

Abstract
Phosphorylation of serine, threonine and tyrosine plays significant roles in cellular signal transduction and in modifying multiple protein functions. Phosphoproteins are coordinated and regulated by a network of kinases, phosphatases and phospho-binding proteins, which modify the phosphorylation states, recognize unique phosphopeptides, or target proteins for degradation. Detailed and complete information on the structure and dynamics of these networks is required to better understand fundamental mechanisms of cellular processes and diseases. High-throughput technologies have been developed to investigate phosphoproteomes in model organisms and human diseases. Among them, mass spectrometry (MS)-based technologies are the major platforms and have been widely applied, which has led to explosive growth of phosphoproteomic data in recent years. New bioinformatics tools are needed to analyze and make sense of these data. Moreover, most research has focused on individual phosphoproteins and kinases. To gain a more complete knowledge of cellular processes, systems biology approaches, including pathways and networks modeling, have to be applied to integrate all components of the phosphorylation machinery, including kinases, phosphatases, their substrates, and phospho-binding proteins. This review presents the latest developments of bioinformatics methods and attempts to apply systems biology to analyze phosphoproteomics data generated by MS-based technologies. Challenges and future directions in this field will be also discussed.
Keywords: Phosphoproteomics, Systems biology, Kinases, Phosphatases, Phospho-binding proteins, Phosphorylation network, Phospho-signaling networks, Mass spectrometry
The reversible phosphorylation of amino acid side chains is one of the most common and important post-translational modifications (PTM) of proteins. Current studies are mainly focused on phosphorylation of serine (Ser), threonine (Thr), and tyrosine (Tyr), though other amino acids can also be phosphorylated such as histidine (His), aspartate (Asp), cysteine (Cys), lysine (Lys) and arginine (Arg). The latter side chain modifications are much less studied due to either lower frequency or experimental difficulties. Protein phosphorylation can result in changes of protein structure, activation/inhibition of protein activities, and promotion/prevention of protein–protein interactions [1,2]. Consequently, protein phosphorylation is one of the key regulatory mechanisms inherent in many important cellular processes, such as cell signaling, growth, and proliferation while abnormal phosphorylation can lead to serious diseases, such as cancer, diabetes and rheumatoid arthritis [3–5]. Thus, a better understanding protein phosphorylation will help to improve our knowledge of important cellular processes, provide a better understanding of disease mechanism, and drive the development of efficient treatments and new biomarker strategies.
More than 30% of human proteins are seen to be phosphorylated, these modifications often occur on multiple distinct sites [6]. Most of phosphoproteins are at substoichiometric concentrations with respect to site occupancy, and the phosphorylation events are usually dynamic and transient. To systematically study phosphoproteins, several high-throughput phosphoproteomic technologies, such as reverse phase protein array, phospho-specific flow cytometry, and mass spectrometry (MS) based technologies have been developed as summarized in recent reviews [7,8]. Among them, MS based technologies have become major platforms that have been routinely applied to identify phosphoproteins and phosphosites at a global, unbiased, and quantifiable level [9–13]. In a typical MS-based phosphoproteomic experiment, the procedure can be divided in four stages: sample preparation, including cell fractionation and protein digestion; enrichment of phosphopeptides via affinity purification; analysis via liquid chromatography (LC) coupled with tandem MS; and finally, localization and quantification of phosphosites/phosphoproteins using bioinformatics approach (summarized in Fig. 1). Recently, several excellent articles have reviewed the technical details of phosphoproteomic experiments, i.e., the first three steps [14–16]. In this review, we focus on the latest developments of bioinformatics approaches to annotate and integrate phosphoproteomic data, including phosphosites identification, comparative study of phosphoproteins, and construction of phosphorylation networks.
Fig. 1.
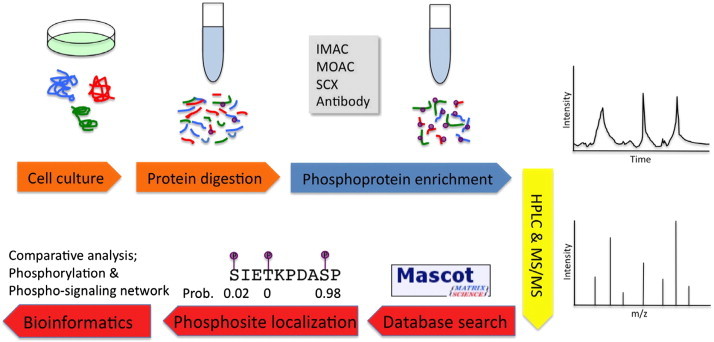
Summary of MS-based phosphoproteomics experiments.
Systems biology is a relative new field that studies the properties and models of biological systems from systematic measurements, i.e., the “omics” data from high-throughput experiments. Systems biology approaches aim to reveal the property and behavior of dynamic and complex biological systems in the system level instead of individual parts [17]. Network construction and modeling are very important parts of systems biology; while signal transduction plays essential roles in the regulation and coordination of networks, and is in many cases mediated by protein phosphorylation [4,5]. Protein kinases and phosphatases are essential components of phosphorylation process: kinases add phosphate groups to their substrates; while protein phosphatases facilitate the reverse reaction. Pathways for protein phosphorylation are large and interconnected networks, involving kinases, phosphatase and their substrates. Large amounts of phosphoproteomics data generated from high-throughput experiments make it possible to construct a range of cell type, tissue, organism, and disease-specific phosphorylation networks, and they have allowed us to investigate the functional phosphorylation associated signaling states. In this review, we will present the recent progress in this exciting field.
1. Phosphosite localization
MS/MS spectra generated from phosphoproteomics experiments are used to identify phosphopeptides by applying database search tools, such as MASCOT [18] and SEQUEST [19]. However, most search engines are not designed or optimized for identification of phosphopeptides, and they don't provide reliable confidence levels for the exact localization of possible phosphosites (i.e., the identification of exact amino acids phosphorylated). This becomes crucial if there are two or more potential phosphosites in detected peptides. The situation becomes even more complicated when the phosphopeptides have low abundance and when intense neutral loss peaks in the MS/MS spectra dominate signals of interest. Statistical methods have been developed to score the reliability of phosphosite localization using one of two following strategies as reviewed in [20]: estimating the probability of each candidate phosphosite based on site-determining ions in MS/MS spectra, examples include the popular A-score [21] and PTM score [9]. Alternate methods include calculating a score based on the difference of database search outputs between different site assignments for a given phosphopeptide, as implemented in Mascot Delta Score [22] and SLIP score [23]. In this section, we present details of these methods, recent developments, and results from the comparison of different phosphosite localization methods.
Gygi and colleagues proposed the A-score, a measurement of the confidence for the correct phosphosite localization in a given peptide that has two or more potential phosphosites [21]. First, based on the peptide sequence, fragment ions that are able to uniquely assign a specific phosphosite are identified and termed “site-determining ions”; then, in the corresponding MS/MS spectrum, the site-determining ions matched peaks are identified and counted for each possible phosphosite; the cumulative binomial probability is calculated using the total number of site-determining ions and the number of matched ions; finally, the probabilities for the top two candidates are used to calculate A-score by formula − 10 × log(P1) + 10 × log(P2), where Pi is the probability for the best two candidates [21]. The PTM sore is calculated in a very similar way [9], the major difference is the selection of ions: all detected ions are used to calculate PTM score; in the case of A-score only “site-determining ions” are used. Taus et al., observed that the density of peaks in different regions is different across each spectrum, and developed phosphoRS, which extends the A-score method by taking this into account [24].
More recently, Nesvizhskii and colleagues developed LuciPHOr, which uses mass accuracy (m/z) and peak intensities for phosphosite localization scoring [25]. LuciPHor is based on the principle that the distribution of peak intensity and m/z for random peaks and peaks with fragment ions are distinguishable, and an odds ratio can be calculated for each peak. There are four steps to compute LuciPHOr score: peak assignment, dynamic training, computing odds ratio, and score calculation. First, peaks with matched ions and random peaks are identified; then, distribution of the intensity and m/z for matched peaks and random peaks are established in the dynamic training step separately. The log odds of a matched peak can be computed using the determined distribution for each peak; a cumulative log odds for a spectrum (i.e. scores for all matched ion in a spectrum are summed up) can be assigned to each candidate site, and the final LuciPHOr score is the difference of cumulative log odds between the top two candidate sites. The authors claimed that LuciPHOr identified more correct phosphosites at equivalent false location rates compared to A-score and Mascot Delta Score [25].
The search engines utilized by phosphoproteomics consider all possible phosphosites in a peptide, and provide an identification score for each phosphopeptide. Thus, the score from a search engine can be used to estimate the site localization reliability. Beausoleil et al. first proposed the normalized Mascot delta score (i.e., the difference between the top two Mascot scores divided by the best Mascot score) for scoring the phosphosite localization and found that its performance was inferior compared with A-score [21]. However, Savitski et al. showed that the Mascot Delta score (MD-score, the difference between the top two Mascot scores) achieves similar performance with A-score, and the performance of normalized Mascot delta score is impaired by the low quality search results [22]. Methods for other search engine and engine-independent methods have been developed [23,26]. One serious drawback of this type method is that the scores of two possible phosphosites are very similar, even equal, for around 10% of the cases. Thus, this method cannot distinguish them.
The performances of different methods for phosphosite localization have been compared in several studies. Marx et al. showed that MD-score, phosphoRS and PTM score have overall similar accuracies, but the results are complementary among these approaches, which implies that there is still room for improvement [27]. This and other studies also demonstrated that localization methods have to be adjusted based on experimental designs (e.g., low vs. high mass accuracy or electron transfer dissociation vs. higher collisional dissociation) [25,27,28]. However, the performances of various phosphosite localization methods have not been validated by extensive experimental examination.
False localization rate (FLR) is a general and useful concept to measure the reliability of phosphosite localization though accurate estimation of the FLR is challenging because peptides can have significantly different uncertainty in phosphosite localization [20]. Furthermore, the incorrect phosphosite localizations are not randomly distributed across peptides; they are often near neighbors to the correct sites. Consequently, the use of decoy sequences, which are commonly used for measuring the reliability of peptide and protein identification, cannot provide accurate estimation for the error of phosphosite localization. To estimate these errors, approaches of phosphorylation estimation in silico have been developed, which computationally phosphorylates amino acid residues that typically do not occur in nature [23,25]. However, the field of FLR calculation is still in its early stages, and more study is critical for improved phosphoproteomics analyses.
2. Phosphoproteomics data resources
After phosphosites are determined by localization methods, many are deposited into public repositories. Due to recent advances in technologies, the amount of phosphoproteomics data in the public depositories has been growing exponentially in recent years [29]. In this section, we will present an overview of data resources in phosphoproteomics including phosphosite databases and phosphorylation networks.
Over the last decade, many public depositories have been developed to store phosphosite information, including Phospho.ELM [30] and phosphoSitePlus [31], which are commonly used in phosphoproteomics studies. Phospho.ELM contains experimentally identified phosphorylation sites in eukaryotic proteins; the current version (April, 2014) is 9.0, which contains 42,573 phosphosites for 8718 phosphoproteins. The current phosphoSitePlus has 144,042 phosphosites for 16,390 proteins, and it also includes other PTM sites, such as 12,300 acetylation sites. There are two major differences between these two resources. 1) Phospho.ELM focuses more on human: the proportions of protein instances are Homo sapiens (62%) and Mus musculus (16%), while the numbers are 51% and 42% for phosphoSitePlus. 2) phosphoSitePlus covers much more journals and papers (over 13,000 papers and 600 different journals), and it adds unpublished results from Cell Signaling Technology research groups. Some general databases also contain phosphorylation information for many proteins, such as HPRD [32] and UniProt [33], while databases for some specific organisms have also been developed, like PhosPhat for Arabidopsis thaliana [34], P3DB for six plants species [35] and phosphoPep for Drosophila [36]. Many post-translational modification (PTM) databases include phosphosite information since it is the most common PTM; examples are Phosida [37], SysPTM [38] and dbPTM [39].
Cataloging phosphosites can help to understand mechanisms of signaling and interactions; however other information is also needed, like the corresponding kinases and phosphatases that are active towards the known phosphosites. These can be used to create wide-ranging phosphorylation networks. The most reliable and extensive information to date is on kinase–substrate networks. High-throughput technologies and computational approaches have been developed to identify these relevant kinase–substrate relationships, and the results are deposited into network databases, like phosphoNetworks and phosphoPOINT [40–42]. PhosphoNetworks contains high-resolution phosphorylation networks derived from protein microarray and MS-based experiments, and it integrates a range of approaches to store, visualize, and analyze phosphorylation data [41]. The phosphorylation networks in PhosphoNetworks connect 230 human kinases with 2591 phosphorylation sites on 652 substrate proteins. Using a computational approach to integrate phosphoproteomics, protein interaction, and gene expression data, Yang et al. established phosphoPOINT, a database of the interactions among kinases, their potential substrates, and interacting proteins [42]. phosphoPOINT includes 4195 phospho-proteins, 518 kinases, and their corresponding protein–protein interaction (PPI) partners.
Complementary to identification of phosphosites by high-throughput experiments, computational methods have been previously developed to predict phosphosites [43]. Many state-of-the-art machine-learning approaches have been used to predict phosphosites, including artificial neural network [43], support vector machine [44], and random forest [45]. Besides protein sequences, some methods also used structural and evolutionary information to improve predictions [37,43,46]. The available methods and challenges in this field have been reviewed and compared for phosphorylation site prediction in recent articles [47,48].
Although phosphoproteomics data have increased dramatically in public repositories, it is important to note that the majority of the data are generated from high-throughput technologies and not validated by methods with higher accuracy. Thus, these databases thus have several problems associated with them aside from potentially inaccurate site identification. First, we do not know the proportion of phosphosites that are functionally active as opposed to sites that are modified by off-target effects of kinases. This is discussed extensively below. Second, phosphosite localization might not be correct if neighboring potential phosphosites exist. Third, there is minimal information on the stoichiometry of phosphosites in current databases, due to the difficulty of quantification. Such information is very important and can provide clues on the significance of the phosphorylation. High-throughput methods to measure phosphorylation stoichiometry have been proposed recently [49], but the performance has not been extensively examined.
3. Functionality and evolution of phosphosites
Over 150,000 phosphosites have been documented in databases, and we expect that the number will continue to increase rapidly. However, the functional effects of most of them have not been investigated with sufficient care to fully understand their significance. Recently, it was proposed that some phosphosites have little or even no biological functional effect at all [50]. This concept can be compared to that of “driver/passenger” mutations in cancer genome [51]. In this respect there may be “driver” phosphorylations that are most important for mediating functional changes as well as “passenger” phosphorylations, which occur due to the normal operation of the cellular machinery but have little or no effect on the regulatory functions. One of the arguments to support this proposal is the lack of evolutionary conservation for some phosphosites. This section presents the recent development on the comparative phosphoproteomics along with the debates about the non-functional phosphorylation.
Landry et al. compared the phosphoproteomes of yeasts and vertebrates and found that phosphoproteomes evolve rapidly in these two lineages, and phosphosites with known functions are much more conserved than those without annotated functions [52]. They estimated that around 65% of phosphosites is not conserved. By comparison, for human vs. mouse orthologs, another study reported that 10–15% phosphosites are found not to be under strong constraint [37]. It was proposed that the non-conserved phosphosites might be non-functional [50,52,53]. However, other studies demonstrate that the functional phosphosites might not be necessarily conserved, and rapidly evolving phosphoproteomes could play important roles in evolution [54]. Furthermore, evolutionary constraints might act at the phosphorylation network level, instead of at the level of individual phosphorylated sequences [55,56].
By analyzing 308 substrates of cyclin-dependent kinas Cdk1 in yeast, Holt et al. found that 90% of phosphosites is in loops and disordered regions of proteins, and the “phosphorylated regions” are conserved across yeast species (i.e., the same loop regions are phosphorylated), but the positions of phosphosites are much less constrained [54]. At least two major mechanisms have been proposed for phosphorylation to regulate protein function: conformation change and phosphopeptide binding [4], as illustrated in Fig. 2. Precise conformation changes require the exact sites to be phosphorylated (i.e., the phosphosites are well conserved across species). On the other hand, phosphopeptide-mediated binding mechanisms may not have such strong requirements and sequence alignments in fact are challenging in these regions. Thus, the authors proposed that Cdk1 modifies substrate functions by phosphopeptide-binding mechanisms: the addition of phosphates to a protein sub-domain disrupts or generates interactions with other proteins and the phosphorylation provides a general guide (or repulsion) for signaling the binding partner [54]. The authors also suggested that the less conserved phosphosites could help to generate new regulatory mechanisms [54].
Fig. 2.
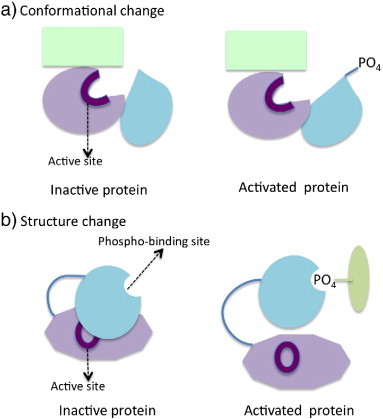
Mechanisms of modulating protein function by phosphorylation.
Tan et al. compared the human phosphoproteome with distantly related species (fly, worm and yeast) and identified 479 phosphosites that were conserved between human and at least one other species, which represents only 11% of 4448 human phosphosites [55]. The corresponding kinases for the 479 conserved phosphosites were identified using a computational approach [57], and conserved kinase–substrate networks (778 phosphorylation events in 698 human proteins) were constructed. Their results demonstrated that part of phosphorylation events is conserved at both sequence and network levels. However, although it is plausible that the conserved sites are functional, the converse is not necessarily true; i.e., the non-conserved sites are not necessarily nonfunctional. Nevertheless, their study suggests the importance of considering the phosphorylation networks instead of phosphosites alone.
4. Phosphorylation and phospho-signaling networks
Phosphorylation events can be portrayed as products of phosphorylation networks, which consist of kinases, their substrates, and their corresponding functional relationships. Many kinase substrates are also kinases, and in many cases, kinases are auto-phosphorylated. Thus, kinases are highly regulated by phosphorylation, and the phosphorylation networks are complex and an important part of signaling pathways. Numerous studies have attempted to construct and characterize phosphorylation networks using both experimental and computational approaches in model organisms and diseases [9,12,13,40,55,58–64]. Significant insights have been gained through these studies. For example, phosphoproteomics studies of stimulated HeLa cells demonstrated that protein phosphorylation can regulate the transcription levels of many downstream targets, and phosphosites are regulated differently within the same protein, suggesting that functional effects of phosphosites within a protein are site-dependent [9]. Another lung cancer phosphoproteomics study found different combination of activated kinases in different cancer patients, implying the importance of the personalized treatments [12]. In a recent review, Liu et al., summarized the methods to construct phosphorylation networks and outlined recent developments of identifying phospho-signatures from phosphorylation networks [65]. To avoid redundancy, this section will focus on recent developments of phosphorylation-associated signaling networks (phospho-signaling networks).
Phospho-signaling networks are the networks and pathways in which phosphorylation plays a significant role in mediation of the signal transduction. Phosphorylation networks are part of phospho-signaling networks in which there are multiple important components, such as protein kinases, phosphatases, and their substrates, and phospho-binding proteins. The relationship among kinases, phosphatases, and phospho-binding proteins is illustrated in Fig. 3. Phosphatases have the opposite function of kinases: they remove the phosphate group from phosphoproteins by hydrolyzing phosphoric acid monoesters into a phosphate ion and a molecule with a free hydroxyl group. Kinases have been the focus of research due to their importance in signaling, while phosphatases were considered more passive housekeeping enzymes and not as important as kinases. However, recent studies clearly show that both kinases and phosphatases are very important for signal transduction: in many cases, kinases control the amplitude of the signal while phosphatases govern the rate and duration of the signal [66–68]. Thus, phosphatase databases and phosphatase–substrate networks have been developed to facilitate the study of phospho-signaling networks [69–72]. Based on the substrates, phosphatases can be divided into tyrosine-specific, serine/threonine-specific, dual specificity phosphatases, and others such as lipid phosphatases. There are 226 human phosphatases known to date, and around 100 of them are classified as tyrosine-specific, implicating that phosphatases are important for phospho-tyrosine related signal transduction [67,69–71,73]. Interestingly, the number of known tyrosine kinases encoded in human genome is similar with the number of tyrosine phosphatases [1] (110 tyrosine kinase, and 100 tyrosine-specific phosphatases). By contrast, only 38 out of 226 human known phosphatases are serine/threonine-specific, but there are more than 400 kinases with known or predicted function to phosphorylate serine/threonine [70]. Protein phosphatase 1 (PP1) and protein phosphatase-2A (PP2A) are responsible for most dephosphorylation reactions counteracting the effects of hundreds of kinases. Their crucial roles in modifying protein functions and in phospho-signaling networks are now beginning to be appreciated [70]. The sequence analysis of phosphatase targets show that the + 1 position is preferred as proline for some phosphatase (CDC14A, CDC14B, SCP, PP5, PP2B, and PP2A), but it is not the exclusive substrate specificity: PP5, PP2A and PP2B recognize other motifs as well [70,74–76]. Furthermore, in general, the substrate specificity is weak for most phosphatases (particularly serine/threonine-specific phosphatases), suggesting the catalytic domains might be guided to act on their substrates by complex mechanisms [77], for example PP2A has multiple regulatory subunits that likely control specificity [78].
Fig. 3.
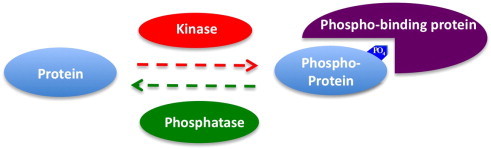
Relationships among kinase, phosphatase and phospho-binding protein. Note that the phosphoprotein and phospho-binding protein could also be a kinase or phosphatase, which makes for a quite sophisticated phospho-signaling network.
Phosphatases and kinases can share substrates and docking sites or even interact directly, and biological insights can be gained through investigating these relationships. For example, in a global protein kinase and phosphatase interaction network of budding yeast, phosphatase CDC14 interacts with around 20 kinases and five other phosphatases, indicating its significance for kinase signaling and DNA damage response [79]. Another study showed that CDC14A/B and cyclin-dependent kinases (CDKs) share several common substrates, confirming that CDC14A/B plays a role in cell cycle regulation [70]. In the case of common substrates, simulation studies demonstrated that the phosphorylation states can reach distinct steady states and the number of such states increases with the number of phosphosites, which highlights the effects of the interactions between kinases and phosphatases [80]. Furthermore, overlapping or common docking sites for kinase and phosphatases suggests a competition mechanism for regulating the phosphorylation states of the shared substrates [81–83].
Phospho-binding proteins are another important component of phospho-signaling networks. One characteristic feature of cellular signaling is the separation of enzyme activity from target recognition. Target recognition is often achieved by the cooperation of proteins with binding domains and proteins with target motif sequences. The Src homology-2 or SH2 domain is one well-characterized peptide-binding domain, and it recognizes and binds phosphotyrosine residues in specific sequence contexts [4]. There are 111 human proteins containing SH2 domains, which can be divided into 38 families based on sequence [84]. Other widespread phospho-binding domains include phosphotyrosine-binding (PTB), 14-3-3, and forkhead-associated (FHA) domains [85–87]. These proteins have diversified functions, such as kinase, phosphatase, transcription factor, and regulation of cytoskeletal architecture and cell adhesion. Thus, they play significant roles in phospho-signaling networks. Proteins without phospho-binding domains can also be affected by phosphorylation, which is the case for proteins with PDZ or SH3 domain. The recognition target of both domains has Ser or Thr, whose phosphorylation can influence the domain interactions and consequently result in uncoupling of signaling [88–90]. Thus, the study of phosphorylation on signaling should go beyond kinase/phosphatase and substrates and apply a systems biology approach that integrates multiple potential interactions, each of which contains a different slice of information [17].
Although the importance of both phosphatases and phospho-binding proteins in signaling pathways has been accepted in recent years [4,73], the studies are still far less comprehensive compared to those of kinases. To complete our knowledge of signaling processes and diseases mechanism, much more work on these important proteins are needed to identify their functions, substrates, and effects using systems biology approaches in particular. Systems biology approaches and the construction of phospho-signaling networks can also reveal the high level properties of systems, improve our understanding of diseases, and help develop new biomarkers and treatments. In a recent study, Koytiger et al. developed a protein microarray technology to systematically investigate interactions between human proteins with SH2 or PTB domain and receptor/adaptor proteins with phosphotyrosines [91,92]. They constructed a protein–protein interaction network, and found that it has a very high degree of connectivity, and many hub nodes are well-know oncogene, moreover, some of them are the primary targets of new anticancer drugs (e.g. EGFR, ERBB2, RET, KIT and ABL1) [92].
5. Integrating phosphoproteomics with other data
Various high throughput technologies have been developed and widely applied to profile different types of cellular components, such as microarray and next generation sequencing for DNA or mRNA, and high-resolution MS for proteins and protein–protein interactions. Combination of these technologies can generate various types of omics data from the same cell type or individual, and integrating diverse omics data can provide significant insights for the mechanism of cellular process or diseases [93,94]. However, integrating diverse type of data is always challenging due to the differences in the technological methods and complexity of cellular process, which include post transcription regulation and post-translational modification [95]. Recently, several studies proposed new methods to integrate phosphoproteomics with other data to investigate signaling pathways. In complex diseases like cancer, mutated genes can dysregulate several pathways simultaneously; this consequently leads to dramatic changes in levels of many genes/proteins, such as mRNA expression, protein abundance, and phosphorylation states [92,96,97]. To identify the dysregulated gene/proteins and pathways in cancer cells, Balbin et al. have attempted to integrate phosphoproteomics with transcriptomics, and proteomics [98]. They developed a novel method to identify proteins and pathways associated with mutated KRAS in non-small cell lung cancer (NSCLC) cell lines. To overcome the low overlap among data sets (only two differentially expressed proteins shared among three data sets at the level of corrected p-value 5%), they designed a metric S to identify transcripts, proteins and phosphoproteins with differential abundances uniquely or consistently seen across different data sets. First, the log-transformed fold changes (LFC) of each protein/gene was computed, and z-transformed for each data set separately. Then, the S score of one protein is the weighted sum of z-transformed LFC; while the weight is a function of the size of data set. Using this metric, 115 differentially abundant proteins were identified at adjusted P-value 5% level. Further pathway analyses based on these 115 proteins suggested an active and targetable subnetwork composing of KRAS, MET, LCK and PAK1 in KRAS-dependent NSCLC cell lines [98].
In another study, phosphosites detected by phosphoproteomics have been integrated with cancer genomics data to study the phosphorylation signaling in cancer. Reimand et al. developed ActiveDriver, a method to identify frequently mutated phosphosites and genes/proteins in cancer [99]. The method is based on linear regression and applies likelihood ratio test to identify genes that have phosphosites with unexpected high mutation rates. ActiveDriver was applied to 87,060 known phosphosites and mutation data from The Cancer Genomics Atlas (TCGA) and identified 150 known or candidate cancer genes whose phosphosites are affected by the mutations [100]. Further analysis showed that those mutations likely led to rewiring of the involved signaling pathways.
Phosphoproteomics has also been integrated with PPI network to reveal network properties of phosphoproteins. Yachie et al. integrated yeast phosphoproteome with protein structure, proteome abundance, and PPI networks to investigate the effects of phosphorylation on protein interaction patterns [101]. They suggest that phosphoproteins have more interactions than other proteins, and the interacting partners of phosphoproteins are more diversified, implying that phosphorylation makes a large contribution to PPI diversity and might have significant roles other than signaling. They also found that interacting proteins tend to be phosphorylated together most likely by the same kinase.
Phosphorylation is only one of dozens frequent PTMs; others include glycosylation, acetylation, methylation, ubiquitination, and SUMOylation. Recent studies revealed extensive crosstalk and interplay between phosphorylation and other PTMs [14,102], for example, the competition between phosphorylation and O-linked glycosylation for Ser and Thr [103–105]; and the promotion/inhibition of ubiquitination by phosphorylation [106]. Recent studies investigated the co-evolution and functional association of PTMs, and constructed a network of associated PTMs that regulates multiple functional states of proteins [107,108]. By integrating protein interaction networks with several PTMs, Woodsmith et al. found that multiple PTMs are enriched in some disordered regions, which have high mutation rate in cancer and can be important for cellular processes [109]. New high throughput methods have developed to enrich phosphorylation, ubiquitylation and/or acetylation for same sample, and enable the integrated analysis for different PTMs [110,111]. However, currently sensitive and robust high throughput approaches are available only for few types of PTM, new reliable methods are needed to study the interplay and crosstalk of all common PTMs.
6. Challenges and future directions
Several challenges still remain to be tackled in the phosphoproteomics field. Due to the limitation of current technologies, many phosphosites have yet to be discovered, and knowledge is limited for the majority of known phosphosites, e.g., lack of functional annotation and abundance information. Moreover, other type of phosphosites, such as phosphohistidine, is much less studied. Histidine phosphorylation was discovered five decades ago, reported to present in both prokaryotes and eukaryotes, and might be more common than phosphotyrosines [112]. Studies showed that it plays significant role in signaling processes in some species and is implicated in certain human diseases. Detection of phosphohistidine is challenging due to its chemical instability [113]. However, recent discovery of phosphohistidine antibodies might provide a valuable method to tackle this challenge [114].
Another challenge is the identification of functionally important phosphosites. Although the proposal of nonfunctional phosphorylation is controversial, some phosphosites likely have more important roles than others. Several computational methods have been developed to distinguish those impotent ones based on the protein structure, but their performance is hard to evaluate [115,116].
As mentioned above, the phospho-signaling network is composed of phosphoproteins, kinases, phosphatases, and phospho-binding proteins. Phosphoproteomics experiments provide large amounts of information about phosphoproteins but minimal information for other components. Computational methods based on motif sequences or PPI network have been developed for prediction of kinases that is associated with phosphosites [57,117], but the method is very limited for the phosphatase and phospho-binding proteins. Consequently, our knowledge of signaling process is incomplete and heavily biased. A lot more work is needed to elucidate the sequence motifs and other factors that affect substrate specificity for phosphatase and interacting partners of phosphoproteins, and these will be the basis for the development of computational methods for prediction.
Acknowledgments
The authors thank Zhenghe John Wang and Danica Wiredja for their comments on the manuscripts. This publication was made possible in part by the Clinical and Translational Science Collaborative of Cleveland (UL1-TR000439) from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research, a grant from the National Libraries of Medicine (R01-LM011247), a grant from the CWRU/UH Center for AIDS Research (P30-AI036219), and a grant from the National Cancer Institute (P30-CA-043703). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- 1.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P. The role of protein phosphorylation in human health and disease—delivered on June 30th 2001 at the FEBS Meeting in Lisbon. Eur J Biochem. 2001;268:5001–5010. doi: 10.1046/j.0014-2956.2001.02473.x. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T., Scott J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 5.Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 6.Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 7.Morris M.K., Chi A., Melas I.N., Alexopoulos L.G. Phosphoproteomics in drug discovery. Drug Discov Today. 2014;19:425–432. doi: 10.1016/j.drudis.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Pierobon M., Wulfkuhle J., Liotta L., Petricoin E. Application of molecular technologies for phosphoproteomic analysis of clinical samples. Oncogene. 2014 doi: 10.1038/onc.2014.16. (in press) [DOI] [PubMed] [Google Scholar]
- 9.Olsen J.V., Blagoev B., Gnad F., Macek B., Kumar C. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 10.Dephoure N., Zhou C., Villen J., Beausoleil S.A., Bakalarski C.E. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayya V., Lundgren D.H., Hwang S.I., Rezaul K., Wu L. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein–protein interactions. Sci Signal. 2009;2:ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- 12.Rikova K., Guo A., Zeng Q., Possemato A., Yu J. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 13.Linding R., Jensen L.J., Ostheimer G.J., van Vugt M.A., Jorgensen C. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roux P.P., Thibault P. The coming of age of phosphoproteomics—from large data sets to inference of protein functions. Mol Cell Proteomics. 2013;12:3453–3464. doi: 10.1074/mcp.R113.032862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Junger M.A., Aebersold R. Mass spectrometry-driven phosphoproteomics: patterning the systems biology mosaic. Wiley Interdiscip Rev Dev Biol. 2014;3:83–112. doi: 10.1002/wdev.121. [DOI] [PubMed] [Google Scholar]
- 16.Kanshin E., Michnick S., Thibault P. Sample preparation and analytical strategies for large-scale phosphoproteomics experiments. Semin Cell Dev Biol. 2012;23:843–853. doi: 10.1016/j.semcdb.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Chuang H.Y., Hofree M., Ideker T. A decade of systems biology. Annu Rev Cell Dev Biol. 2010;26(26):721–744. doi: 10.1146/annurev-cellbio-100109-104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perkins D.N., Pappin D.J.C., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 19.Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 20.Chalkley R.J., Clauser K.R. Modification site localization scoring: strategies and performance. Mol Cell Proteomics. 2012;11:3–14. doi: 10.1074/mcp.R111.015305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beausoleil S.A., Villen J., Gerber S.A., Rush J., Gygi S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 22.Savitski M.M., Lemeer S., Boesche M., Lang M., Mathieson T. Confident phosphorylation site localization using the Mascot Delta Score. Mol Cell Proteomics. 2011;10(M110):003830. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker P.R., Trinidad J.C., Chalkley R.J. Modification site localization scoring integrated into a search engine. Mol Cell Proteomics. 2011;10(M111):008078. doi: 10.1074/mcp.M111.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taus T., Kocher T., Pichler P., Paschke C., Schmidt A. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 25.Fermin D., Walmsley S.J., Gingras A.C., Choi H., Nesvizhskii A.I. LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using modified target–decoy approach. Mol Cell Proteomics. 2013;12:3409–3419. doi: 10.1074/mcp.M113.028928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaudel M., Breiter D., Beck F., Rahnenfuhrer J., Martens L. D-score: a search engine independent MD-score. Proteomics. 2013;13:1036–1041. doi: 10.1002/pmic.201200408. [DOI] [PubMed] [Google Scholar]
- 27.Marx H., Lemeer S., Schliep J.E., Matheron L., Mohammed S. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat Biotechnol. 2013;31:557–564. doi: 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- 28.Wiese H., Kuhlmann K., Wiese S., Stoepel N.S., Pawlas M. Comparison of alternative MS/MS and bioinformatics approaches for confident phosphorylation site localization. J Proteome Res. 2014;13(2):1128–1137. doi: 10.1021/pr400402s. [DOI] [PubMed] [Google Scholar]
- 29.Lemeer S., Heck A.J. The phosphoproteomics data explosion. Curr Opin Chem Biol. 2009;13:414–420. doi: 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 30.Dinkel H., Chica C., Via A., Gould C.M., Jensen L.J. Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S. Human protein reference database—2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apweiler R., Bateman A., Martin M.J., O'Donovan C., Magrane M. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durek P., Schmidt R., Heazlewood J.L., Jones A., MacLean D. PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Res. 2010;38:D828–D834. doi: 10.1093/nar/gkp810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J., Agrawal G.K., Thelen J.J., Xu D. P3DB: a plant protein phosphorylation database. Nucleic Acids Res. 2009;37:D960–D962. doi: 10.1093/nar/gkn733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodenmiller B., Malmstrom J., Gerrits B., Campbell D., Lam H. PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol Syst Biol. 2007;3:139. doi: 10.1038/msb4100182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gnad F., Ren S., Cox J., Olsen J.V., Macek B. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007;8:R250. doi: 10.1186/gb-2007-8-11-r250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Xing X., Ding G., Li Q., Wang C. SysPTM: a systematic resource for proteomic research on post-translational modifications. Mol Cell Proteomics. 2009;8:1839–1849. doi: 10.1074/mcp.M900030-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu C.T., Huang K.Y., Su M.G., Lee T.Y., Bretana N.A. DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013;41:D295–D305. doi: 10.1093/nar/gks1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newman R.H., Hu J., Rho H.S., Xie Z., Woodard C. Construction of human activity-based phosphorylation networks. Mol Syst Biol. 2013;9:655. doi: 10.1038/msb.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J., Rho H.S., Newman R.H., Zhang J., Zhu H. PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics. 2014;30:141–142. doi: 10.1093/bioinformatics/btt627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang C.Y., Chang C.H., Yu Y.L., Lin T.C., Lee S.A. PhosphoPOINT: a comprehensive human kinase interactome and phospho-protein database. Bioinformatics. 2008;24:i14–i20. doi: 10.1093/bioinformatics/btn297. [DOI] [PubMed] [Google Scholar]
- 43.Blom N., Gammeltoft S., Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 44.Dou Y., Yao B., Zhang C. PhosphoSVM: prediction of phosphorylation sites by integrating various protein sequence attributes with a support vector machine. Amino Acids. 2014;46:1459–1469. doi: 10.1007/s00726-014-1711-5. [DOI] [PubMed] [Google Scholar]
- 45.Trost B., Kusalik A. Computational phosphorylation site prediction in plants using random forests and organism-specific instance weights. Bioinformatics. 2013;29:686–694. doi: 10.1093/bioinformatics/btt031. [DOI] [PubMed] [Google Scholar]
- 46.Brinkworth R.I., Breinl R.A., Kobe B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc Natl Acad Sci U S A. 2003;100:74–79. doi: 10.1073/pnas.0134224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trost B., Kusalik A. Computational prediction of eukaryotic phosphorylation sites. Bioinformatics. 2011;27:2927–2935. doi: 10.1093/bioinformatics/btr525. [DOI] [PubMed] [Google Scholar]
- 48.Xue Y., Gao X., Cao J., Liu Z., Jin C. A summary of computational resources for protein phosphorylation. Curr Protein Pept Sci. 2010;11:485–496. doi: 10.2174/138920310791824138. [DOI] [PubMed] [Google Scholar]
- 49.Wu R., Haas W., Dephoure N., Huttlin E.L., Zhai B. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods. 2011;8:677–683. doi: 10.1038/nmeth.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lienhard G.E. Non-functional phosphorylations? Trends Biochem Sci. 2008;33:351–352. doi: 10.1016/j.tibs.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Marx V. Cancer genomes: discerning drivers from passengers. Nat Methods. 2014;11:375–379. doi: 10.1038/nmeth.2891. [DOI] [PubMed] [Google Scholar]
- 52.Landry C.R., Levy E.D., Michnick S.W. Weak functional constraints on phosphoproteomes. Trends Genet. 2009;25:193–197. doi: 10.1016/j.tig.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Levy E.D., Michnick S.W., Landry C.R. Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Philos Trans R Soc Lond B Biol Sci. 2012;367:2594–2606. doi: 10.1098/rstb.2012.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holt L.J., Tuch B.B., Villen J., Johnson A.D., Gygi S.P. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan C.S., Bodenmiller B., Pasculescu A., Jovanovic M., Hengartner M.O. Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal. 2009;2:ra39. doi: 10.1126/scisignal.2000316. [DOI] [PubMed] [Google Scholar]
- 56.Tan C.S., Jorgensen C., Linding R. Roles of “junk phosphorylation” in modulating biomolecular association of phosphorylated proteins? Cell Cycle. 2010;9:1276–1280. doi: 10.4161/cc.9.7.11066. [DOI] [PubMed] [Google Scholar]
- 57.Miller M.L., Jensen L.J., Diella F., Jorgensen C., Tinti M. Linear motif atlas for phosphorylation-dependent signaling. Sci Signal. 2008;1:ra2. doi: 10.1126/scisignal.1159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drake J.M., Graham N.A., Stoyanova T., Sedghi A., Goldstein A.S. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A. 2012;109:1643–1648. doi: 10.1073/pnas.1120985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koivomagi M., Ord M., Iofik A., Valk E., Venta R. Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol. 2013;20:1415–1424. doi: 10.1038/nsmb.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song C.X., Ye M.L., Liu Z.X., Cheng H., Jiang X. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol Cell Proteomics. 2012;11:1070–1083. doi: 10.1074/mcp.M111.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y., Kweon H.K., Shively C., Kumar A., Andrews P.C. Towards systematic discovery of signaling networks in budding yeast filamentous growth stress response using interventional phosphorylation data. PLoS Comput Biol. 2013;9 doi: 10.1371/journal.pcbi.1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Damle N.P., Mohanty D. Deciphering kinase–substrate relationships by analysis of domain-specific phosphorylation network. Bioinformatics. 2014;30(12):1730–1738. doi: 10.1093/bioinformatics/btu112. [DOI] [PubMed] [Google Scholar]
- 64.Varjosalo M., Keskitalo S., Van Drogen A., Nurkkala H., Vichalkovski A. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. doi: 10.1016/j.celrep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 65.Liu Z., Wang Y., Xue Y. Phosphoproteomics-based network medicine. FEBS J. 2013;280:5696–5704. doi: 10.1111/febs.12380. [DOI] [PubMed] [Google Scholar]
- 66.Hornberg J.J., Bruggeman F.J., Binder B., Geest C.R., de Vaate A.J. Principles behind the multifarious control of signal transduction. ERK phosphorylation and kinase/phosphatase control. FEBS J. 2005;272:244–258. doi: 10.1111/j.1432-1033.2004.04404.x. [DOI] [PubMed] [Google Scholar]
- 67.Tonks N.K. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 68.Heinrich R., Neel B.G., Rapoport T.A. Mathematical models of protein kinase signal transduction. Mol Cell. 2002;9:957–970. doi: 10.1016/s1097-2765(02)00528-2. [DOI] [PubMed] [Google Scholar]
- 69.Liberti S., Sacco F., Calderone A., Perfetto L., Iannuccelli M. HuPho: the human phosphatase portal. FEBS J. 2013;280:379–387. doi: 10.1111/j.1742-4658.2012.08712.x. [DOI] [PubMed] [Google Scholar]
- 70.Li X., Wilmanns M., Thornton J., Kohn M. Elucidating human phosphatase–substrate networks. Sci Signal. 2013;6:rs10. doi: 10.1126/scisignal.2003203. [DOI] [PubMed] [Google Scholar]
- 71.Sacco F., Perfetto L., Castagnoli L., Cesareni G. The human phosphatase interactome: an intricate family portrait. FEBS Lett. 2012;586:2732–2739. doi: 10.1016/j.febslet.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hatzihristidis T., Liu S., Pryszcz L., Hutchins A.P., Gabaldon T. PTP-central: a comprehensive resource of protein tyrosine phosphatases in eukaryotic genomes. Methods. 2014;65:156–164. doi: 10.1016/j.ymeth.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 73.Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 74.Gray C.H., Good V.M., Tonks N.K., Barford D. The structure of the cell cycle protein Cdc14 reveals a proline-directed protein phosphatase. EMBO J. 2003;22:3524–3535. doi: 10.1093/emboj/cdg348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ham B.M., Jayachandran H., Yang F., Jaitly N., Polpitiya A.D. Novel Ser/Thr protein phosphatase 5 (PP5) regulated targets during DNA damage identified by proteomics analysis. J Proteome Res. 2010;9:945–953. doi: 10.1021/pr9008207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ulloa L., Dombradi V., Diaznido J., Szucs K., Gergely P. Dephosphorylation of distinct sites on microtubule-associated protein Map1b by protein phosphatases 1, 2a and 2b. FEBS Lett. 1993;330:85–89. doi: 10.1016/0014-5793(93)80925-k. [DOI] [PubMed] [Google Scholar]
- 77.Tiganis T., Bennett A.M. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seshacharyulu P., Pandey P., Datta K., Batra S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Breitkreutz A., Choi H., Sharom J.R., Boucher L., Neduva V. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thomson M., Gunawardena J. Unlimited multistability in multisite phosphorylation systems. Nature. 2009;460:274–277. doi: 10.1038/nature08102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirschi A., Cecchini M., Steinhardt R.C., Schamber M.R., Dick F.A. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat Struct Mol Biol. 2010;17:1051–1057. doi: 10.1038/nsmb.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanoue T., Adachi M., Moriguchi T., Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 83.Salazar C., Hofer T. Competition effects shape the response sensitivity and kinetics of phosphorylation cycles in cell signaling. Ann N Y Acad Sci. 2006;1091:517–530. doi: 10.1196/annals.1378.093. [DOI] [PubMed] [Google Scholar]
- 84.Liu B.A., Shah E., Jablonowski K., Stergachis A., Engelmann B. The SH2 domain-containing proteins in 21 species establish the provenance and scope of phosphotyrosine signaling in eukaryotes. Sci Signal. 2011;4:ra83. doi: 10.1126/scisignal.2002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yaffe M.B. Phosphotyrosine-binding domains in signal transduction. Nat Rev Mol Cell Biol. 2002;3:177–186. doi: 10.1038/nrm759. [DOI] [PubMed] [Google Scholar]
- 86.Yaffe M.B., Elia A.E. Phosphoserine/threonine-binding domains. Curr Opin Cell Biol. 2001;13:131–138. doi: 10.1016/s0955-0674(00)00189-7. [DOI] [PubMed] [Google Scholar]
- 87.Gong W., Zhou D., Ren Y., Wang Y., Zuo Z. PepCyber:P ~ PEP: a database of human protein protein interactions mediated by phosphoprotein-binding domains. Nucleic Acids Res. 2008;36:D679–D683. doi: 10.1093/nar/gkm854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen N.A., Brenman J.E., Snyder S.H., Bredt D.S. Binding of the inward rectifier K + channel Kir 2.3 to PSD-95 is regulated by protein kinase A phosphorylation. Neuron. 1996;17:759–767. doi: 10.1016/s0896-6273(00)80207-x. [DOI] [PubMed] [Google Scholar]
- 89.Chen D., Waters S.B., Holt K.H., Pessin J.E. SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J Biol Chem. 1996;271:6328–6332. doi: 10.1074/jbc.271.11.6328. [DOI] [PubMed] [Google Scholar]
- 90.Kuriyan J., Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 91.Kaushansky A., Allen J.E., Gordus A., Stiffler M.A., Karp E.S. Quantifying protein–protein interactions in high throughput using protein domain microarrays. Nat Protoc. 2010;5:773–790. doi: 10.1038/nprot.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koytiger G., Kaushansky A., Gordus A., Rush J., Sorger P.K. Phosphotyrosine signaling proteins that drive oncogenesis tend to be highly interconnected. Mol Cell Proteomics. 2013;12:1204–1213. doi: 10.1074/mcp.M112.025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen R., Mias G.I., Li-Pook-Than J., Jiang L., Lam H.Y. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang S.S.C., Clarke D.C., Gosline S.J.C., Labadorf A., Chouinard C.R. Linking proteomic and transcriptional data through the interactome and epigenome reveals a map of oncogene-induced signaling. PLoS Comput Biol. 2013;9 doi: 10.1371/journal.pcbi.1002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Landry C.R., Levy E.D., Abd Rabbo D., Tarassov K., Michnick S.W. Extracting insight from noisy cellular networks. Cell. 2013;155:983–989. doi: 10.1016/j.cell.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 96.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carretero J., Shimamura T., Rikova K., Jackson A.L., Wilkerson M.D. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Balbin O.A., Prensner J.R., Sahu A., Yocum A., Shankar S. Reconstructing targetable pathways in lung cancer by integrating diverse omics data. Nat Commun. 2013;4:2617. doi: 10.1038/ncomms3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reimand J., Bader G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reimand J., Wagih O., Bader G.D. The mutational landscape of phosphorylation signaling in cancer. Sci Rep. 2013;3:2651. doi: 10.1038/srep02651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yachie N., Saito R., Sugiyama N., Tomita M., Ishihama Y. Integrative features of the yeast phosphoproteome and protein–protein interaction map. PLoS Comput Biol. 2011;7:e1001064. doi: 10.1371/journal.pcbi.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Venne A.S., Kollipara L., Zahedi R.P. The next level of complexity: crosstalk of posttranslational modifications. Proteomics. 2014;14:513–524. doi: 10.1002/pmic.201300344. [DOI] [PubMed] [Google Scholar]
- 103.Zeidan Q., Hart G.W. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22. doi: 10.1242/jcs.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Butt A.M., Khan I.B., Hussain M., Idress M., Lu J. Role of post translational modifications and novel crosstalk between phosphorylation and O-beta-GlcNAc modifications in human claudin-1,-3 and -4. Mol Biol Rep. 2012;39:1359–1369. doi: 10.1007/s11033-011-0870-7. [DOI] [PubMed] [Google Scholar]
- 105.Kamemura K., Hayes B.K., Comer F.I., Hart G.W. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J Biol Chem. 2002;277:19229–19235. doi: 10.1074/jbc.M201729200. [DOI] [PubMed] [Google Scholar]
- 106.Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 107.Minguez P., Parca L., Diella F., Mende D.R., Kumar R. Deciphering a global network of functionally associated post-translational modifications. Mol Syst Biol. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beltrao P., Bork P., Krogan N.J., van Noort V. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol. 2013;9:714. doi: 10.1002/msb.201304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Woodsmith J., Kamburov A., Stelzl U. Dual coordination of post translational modifications in human protein networks. PLoS Comput Biol. 2013;9:e1002933. doi: 10.1371/journal.pcbi.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Swaney D.L., Beltrao P., Starita L., Guo A., Rush J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat Methods. 2013;10:676–682. doi: 10.1038/nmeth.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mertins P., Qiao J.W., Patel J., Udeshi N.D., Clauser K.R. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ciesla J., Fraczyk T., Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol. 2011;58:137–148. [PubMed] [Google Scholar]
- 113.Kee J.M., Muir T.W. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem Biol. 2012;7:44–51. doi: 10.1021/cb200445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kee J.M., Oslund R.C., Perlman D.H., Muir T.W. A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat Chem Biol. 2013;9:416–421. doi: 10.1038/nchembio.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beltrao P., Albanese V., Kenner L.R., Swaney D.L., Burlingame A. Systematic functional prioritization of protein posttranslational modifications. Cell. 2012;150:413–425. doi: 10.1016/j.cell.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Akiva E., Friedlander G., Itzhaki Z., Margalit H. A dynamic view of domain–motif interactions. PLoS Comput Biol. 2012;8:e1002341. doi: 10.1371/journal.pcbi.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Linding R., Jensen L.J., Pasculescu A., Olhovsky M., Colwill K. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008;36:D695–D699. doi: 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]