hardy-Weinberg equilibrium (HWE) <10−4 and call rate >95% in cases and controls).
For an enrichment test for exonic SNPs in 77 newly identified genes (P <0.05 in a single-variant test of sequencing data), we imputed common and low-frequency variants (MAF >0.5%) from the Korean GWAS data (800 RA cases and 3,999 controls) by ShapeIt and IMPUTE2 with the 1000 Genomes Phase I reference panel. We performed logistic regressions for 1,000-times permuted phenotypes with the top 10 PCs as covariates by PLINK. Then, the numbers of genic, exonic, nonsynonymous, or synonymous variants reaching the Pthreshold <0.05 between observed and permuted data were compared by using a Fisher’s exact test.
In a gene-based analysis of rare coding variants (MAF <5%), we performed both nonburden testing (optimal sequence kernel association test (SKAT-O)) [26] and burden testing (SCORE-seq) [27,28]. Weighted analysis was performed for rare nonsynonymous variants using SIFT [29], PolyPhen2 [30], and CAROL [31] scores. Statistical significance was determined by using 1,000,000 case-control permutations.
In pathway-based enrichment test of NGS data, we generated 1,000 permuted phenotype sets and their disease association P values for each SNV by logistic regression with adjustment for the top 10 PCs. To eliminate linkage disequilibrium (LD)-derived enrichment bias, we clumped the set of SNVs (r2 <0.4) in order of statistical significance. Then, we compared the number of SNV with P <Pthreshold between permuted datasets and empirical datasets by Fisher’s exact tests. Genes in each functional pathway were obtained from Ontology and KEGG.
Ethics approval
Ethics approval was granted by the institutional ethics committee of Hanyang University in the Republic of Korea.
Patient and control consents were obtained.
Results
After stringent quality control on the 666 targeted genes (Figure S1 in Additional file 1) as well as the sequenced samples (see Materials and Methods), we obtained a final dataset for analysis that consisted of 10,588 exonic variants from 398 genes in 1,217 RA cases (age = 41.9 ± 12.8 (mean age ± standard deviation (SD)); female = 87.4%) and 717 controls (age = 35.1 ± 10.7 (mean age ± SD); female = 85.3%) (Table 1).
The majority of SNVs were rare (90.6% with MAF <5%), and 6,605 SNVs were novel which were not identified in the 1000 Genome Project dataset (Figure S4 in Additional file 1). The transition/transversion (Ti/Tv) rate was 2.93 in RA cases and 2.92 in controls, which indicates good quality control based on expected human mutation types (Table S2 in Additional file 1). We note that a high concordance rate was observed between genotype calls from sequencing versus other genotyping methods such as GWAS and iCHIP by non-reference sensitivity [32] and non-reference discrepancy rate [33] (Table S3 in Additional file 1). In addition, validation using a TaqMan assay for a selected 37 SNVs showed high concordance rates with sequencing data (99.3%).
We used three different strategies for analysis of variants that passed the stringent quality control: (1) single-variant association test, (2) gene-based test for rare variants of which MAF are less than 0.05, and (3) pathway-based association enrichment test (Figure 1). Regarding the single-variant association test, we further perform a meta-analysis using the current NGS data and previous GWAS/iCHIP data from independent Koreans, and evaluate that exonic SNPs in the genes associated with RA in the NGS data are enriched for RA association in imputed GWAS dataset.
Figure 1.
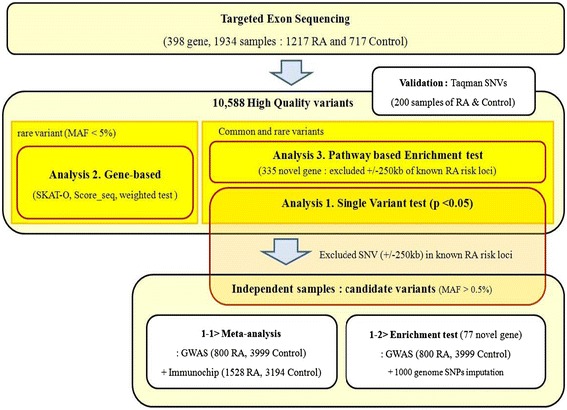
Description of the study design. We analyzed data from protein-coding variants within targeted genes using three different strategies for analysis.
In the first single-variant association analysis using 10,588 SNVs in 1,217 cases and 717 controls, we identified thirteen nonsynonymous variants with P <0.01, none of which reached the significance threshold after Bonferroni correction (Table S4 in Additional file 1). To compensate for the limited power that may have resulted in a lack of significant associations, we performed a meta-analysis for 108 coding SNVs (located in 89 genes) that were associated with a P value less than 0.05 in the single-variant association test. We used two independent Korean genomic data in the meta-analysis with NGS data; one was Korean RA GWAS dataset [4] (n = 1,099) combined with independent Korean control data (n = 3,700) genotyped by Illumina HumanOmni1-Quad BeadChip, and the other was Korean iCHIP data from 4,722 independent case-control subjects [8].
We focused on novel genes in the meta-analysis dataset of 3,580 RA cases and 7,938 controls, excluding any known GWAS or iCHIP signals. However, neither common nor low-frequency novel variants achieved genome-wide significance (P <1.0 × 10−8), with an SNV (rs1088680) within PRKCH showing the strongest association (P = 3.16 × 10−5) (Table S5 in Additional file 1).
Next, in an attempt to investigate whether an aggregate effect of 89 risk genes identified in NGS (P <0.05) exists or not, we performed an enrichment analysis for RA associations in all risk genes excluding 12 known RA risk loci using an independent dataset from our GWAS. Association of 41,454 SNVs within the 77 genes in the imputed Korean GWAS data was compared with the results after 1,000 case-control permutations by Fisher’s exact tests (Figure 2). To eliminate LD-derived bias, we clumped the set of independent SNVs (r2 <0.4).
Figure 2.
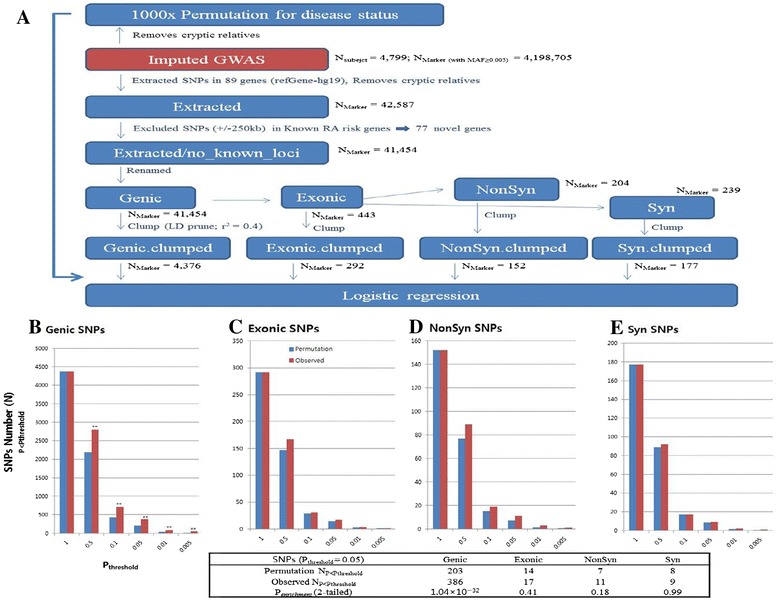
Enrichment analysis of 77 novel genes with nominal signal on exon sequencing in the GWAS dataset. (A) We performed logistic regressions including 10 principal components using 1,000-times permuted phenotypes. The numbers of (B) genic, (C) exonic, (D) nonsynonymous (NonSyn), and (E) synonymous (Syn) variants reaching the P <0.05 threshold following 1,000 permutations are shown. Significant enrichment of SNVs using the P <0.05 threshold was assessed using Fisher’s exact tests (** P <0.01). These enrichment signals were driven by each group (P enrichment (genic) = 1.04 × 10−32, P enrichment (exon) = 0.41, P enrichment (NonSyn) = 0.18, and P enrichment (Syn) = 0.98 at P threshold = 0.05).
Although the significant signal appeared to be driven by 4,376 genic variants (observed NP<Pthreshold/Ntotal = 386/4376; Penrichment (genic) = 1.04 × 10−32), we found no evidence for enriched association at coding variants only (exon (observed NP<Pthreshold/Ntotal = 17/292; Penrichment = 0.41 at Pthreshold = 0.05), nonsynonymous (11/152; Penrichment = 0.18), and synonymous (9/177; Penrichment = 0.99)) (Figure 2).
In the second analysis, we performed gene-based analysis of rare coding variant (MAF <5%) using nonburden tests (optimal sequence kernel association test (SKAT-O)) [26], burden tests (SCORE-seq) [28], and weighted tests with SIFT [29], PolyPhen2 [30], and CAROL [31] scores for the functional effects of the variants. A total of 17 genes had a nominal burden signal of association (P <0.05), most of which had two or more nonsynonymous rare variants, although they did not reach the threshold for significance after Bonferroni correction (P <1.2 × 10−4) (Table 2, Figure S5 in Additional file 1). For VSTM1, a top gene driven by the gene-based test, we further validated eight rare variants in VSTM1 using Sanger sequencing with the same samples that were heterozygous for any of those variants in the initial sequencing stage; 29 of 31 samples were validated as heterozygous (false-positive rate = 6.45% (2/31)). The following analysis using only these validated samples showed that a set of the validated seven nonsynonymous variants of VSTM1 conferred a protective role in RA (P = 4.55 × 10−3 in SKAT-O, P = 7.80 × 10−4 in SCORE-seq). The four variants of VSTM1 were primarily within the immunoglobulin-like domain among the coding regions. The two variants, A33T at the domain and D122N closed to the domain, were thought to be deleterious variants by PolyPhen2, suggesting that these may have a functionally protective role in RA (Figure 3).
Table 2.
Gene-based tests of rare nonsynonymous variants in RA
| GENE | Chr | Gene-based test * | ||
|---|---|---|---|---|
| N marker | SKAT-O (nonburden test) P value | SCORE_seq (burden test) | ||
| P value | ||||
| VSTM1 | 19 | 7 | 4.55 × 10−3 | 7.80 × 10−4 |
| KPRP | 1 | 8 | 6.38 × 10−3 | 6.51× 10−3 |
| C6orf99 | 6 | 5 | 0.05155 | 0.01669 |
| PARD3 | 10 | 21 | 0.14496 | 0.01887 |
| PYGL | 14 | 10 | 0.02287 | 0.02057 |
| ARHGAP26 | 5 | 9 | 0.01939 | 0.02114 |
| NCF2 | 1 | 8 | 0.02574 | 0.02159 |
| CCR6 | 6 | 7 | 0.01131 | 0.02776 |
| TRAF6 | 11 | 8 | 0.04621 | 0.02876 |
| GRIN2B | 12 | 6 | 0.20806 | 0.02966 |
| SNTB1 | 8 | 6 | 0.00987 | 0.03395 |
| PTCD3 | 2 | 11 | 0.14462 | 0.03772 |
| CA8 | 8 | 3 | 0.17982 | 0.03792 |
| NRXN3 | 14 | 8 | 0.12536 | 0.03905 |
| CPEB4 | 5 | 7 | 0.05104 | 0.04010 |
| CTNNA3 | 10 | 16 | 0.10312 | 0.04079 |
| KRT24 | 17 | 5 | 0.02866 | 0.04837 |
*We defined rare nonsynonymous variants as MAF <5% in both cases and controls. We selected 347 genes with two or more rare nonsynonymous variants for gene-based tests. RA, rheumatoid arthritis; Chr, chromosome; Nmarker, number of rare nonsynonymous variants for each gene.
Figure 3.
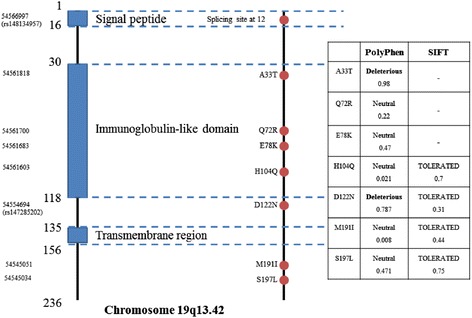
Rare variants of VSTM1 identified by exon sequencing. The seven nonsynonymous variants of VSTM1 that were validated by the Sanger sequencing method were primarily singletons driven from controls except for A33T (RA (n = 3), control (n = 5)). The four nonsynonymous variants of VSTM1 lie within the coding regions of the immunoglobulin-like domain.
In the third analysis, we conducted a pathway enrichment analysis of coding variants (nonsynonymous and synonymous) within 335 novel genes using Ontology and KEGG, which were obtained after excluding the known RA risk loci (+/-250 kb) from the initial 398 genes in NGS. In this analysis of both common and rare variants, we observed weak but significant evidence of overall enrichment of coding SNVs (Penrichment (nonsynonymous) = 8.55 × 10-4 and Penrichment (synonymous) = 0.166 at Pthreshold = 0.05) (data not shown) for the 335 genes. However, in the analysis for each pathway, we did not identify any pathway in which a significant enrichment for coding variants for RA existed at Pthreshold = 0.05.
Discussion
This study does not support our hypothesis that the substantial proportion of missing heritability for RA can be inferred from rare coding variants. Among the 10,588 candidate variants of 398 genes analyzed in a cohort of 1,217 RA cases and 717 controls, 13 single nonsynonymous variants showed only nominal significance with a P value less than 0.01. Several genes found in the gene-based analysis also showed only weak association with RA.
Strikingly, this lack of a significant effect is consistent with that observed by Diogo et al. [17] in Caucasian population, in which most of the 25 candidate genes subjected to deep exon sequencing did not harbor rare coding variants contributing to risk of RA despite some evidence of accumulation of rare missense variants in gene-based test. Although 16 genes were overlapped between our Korean study and the Caucasian study [17], we could not find any significant single coding variant or gene that was shared in both ethnic groups. Taken together, rare functional variants may have very weak contribution to development of RA.
Besides, recent large-scale sequencing studies of six common autoimmune diseases (autoimmune thyroid disease, Crohn’s disease, celiac disease, psoriasis, multiple sclerosis, and type 1 diabetes) showed a negligible impact of rare autoimmune-locus coding variants on unexplained heritability (<3%) [34].
A possible reason for this negative finding may be the limited number of targeted genes that were sequenced, which were 398 genes in the current study. Rare coding variants of the remaining genes across whole genome could participate in the missing genetic contribution to RA. The other potential reason may be genetic heterogeneity for RA, in which each individual or small subset of RA may have their particular rare causal variants. The current study revealed a lot of examples in which a specific rare variant was observed in only one or two individuals among 1,934 subjects. To discover these lots of ‘private’ causal variants that are susceptible only in small subsets of RA patients, deep sequencing of whole exome from very large RA cases, approximately thousands of individuals, would be required.
VSTM1, a top signal gene driven by a gene-based test, is a glycoprotein primarily expressed in immune tissues, which can promote the differentiation and activation of Th17 cells [35]. In addition, two variants of VSTM1 (A33T, D122N) found in the current study were deleterious variants by PolyPhen2. Therefore, it is certainly worth replicating these variants or performing deep sequencing of the entire VSTM1 gene from an independent larger population, especially in Caucasians.
There are several limitations of this study. First, we performed a targeted exon sequencing study, which tends to generate more biased data than whole-exome or whole-genome sequencing studies. Additionally, we did not attempt to validate all rare variants identified by alternative methods, but rather performed it only for selected variants such as Sanger sequencing of rare nonsynonymous variants of VSTM1 and TaqMan genotyping of 37 variants from 200 samples. Finally, we included approximately 2,000 subjects for sequencing, which might not be enough of a sample size to discover rare variants. Consequently, this lack of power may lead most of the analysis in the study, such as the single-variant association analysis, gene-based tests, and enrichment tests, to be less significant with weak association.
However, to our knowledge, this study represents the largest sequencing study that evaluated the largest number of candidate genes with the largest case controls for RA until now. Despite negative findings, further replication of the possible single variants or rare variants in the study will be of some interest.
Conclusions
We were unable to identify rare coding variants with large effect of 398 targeted genes. Despite much anticipation regarding missing heritability, our study raises skepticism about next-generation sequencing of targeted genes in order to discover rare variants with large effect for complex traits like RA. With the advance of genetic technology such as capturing, sequencing of targeted genes, and whole-exome or genome sequencing with a lot of subjects could define more details about the genetic architecture of RA in future.
Acknowledgements
The authors are grateful to their colleagues for assistance with sample preparation, data collection and biospecimens from the Korean Biobank Project, which is supported by the Korea Center for Disease Control and Prevention at the Korea National Institute of Health. This study was supported by a grant from the Korea Healthcare Technology R&D Project [A111218-11-GM01, HI12C1834 (A121983)], Ministry for Health and Welfare, Republic of Korea.
Abbreviations
- Chr
chromosome
- db
database
- GWAS
genome-wide association studies
- HLA
human leukocyte antigen
- HWE
Hardy-Weinberg equilibrium
- iCHIP
immunochip
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MHC
major histocompatibility complex
- NGS
next-generation sequencing
- OR
odds ratio
- PC
principal component
- RA
rheumatoid arthritis
- SD
standard deviation
- SEM
standard error of the mean
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
Additional file
Targeted gene coverage rate (percentage) of coding variants sequenced within 398 genes via exon sequencing. Table S2. Quality metrics for exon sequencing of 398 targeted genes. Table S3. Concordance of targeted exon sequencing (NGS) with other datasets (GWAS, iCHIP), and validation data (Taqman). Table S4. Single-variant test: results for exon sequencing of nonsynonymous variants (P <0.01). Table S5. Meta-analysis of single-association tests for coding variants (NGS + GWAS + iCHIP). Figure S1. A multifaceted approach in selecting target genes for resequencing in RA. Figure S2. Targeted exon sequencing pipeline. Figure S3. Principle component analysis for targeted exon sequencing. Figure S4. High-quality variants identified by targeted exon sequencing. Figure S5. Gene-based analysis of rare nonsynonymous variants in RA.
Footnotes
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
SYB contributed to the data collection and analysis, interpretation of data, manuscript writing, critical revision, and final approval of manuscript. YJN contributed to the data analysis, critical revision, and final approval of manuscript. KK contributed to the data analysis, was involved in drafting the manuscript, critical revision, and final approval of manuscript. YBJ contributed to the data collection, critical revision, and final approval of manuscript. YP contributed to the data analysis, critical revision, and final approval of manuscript. JL contributed to the data analysis, critical revision, and final approval of manuscript. SYL contributed to the data analysis, critical revision, and final approval of manuscript. AAA contributed to the data analysis, critical revision, and final approval of manuscript. JJ contributed to the data analysis, critical revision, and final approval of manuscript. HR contributed to the data analysis, critical revision, and final approval of manuscript. JYL contributed to the data interpretation, critical revision, and final approval of manuscript. BGH contributed to the data interpretation, critical revision, and final approval of manuscript. SMA contributed to the study design, critical revision, and final approval of manuscript. SW contributed to the data analysis, critical revision, and final approval of manuscript. HSL contributed to the interpretation of data, manuscript writing, critical revision, and final approval of manuscript. SCB contributed to the data collection and analysis, interpretation of data, manuscript writing, critical revision, and final approval of manuscript. SYB, HSL, and SCB have full access to all data in this study and take responsibility for the integrity and analysis of data. All authors read and approved the final manuscript.
Contributor Information
So-Young Bang, Email: sybang@hanyang.ac.kr.
Young-Ji Na, Email: na.youngji@gmail.com.
Kwangwoo Kim, Email: ATGC@hanyang.ac.kr.
Young Bin Joo, Email: addmedic@hanmail.net.
Youngho Park, Email: messenger05@nate.com.
Jaemoon Lee, Email: wwwljmcokr@gmail.com.
Sun-Young Lee, Email: 209abcde@gmail.com.
Adnan A Ansari, Email: aaansari_1989@yahoo.com.
Junghee Jung, Email: junghee@macrogen.com.
Hwanseok Rhee, Email: hwanseok@macrogen.com.
Jong-Young Lee, Email: Leeleejy63@gmail.com.
Bok-Ghee Han, Email: bokghee@nih.go.kr.
Sung-Min Ahn, Email: ahnsungmin@gmail.com.
Sungho Won, Email: sunghow@gmail.com.
Hye-Soon Lee, Email: lhsberon@hanyang.ac.kr.
Sang-Cheol Bae, Email: scbae@hanyang.ac.kr.
References
- 1.MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 3.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FAS, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJH, Costenbader KH, Criswell LA, Crusius JBA, Cui J, de Bakker PIW, De Jager PL, Ding B, Emery P, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freudenberg J, Lee HS, Han BG, Shin HD, Kang YM, Sung YK, Shim SC, Choi CB, Lee AT, Gregersen PK, Bae SC. Genome-wide association study of rheumatoid arthritis in Koreans: population-specific loci as well as overlap with European susceptibility loci. Arthritis Rheum. 2011;63:884–893. doi: 10.1002/art.30235. [DOI] [PubMed] [Google Scholar]
- 5.Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, Westra HJ, Fehrmann RS, Kurreeman FA, Thomson B, Gupta N, Romanos J, McManus R, Ryan AW, Turner G, Brouwer E, Posthumus MD, Remmers EF, Tucci F, Toes R, Grandone E, Mazzilli MC, Rybak A, Cukrowska B, Coenen MJ, Radstake TR, van Riel PL, Li Y, de Bakker PI, Gregersen PK, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAllister K, Eyre S, Orozco G. Genetics of rheumatoid arthritis: GWAS and beyond. OA Rheumatol Res Rev. 2011;3:31–46. doi: 10.2147/OARRR.S14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, Zhernakova A, Stahl E, Viatte S, McAllister K, Amos CI, Padyukov L, Toes RE, Huizinga TW, Wijmenga C, Trynka G, Franke L, Westra HJ, Alfredsson L, Hu X, Sandor C, de Bakker PI, Davila S, Khor CC, Heng KK, Andrews R, Edkins S, Hunt SE, Langford C, Symmons D, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim K, Bang SY, Lee HS, Cho SK, Choi CB, Sung YK, Kim TH, Jun JB, Yoo DH, Kang YM, Kim SK, Suh CH, Shim SC, Lee SS, Lee J, Chung WT, Choe JY, Shin HD, Lee JY, Han BG, Nath SK, Eyre S, Bowes J, Pappas DA, Kremer JM, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Arlestig L, Okada Y, Diogo D, et al: High-density genotyping of immune loci in Koreans and Europeans identifies eight new rheumatoid arthritis risk loci.Ann Rheum Dis 2014, [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 9.Raychaudhuri S. Recent advances in the genetics of rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:109–118. doi: 10.1097/BOR.0b013e328336474d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee H-S, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, Siminovitch KA, Bae S-C, Plenge RM, Gregersen PK, de Bakker PIW. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, Voight BF, Kraft P, Chen R, Kallberg HJ, Kurreeman FA, Diabetes Genetics R, Meta-analysis C, Myocardial Infarction Genetics C, Kathiresan S, Wijmenga C, Gregersen PK, Alfredsson L, Siminovitch KA, Worthington J, de Bakker PI, Raychaudhuri S, Plenge RM. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44:483–489. doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 2013;9:141–153. doi: 10.1038/nrrheum.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, Latiano A, Goyette P, Green T, Halfvarson J, Haritunians T, Korn JM, Kuruvilla F, Lagace C, Neale B, Lo KS, Schumm P, Torkvist L, Dubinsky MC, Brant SR, Silverberg MS, Duerr RH, Altshuler D, Gabriel S, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, Cleynen I, Colombel JF, de Rijk P, Dewit O, Finkel Y, Gassull MA, Goossens D, Laukens D, Lemann M, Libioulle C, O'Morain C, Reenaers C, Rutgeerts P, Tysk C, Zelenika D, Lathrop M, Del-Favero J, Hugot JP, de Vos M, Franchimont D, Vermeire S, Louis E, Georges M. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 16.Bonnefond A, Clement N, Fawcett K, Yengo L, Vaillant E, Guillaume JL, Dechaume A, Payne F, Roussel R, Czernichow S, Hercberg S, Hadjadj S, Balkau B, Marre M, Lantieri O, Langenberg C, Bouatia-Naji N, Charpentier G, Vaxillaire M, Rocheleau G, Wareham NJ, Sladek R, McCarthy MI, Dina C, Barroso I, Jockers R, Froguel P. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44:1297–1301. doi: 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diogo D, Kurreeman F, Stahl Eli A, Liao Katherine P, Gupta N, Greenberg Jeffrey D, Rivas Manuel A, Hickey B, Flannick J, Thomson B, Guiducci C, Ripke S, Adzhubey I, Barton A, Kremer Joel M, Alfredsson L, Sunyaev S, Martin J, Zhernakova A, Bowes J, Eyre S, Siminovitch Katherine A, Gregersen Peter K, Worthington J, Klareskog L, Padyukov L, Raychaudhuri S, Plenge Robert M. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet. 2013;92:15–27. doi: 10.1016/j.ajhg.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 19.Okada Y, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Kawaguchi T, Stahl EA, Kurreeman FA, Nishida N, Ohmiya H, Myouzen K, Takahashi M, Sawada T, Nishioka Y, Yukioka M, Matsubara T, Wakitani S, Teshima R, Tohma S, Takasugi K, Shimada K, Murasawa A, Honjo S, Matsuo K, Tanaka H, Tajima K, Suzuki T, Iwamoto T, Kawamura Y, et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet. 2012;44:511–516. doi: 10.1038/ng.2231. [DOI] [PubMed] [Google Scholar]
- 20.Lee HS, Kim T, Bang SY, Na YJ, Kim I, Kim K, Kim JH, Chung YJ, Shin HD, Kang YM, Shim SC, Suh CH, Park YB, Kim JS, Kang C, Bae SC. Ethnic specificity of lupus-associated loci identified in a genome-wide association study in Korean women. Ann Rheum Dis. 2014;73:1240–1245. doi: 10.1136/annrheumdis-2012-202675. [DOI] [PubMed] [Google Scholar]
- 21.Bult CJ, Eppig JT, Kadin JA, Richardson JE, Blake JA. The Mouse Genome Database (MGD): mouse biology and model systems. Nucleic Acids Res. 2008;36:D724–D728. doi: 10.1093/nar/gkm961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Do R, Kathiresan S, Abecasis GR. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum Mol Genet. 2012;21:R1–R9. doi: 10.1093/hmg/dds387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, Christiani DC, Wurfel Mark M, Lin X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, Kathiresan S, Purcell SM, Roeder K, Daly MJ. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin D-Y, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopes MC, Joyce C, Ritchie GR, John SL, Cunningham F, Asimit J, Zeggini E. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51. doi: 10.1159/000334984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Q, Guo Y, Li J, Long J, Zhang B, Shyr Y. Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genomics. 2012;13:S8. doi: 10.1186/1471-2164-13-S8-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunt KA, Mistry V, Bockett NA, Ahmad T, Ban M, Barker JN, Barrett JC, Blackburn H, Brand O, Burren O, Capon F, Compston A, Gough SCL, Jostins L, Kong Y, Lee JC, Lek M, MacArthur DG, Mansfield JC, Mathew CG, Mein CA, Mirza M, Nutland S, Onengut-Gumuscu S, Papouli E, Parkes M, Rich SS, Sawcer S, Satsangi J, Simmonds MJ, et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature. 2013;498:232–235. doi: 10.1038/nature12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, Zhang Y, Wang P, Li T, Fu W, Mo X, Shi T, Zhang Z, Chen Y, Ma D, Han W. VSTM1-v2, a novel soluble glycoprotein, promotes the differentiation and activation of Th17 cells. Cell Immunol. 2012;278:136–142. doi: 10.1016/j.cellimm.2012.07.009. [DOI] [PubMed] [Google Scholar]